

Abstract
Background: Germline mutations in mismatch repair (MMR) genes are found in only about half of clinically diagnosed families with hereditary non-polyposis colorectal cancer syndrome (HNPCC) (or Lynch syndrome). Early identification of gene carriers is essential to reduce cancer incidence and overall mortality.
Aims: Recent evidence indicates an increase in size and number of sebaceous glands following activation of the hedgehog pathway, a crucial signalling pathway for animal development that is aberrantly activated in several types of cancer. Here we sought to assess a possible association between Fordyce granules (FGs—that is, ectopic sebaceous glands on the oral mucosa) and HNPCC.
Methods: A total of 15 members of five different genetically unrelated HNPCC kindreds (MLH1 gene mutation n = 8; undetectable MLH1 protein at immunochemistry n = 4; clinical diagnosis n = 3) and 630 genetically unrelated age and sex matched healthy controls were examined. Following examination of the oral mucosa surface, subjects were categorised as either FGs positive or FGs negative.
Results: Evidence of FGs was significantly associated with HNPCC (13/15 (86.7%) affected patients v 6/630 (0.95%) controls; p<0.0001), with a relative risk of 91.0 (95% confidence interval 40.05–206.76). The observed difference remained significant when carriers of germline mutations in MMR genes were considered (8/15 v 6/630; p<0.0001). The most common site for the FGs in HNPCC patients was the lower gingival and vestibular oral mucosa.
Conclusions: Our findings suggest that a previously unrecognised activation of the sebaceous glands system occurs in HNPCC. The observation could be of value for attending physicians in identifying affected families and/or increase the accuracy of the currently available molecular genetics screenings.
Keywords: colorectal neoplasms, hereditary non-polyposis/diagnosis, fordyce granules, sebaceous glands, physical examination
Hereditary non-polyposis colorectal cancer syndrome (HNPCC) (or Lynch syndrome) is the most common form of hereditary colorectal cancer (CRC),1,2 caused by autosomal dominantly inherited germline mutations in mismatch repair (MMR) genes. Mutation carriers have a lifetime risk of >80% of developing CRC, together with an excess of extracolonic cancers at an early age.3,4MLH1 and MSH2 accounts for almost 90% of identified mutations while MSH6 accounts for approximately 10%; mutations in PMS2, MLH3, and EXO1 have also been reported.1,2 Early identification of HNPCC gene carriers is essential to reduce cancer incidence and overall mortality.1,3,4 However, the germline mutations in MMR genes can be established in only about half of clinically diagnosed HNPCC families,1 while with the single exception of the rare Muir-Torre syndrome (MTS) variant (OMIM #158230),5,6 no obvious phenotypical physical stigmata aiding in the diagnosis exist. As a consequence, the actual frequency of HNPCC in the general population remains unknown. Theoretical reasons indicate a prevalence of 1:350 to 1:1700, while more conservative estimates are 2–7% of the total CRC cases, which suggests that HNPCC is currently underdiagnosed,4 with devastating clinical consequences.
Fordyce granules (FGs)7,8 (that is, intraoral ectopic sebaceous glands) are thought to be benign common lesions on the oral mucosa of adults subjects. Their prevalence rates increase from childhood (0.2%)9 to adulthood (0.97%), and rates are slightly higher in adult males (1.77%) than females (0.52%).10 No correlations between FGs and systemic diseases are known to date. Recent evidence indicates an increase in size and number of sebaceous glands following activation of the hedgehog (Hh) pathway,11 a crucial signalling pathway for animal development that is aberrantly activated in several types of cancer.12,13 Here we sought to assess a possible association between HNPCC and FGs.
METHODS
A total of 15 surviving members of five different genetically unrelated HNPCC kindreds (seven males, eight females; age at examination 53.7 (SD15.1) years; family 1, n = 4; family 2, n = 4; family 3, n = 1; family 4, n = 3; family 5, n = 3) originating from the Taranto and Brindisi areas of southern Italy were examined. All families fulfilled the Amsterdam criteria, and all patients had a history of proven cancer at the time of examination (CRC, n = 11 (73.3%); other cancers, n = 4).
Given the specific aim of the present study, only subjects who agreed to undergo HNPCC genetic molecular studies were invited to participate, while subjects with undetermined disease status were excluded.
Mean age at cancer onset was 41.5 years (SD14.1). High molecular weight DNA was extracted from frozen tissues or blood according to standard methods. Microsatellite instability (MSI) was assessed using five polymorphic markers: BAT25, BAT26, D2S123, D5S346, and D18S858. MSI was considered to be present when two or more of the five markers showed instability. The promoter regions and each exon of the MLH1 and MSH2 genes from genomic DNA were individually amplified and subsequently sequenced directly. Germline mutations in MMR genes were defined according to standard nomenclature recommendations.14
Immunohistochemical analysis was performed as previously described.15 The panel of immunostaining encompassed MSH2 (FE11, diluted 1:200; Oncogene Research Products, Boston, Massachusetts, USA) and MLH1 (G168–728, diluted 1:100; Pharmingen, San Diego, California, USA). Sections (5 µm) containing both tumour tissue and normal colonic mucosa (as internal control) were cut. After deparaffination and rehydration, including a step of endogenous peroxidase block with methanol-peroxidase for 30 minutes, sections were microwaved for non-enzymatic epitope retrieval in citrate buffer solution (pH 6.0) at 750 W for five minutes (three times). Slides were left to cool in the buffer solution for 20 minutes. Monoclonal antibodies anti-MSH2 and anti-MLH1 were incubated for 60 minutes. Slides were incubated with the primary antibodies for 30 minutes. Immunostaining was carried out using the streptavidin-biotin-peroxidase amplification system in an automatic immunostainer (Techmate 500; Dako, Glostrup, Denmark). CRCs were judged to be negative for expression only if they lacked staining in a sample in which normal colonocytes and stroma were stained. The results were not considered in the event of no immunostaining of normal tissues.
A total of 630 genetically unrelated age and sex matched healthy controls (300 males, 330 females; mean age 54.0 (SD 16.1) years) were also recruited. The oral mucosa surface was examined by an experienced odontostomatologist, who was unaware of the clinical and genetic data of the subjects, and categorised as either FGs positive or FGs negative. No cases of CRCs were present in the family history and pedigrees of the controls. High resolution photographs of the selected areas were acquired using either a Yashica Dental Eye photocamera with an automated on-axis flashbulb, emitting standard photographic white light, and a 55 mm, f 1:4 Yashica lens (Yashica-Kyocera Co., Kyoto, Japan) or a Nikon Coolpix 4500 digital camera (4 Mpixels, 4× optical + 4× digital zoom, 16 Mb card; Nikon, Japan). We obtained approval from our local human investigation committee, and written informed consent for examination and photographic documentation from all subjects before inclusion in the study.
Data analysis
A coefficient of agreement for nominal scales (κ)16 was applied to assess interobserver variations in detection of FGs. A κ value <0.20 was rated as poor inter-rater agreement, 0.21–0.40 as fair, 0.41–0.60 as moderate, 0.61–0.80 as good, and 0.81–1.0 as very good.17
Differences in the proportions of FGs between HNPCC patients or carriers of germline mutations in MMR genes versus controls were assessed using the χ2 statistic, and relative risk was calculated. The effects of population size on type I (α) (that is, the probability of rejecting a true null hypothesis) and type II (β) (that is, the probability of accepting a false null hypothesis) statistical errors in the data interpretation were examined using a sampling size algorithm. Given the reported proportions of FGs in the HNPCC and control subjects, the minimal required sample size was n = 5 for each group for type I/α and type II/β statistical errors of p = 0.01. A two sided p value of <0.05 was considered to be statistically significant. The MedCalc version 7.5 statistical software package (MedCalc Software, Mariakerke, Belgium) was used.
RESULTS
Germline mutations in MMR genes were detectable in 8/15 cases (53.5%) (that is, 3/5 families) and identified as MLH1 mutations, including the c.210_213delAGAA in exon 3 (n = 3 patients) and c.545+1G>A in intron 6 (n = 5 patients) mutations. MLH1 protein was undetectable at immunohistochemistry in four patients (26.7%) and the diagnosis was clinical in three patients (20%). MSI was present in 10/11 examined CRCs (90.9%).
Clinical examination was found to be reproducible in identifying FGs, with a κ value for interobserver agreement (mean (SEM)) of 0.936 (0.045) (95% confidence interval (CI) 0.847–1.0), indicating a high degree of concordance. FGs (fig 1 ▶) were evident in 13/15 (86.7%) affected patients versus 6/630 (0.95%) controls (difference = 77.18%; 95% CI 59.84–94.51; χ2 = 79.45, df = 1; p<0.0001), with a relative risk of 91.0 (95% CI 40.05–206.76). The difference in FGs proportions remained significant only when carriers of germline mutations in MMR genes were accounted for (8/15 HNPCC v 6/630 controls; difference = 43.81%, 95% CI 18.46–69.16, χ2 = 25.36, df = 1; p<0.0001; relative risk 56.0, 95% CI 22.17–141.42).
Figure 1.
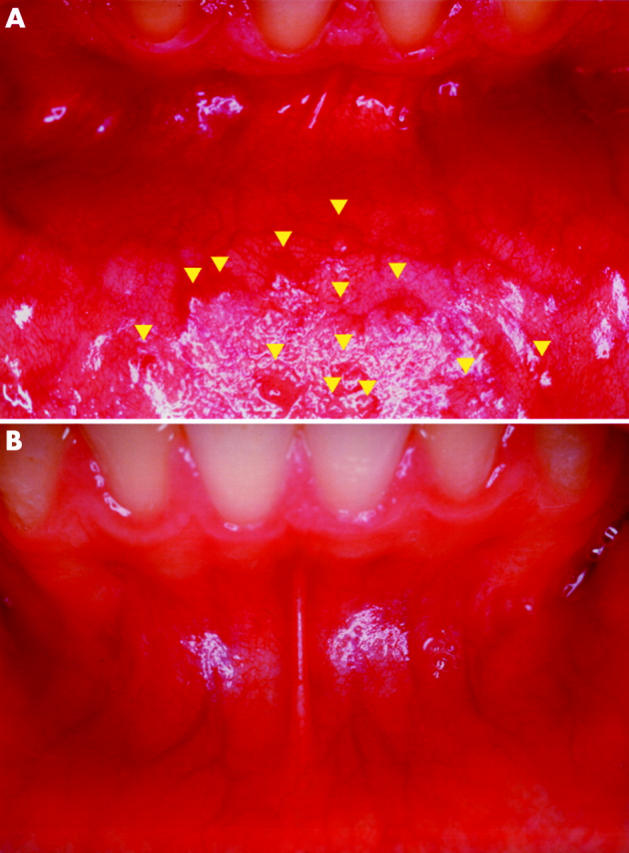
(A) Fordyce granules (FGs) (arrowheads) in the oral vestibular mucosa of a patient with hereditary non-polyposis colorectal cancer carrying the c.545+1G>A in intron 6 MLH1 germline mutation. FGs appear as rice-like yellow-white papules with a maximum diameter of ∼1–3 mm, surrounded by normal mucosa. Local elevation of the epithelium is clearly recognisable. (B) Corresponding oral vestibular mucosa in a healthy control subject.
Both FGs negative HNPCC patients had a clinical diagnosis, without detectable germline mutations in MMR genes. Maximum diameter of FGs was estimated as 1.54 (0.64) mm (95% CI 1.33–1.74 mm; range 0.8–3.0 mm), with the most common site for FGs being the lower gingival and vestibular oral mucosa in HNPCC patients compared with the mandibular retromolar pad and the upper lip vermilion in controls. Sensitivity and specificity of FGs as a diagnostic test for HNPCC were 86.7% (95% CI 59.5–98) and 99% (95% CI 97.9–99.6) for cases and controls. Positive and negative predictive values were 68.4% and 99.7%, respectively.
DISCUSSION
Our findings suggest that FGs may be associated with systemic disease, and that a previously unrecognised activation of the sebaceous glands system may occur in individuals carrying HNPCC gene mutations and/or patients with Amsterdam criteria positive CRC. This observation may be of help in identifying affected families. In the present study, the frequency of recognisable FGs was found to be approximately 50–90-fold higher in HNPCC patients than in controls.
At present, FGs are thought to be benign lesions on the oral surface in healthy individuals.8,9,10 Demonstration of cancers originating from FGs is rarely reported in the literature.18 The average frequency of FGs has been reported to increase by ∼4.85-fold from childhood9 to adulthood.10 The mechanisms underlying the age dependent changes in the frequency of FGs remain unclear. As a consequence, the frequency of the observed FGs sign as a function of age at examination and/or development of CRC in HNPCC patients needs further investigation.
Moreover, the question of the biological specificity of the FGs sign in HNPCC remains to be addressed. Evidence of FGs in CRC negative HNPCC individuals and lack of FGs in patients with sporadic (Amsterdam negative) CRCs (S Parrini et al, unpublished data) seem to suggest a link between ectopic sebaceous glands and the MMR gene defect, rather than simple coexistence of the CRC itself.
The biological significance of the observed clinical association remains unclear to date. However, Hh, a signalling pathway regulating a variety of developmental processes, including vasculogenesis, has been shown to increase the size and number of sebaceous glands.11,12 Thus a conceivable working hypothesis is that Hh pathway activation may coexist in HNPCC, leading to sebaceous gland development, and recognisable FGs. In this regard, a specific role for the Hh signalling pathway in colonic cancerogenesis has been previously reported,19–21 lending support to this theory. In view of this clinical observation, it would be advisable to test the role of the Hh signalling pathway in HNPCC cancers. Interestingly, the only recognisable physical sign on clinical examination for HNPCC is skin changes related to neoplasms originating from the skin sebaceous glands in MTS, a rare variant of HNPCC.5
A strong correlation between MSH2 mutations and MTS has been recently reported.22,23 MTS is a cancer predisposing genodermatosis characterised by sebaceous gland tumours, including sebaceous adenomas, sebaceous epitheliomas, and sebaceous carcinomas, and less commonly keratoacantomas.24 However, MLH1 mutations have also been found in at least two families.25,26 Interestingly, in both reports a MLH1 deletion was present and found to be similar to that identified in our family 1. Moreover, phenotypic variation seems to be present, even among carriers of the same MSH2 mutations. For instance, the splicing mutation affecting exon 5 identified in three MTS families is one of the most common HNPCC molecular defects,27 while no information regarding cutaneous tumours in the majority of families with the MSH2 mutation reported in the literature is available.23 Taken together, the reported evidence, along with other recent studies,28 would support the hypothesis that the molecular basis of MTS does not substantially differ from that of HNPCC.
As the Hh pathway has been shown to play a key role in cancer related angiogenesis,29,30 our previous report on increased microvascular complexity in the oral mucosa of patients with HNPCC 4,31 further supports this hypothesis.
In view of our findings, an interesting future study would be a comparison between HNPCC patients and non-HNPCC patients carrying CRCs with documented MSI, although in the latter cases both mutations in MMR genes are usually somatic. In these patients a distinct molecular pathway has been demonstrated to be caused by a defect in DNA MMR genes,32 although the concept that the first mutation is germinal in HNPCC patients and somatic in sporadic MSI colorectal tumours should be relevant in determining the associated phenotypic features.
Further prospective research in a larger population is certainly needed to confirm the reported clinical association, and to verify the predictive value of FGs in detecting HNPCC. Whether this clinical observation could be helpful in increasing the rate of HNPCC diagnosis in affected families warrants further investigation.
Abbreviations
HNPCC, hereditary non-polyposis colorectal cancer
MMR, mismatch repair
CRC, colorectal carcinoma
FGs, Fordyce granules
Hh, hedgehog
MSI, microsatellite instability
MTS, Muir-Torre syndrome
Conflict of interest: None declared.
REFERENCES
- 1.Lynch HT, Lynch JF. Lynch syndrome: history and current status. Dis Markers 2004;20:181–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med 2003;348:919–32. [DOI] [PubMed] [Google Scholar]
- 3.Peltomaki P, Vasen H. Mutations associated with HNPCC predisposition-Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers 2004;20:269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roy HK, Lynch HT. Diagnosing Lynch syndrome: Is the answer in the mouth? Gut 2003;52:1665–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch HT, Lynch PM, Pester JA, et al. Sebaceous neoplasia and visceral cancer (Torre’s syndrome) and its relationship to the cancer family syndrome. In: Lynch HT, Fusaro RM, eds. Cancer-associated genodermatoses. New York: van Nostrand Reinhold, 1982:366–93.
- 6.Online Mendelian Inheritance in Man. OMIM. MIM Number #158320. Date last edited: 08/27/2004, Baltimore, Md: Johns Hopkins University. Available at: http//www.ncbi.nlm.nih.gov/omim/ (accessed 23/6/2005).
- 7.Fordyce J. A peculiar affection of the mucous membrane of the lip and oral cavity. J Cutan Genito-Urin Dis 1896;14:413–19. [Google Scholar]
- 8.Daley T. Pathology of intraoral sebaceous glands. J Oral Pathol Med 1993;22:241–5. [DOI] [PubMed] [Google Scholar]
- 9.Sedano HO, Carreon Freyre I, Garza de la Garza ML, et al. Clinical orodental abnormalities in Mexican children. Oral Surg Oral Med Oral Pathol 1989;68:300–11. [DOI] [PubMed] [Google Scholar]
- 10. http://www.maxillofacialcenter.com/BondBook/softtissue/fordycegran.html (accessed 23/6/2005).
- 11.Allen M, Grachtchouk M, Sheng H, et al. Hedgehog signaling regulates sebaceous gland development. Am J Pathol 2003;163:2173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science 2004;304:1755–9. [DOI] [PubMed] [Google Scholar]
- 13.Watkins DN, Peacock CD. Hedgehog signalling in foregut malignancy. Biochem Pharmacol 2004;68:1055–60. [DOI] [PubMed] [Google Scholar]
- 14. http://www.hgvs.org/mutnomen/ (accessed 23/6/2005).
- 15.Valentini AM, Renna L, Armentano R, et al. Mismatch repair, p53 and β-catenin proteins in colorectal cancer. Anticancer Res 2002;22:2083–8. [PubMed] [Google Scholar]
- 16.Cohen J. A coefficient of agreement for nominal scales. Educ Psychol Meas 1960;20:37–46. [Google Scholar]
- 17.Altman DG. Practical statistics for medical research. London: Chapman and Hall, 1991.
- 18.Alawi F, Siddiqui A. Sebaceous carcinoma of the oral mucosa: case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2005;99:79–84. [DOI] [PubMed] [Google Scholar]
- 19.Oniscu A, James RM, Morris RG, et al. Expression of Sonic hedgehog pathway genes is altered in colonic neoplasia. J Pathol 2004;203:909–17. [DOI] [PubMed] [Google Scholar]
- 20.Qualtrough D, Buda A, Gaffield W, et al. Hedgehog signalling in colorectal tumour cells: induction of apoptosis with cyclopamine treatment. Int J Cancer 2004;110:831–7. [DOI] [PubMed] [Google Scholar]
- 21.Berman DM, Karhadkar SS, Maitra A, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003;425:846–51. [DOI] [PubMed] [Google Scholar]
- 22.Mangold E, Pagenstecher C, Leister M, et al. A genotype-phenotype correlation in HNPCC: strong predominance of MSH2 mutations in 41 patients with Muir-Torre syndrome. J Med Genet 2004;41:567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucci-Cordisco E, Zito I, Gensini F, et al. Hereditary nonpolyposis colorectal cancer and related conditions. Am J Med Genet 2003;122A:325–34. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz RA, Torre DP. The Muir-Torre syndrome: a 25 year retrospect. J Am Acad Dermatol 1995;33:90–104. [DOI] [PubMed] [Google Scholar]
- 25.Kruse R, Rutten A, Lamberti C, et al. Muir-Torre phenotype has a frequency of DNA mismatch-repair-gene mutations similar to that in hereditary nonpolyposis colorectal cancer families defined by the Amsterdam Criteria. Am J Hum Genet 1998;63:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bapat BV, Madlensky L, Temple LK, et al. Family history characteristics, tumor microsatellite instability and germline MSH2 and MLH1 mutations in hereditary colorectal cancer. Hum Genet 1999;104:167–76. [DOI] [PubMed] [Google Scholar]
- 27.Desai DC, Lockman JC, Chadwick RB, et al. Recurrent germline mutation in MSH2 arises frequently de novo. J Med Genet 2000;37:646–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barana D, Cetto GL, Oliani C, et al. Spectrum of genetic alterations in Muir-Torre syndrome is the same as in HNPCC. Am J Med Genet 2004;125A:318–9. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan DC, Bicknell R. New molecular pathways in angiogenesis. Br J Cancer 2003;89:228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olsen CL, Hsu PP, Glienke J, et al. Hedgehog-interacting protein is highly expressed in endothelial cells but down-regulated during angiogenesis and in several human tumors. BMC Cancer 2004;4:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Felice C, Latini G, Bianciardi G, et al. Abnormal vascular network complexity: A new phenotypical marker in Lynch cancer family syndrome II. Gut 2003;12:1764–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996;87:159–70. [DOI] [PubMed] [Google Scholar]