

Abstract
Background and methods: Cytosin-guanosin dinucleotide (CpG) motifs of bacterial DNA are known to be potent activators of innate immunity. We have shown previously that administration of CpG containing oligodeoxynucleotide (CpG-ODN) to mice before the onset of dextran sodium sulphate induced colitis ameliorated colitis and inhibited induction of proinflammatory cytokines. To investigate the possible involvement of CD4+ T cells in the prophylactic CpG-ODN effects, we used the SCID transfer model of colitis.
Results: CD4+CD62L+ T cells from CpG-ODN treated donors did not induce significant intestinal inflammation in SCID recipients, in contrast with control cells. Additionally, cotransfer of these cells with CD4+CD62L+ cells from normal mice protected recipient animals from colitis, indicating regulatory activity. Also, CD4+CD62L+ cells from toll-like receptor 9 deficient animals induced a significantly more severe colitis in SCID recipients than cells from wild-type littermate controls, suggesting a similar protective role of “endogenous” bacterial DNA leading to a less “aggressive” phenotype of these cells. There was no detectable difference in regulatory T cell surface markers between aggressive and attenuated cell pools but attenuated cell pools showed reduced proliferation in vitro and in vivo and produced less interferon γ, interleukin (IL)-5, and IL-6 after anti-CD3 stimulation.
Conclusions: Collectively, our data support the concept that both endogenous bacterial DNA and exogenously supplied CpG motifs of bacterial DNA induce regulatory properties in CD4+ T cells. Therefore, bacterial DNA derived from the normal gut flora may contribute essentially to the homeostasis between effector and regulatory immune mechanisms in healthy individuals to protect them from chronic intestinal inflammation.
Keywords: experimental colitis, CpG motifs, bacterial DNA, SCID transfer model of colitis
The pathogenesis of chronic inflammatory bowel disease (IBD) is still unknown. Its aetiology is complex and multifactorial, involving genetic and environmental factors.1,2 There is abundant evidence that the resident intestinal flora plays a critical role in the initiation and perpetuation of chronic intestinal inflammation, as demonstrated in numerous genetic mouse and rat models of spontaneous colitis who fail to develop disease under germ free conditions.3–9 On the other hand, there are several observations suggesting that an “over clean” environment, especially in early childhood, increases the risk of IBD.10 Recently, mutations in the NOD2 gene were found to be associated with Crohn’s disease.11 The NOD2 gene product is thought to operate as an intracellular receptor for microbial constituents, such as muramyl dipeptide,12,13 resulting in nuclear factor κB activation in monocytes.14 Hence there is cumulative convincing evidence for a multifaceted role of bacteria and their products in the development of chronic intestinal inflammation.
Some years ago, the importance of bacterial DNA as an activating product of the vertebrate immune system was recognised.15 As shown, cytosin-guanosin dinucleotide (CpG) sequence motifs composed of unmethylated CpG dinucleotides are the immunostimulatory component of bacterial DNA.16 Oligonucleotides (ODN) containing CpG motifs can directly or indirectly activate dendritic cells, murine macrophages, B lymphocytes, natural killer cells, and T lymphocytes. In response to this stimulus, cells start to proliferate, process, and present antigens, and start to secrete a variety of proinflammatory cytokines, including interleukin (IL)-6, IL-12, tumour necrosis factor (TNF), and interferon γ (IFN-γ), eventually leading to strong induction of a Th1 skewed immune response.17 Bacterial DNA differs in three main respects from vertebrate DNA. Firstly, CpG motifs occur at random in bacterial DNA (1/16) and are rare in vertebrate DNA (1/60).18 Secondly, cytosines in vertebrate DNA are mostly methylated in contrast with bacterial DNA. Thirdly, in vertebrate DNA there are significant amounts of motifs which can suppress the stimulatory effects of CpG motifs.19 Based on these differences, the vertebrate immune system has developed mechanisms to specifically recognise bacterial DNA. Recent findings indicate that the toll-like receptor (TLR) 9 is critical for recognition of CpG motifs of bacterial DNA20 and to date all CpG motif induced effects observed were dependent on TLR9 ligation.21
Synthetic unmethylated CpG-ODN mimic the immune activating properties of bacterial DNA and were successfully used as an adjuvant in immunisation studies22,23 and in models of allergic diseases.24 The effects of CpG-ODN in experimental colitis are divergent. We and others showed previously that application of CpG motifs during ongoing colitis leads to IFN-γ dependent exacerbation25,26 whereas prophylactic CpG-ODN administration inhibited disease.26,27 We had observed that, after CpG-ODN pretreatment, IL-10 production was increased and IFN-γ secretion was decreased following colitis induction,26 suggesting a strong immune modulatory effect. Because both cytokines are indicators of T helper cell polarisation, we speculated that induction of specific T cell subsets was involved in these protective immune mechanisms. To test this hypothesis, we used the SCID transfer model of colitis which is controlled by defined CD4+ T cell subsets.
MATERIALS AND METHODS
Mice
C.B-17 SCID and Balb/c mice, weighing 20–22 g (Charles River, Germany), were used for the experiments and housed in a conventional facility. TLR9 deficient mice were a kind gift from Dr Shizuo Akira, (Osaka, Japan). All mice were F1 progeny of 129/Ola × C57BL/6. As controls, age matched littermates were used. Cells isolated from Balb/c donors or TLR9−/− mice or littermate controls were transferred into SCID mice. No signs of graft versus host disease were seen. The animal studies were approved by the local institutional review board.
Oligodeoxynucleotides
Phosphothioate stabilised ODN were obtained from Metabion (Martinsried, Germany). The sequence of the ODN is as follows:
CpG-ODN: “ODN1668”28 “5′-TCC ATG ACG TTC CTG ATG CT-3′”.
GpG-ODN: “ODN1668G” “5′-TCC ATG AGG TTC CTG ATG CT-3′” (control).
CD4+CD62L+ T cell transfer model of colitis
Splenic CD4+CD62L+ T cells from Balb/c mice were isolated as described previously, with slight modifications.29 In brief, CD4+ T cells were purified from spleen mononuclear cells of healthy mice by negative depletion of other cell types using anti-CD8, anti-MHC-II, anti-B220, and anti-CD11b cells antibodies (purchased from Pharmingen, Hamburg, Germany), and antirat-IgG immunomagnetic microbeads (Miltenyi Biotech, Bergisch Gladbach, Germany). The resulting CD4+ T cells were further separated by immunomagnetic beads into CD62L+ and CD62L− T cells. The former cells (purity >95%) showed high expression of CD45RB by FACS analysis. CD4+CD62L+ T cells (0.25×106) were resuspended in 200 μl of sterile phosphate buffered saline (PBS) and injected intraperitoneally in recipient C.B.-17 SCID or RAG1 deficient mice. In the cotransfer experiments, 0.25×106 CD4+CD62L+ cells from untreated donors plus 0.25×106 CD4+CD62L+ cells from CpG-ODN treated donors were injected. Colitis activity was monitored by weight changes and histological analysis, as specified below. For experiments with CpG-ODN or GpG-ODN treated donor animals, mice received 10 μg of ODN in 100 μl sterile PBS intraperitoneally for five days prior to isolation of CD4+CD62L+ T cells.
Histological examination
Cross sections of the rectum were fixed in 10% buffered formalin and stained with haematoxylin-eosin. To quantify the histological damage in gut tissue, a new score was established, as shown in table 1 ▶. The predominant feature of microscopic inflammation in transfer colitis is the mononuclear cell infiltration limited to the mucosa and the consecutive mucosal damage (loss of goblet cells, loss of crypts). Both features were independently graded from 0 to 4 and the mean score was noted. Signs of severe colitis may include occasionally crypt abscesses, extension of the inflammatory infiltrate to the submucosa, and mucosal extravascular haemorrhage. Each of these signs was acknowledged by a 0.5 increase in histology score, with a maximum limited to 4. Histological analysis was performed by two investigators in a blinded fashion.
Table 1.
Histological score for the SCID transfer model of colitis
| Score | 0 | 1 | 2 | 3 | 4 | + 0.5 |
| Inflammatory infiltrate | None | Focal | Cirumferential mild | Cirumferential moderate | Cirumferential severe | Crypt abscesses, submucosal infiltration, mucosal haemorrhage |
| Mucosal architecture | Intact | Intact | Intact | Damaged | Loss |
Incubation of CD4+CD62L+T cells
Cells were isolated as described above and then incubated at a concentration of 106 cells/ml cell culture medium (RPMI-1640, 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin from Gibco-BRL (Eggenstein, Germany)) and β-mercaptoethanol 3×10−5M (Sigma, Deisenhofen, Germany). Cells were stimulated over 24 hours in anti-CD3 (2.5 μg/well) (mAb, 145-2C11; BD Pharmingen) coated wells and cytokine levels were measured in the supernatant by ELISA (all from Endogene, Woburne, Massachusetts, USA) using four wells per condition.
Proliferation assay
Isolated cells were incubated in 96 well plates coated previously with anti-CD3 (2.5 μg/well). After 48 hours cells were pulsed with 0.5 μCi/well (3H)thymidine for 16 hours and counts per minute were measured using a scintillation counter.
Isolation and incubation of mesenteric lymph node cells
Mesenteric lymph nodes (pooled from each group of mice) were collected under sterile conditions in ice cold medium and lymph nodes were mechanically disrupted and filtered through a cell strainer (70 μm). Cells (2×105/well) were incubated in 200 μl culture medium over 24 hours and cytokine levels were measured in the supernatant by ELISA (all from Endogene) using four wells per condition.
FACS analysis
Samples were analysed using two colour staining. Briefly, isolated lymphocytes were preincubated with 40 μg/ml of 2.4G2 monoclonal antibody with 10% fetal calf serum to block FcRs and then stained with both FITC and PE conjugated monoclonal antibodies. Cells were washed and analysed by flow cytometry using Epics XL-MCL (Coulter Electronics, Krefeld, Germany). All steps were performed at 4°C to prevent receptor internalisation.
Quantitative RT-PCR (Light Cycler)
An additional colonic tissue specimen (1 cm) was harvested for quantitative reverse transcription-polymerase chain reaction (RT-PCR) and transferred to ice cold RNAlater solution (Ambion, Huntingdon, UK). RNA was extracted using the RNeasy-kit (Qiagen, Hilden, Germany) and the Qiagen Shredder-kit following the manufacturer’s recommendations. RNA was transcribed using the Promega (Mannheim, Germany) Reverse Transcription System, as recommended by the manufacturer. Quantification of cytokine mRNA was performed using a Light Cycler (Roche, Molecular Systems, Mannheim, Germany). For standardisation, β-actin was amplified (primer pair: 5′-TGG AAT CCT GTG GCA TCC ATG AAA C-3′ and 5′-TAA AAC GCA GCT CAG TAA CAG TCC G-3′).
Primers and conditions
IFN-γ: 5′-TGG AGG AAC TGG CAA AAG GAT GGT-3′ and 5′-TTG GGA CAA TCT CTT CCC CAC-3′, annealing temperature at 62°C, 3 mM MgCl2.
IL-6: 5′-TGG AGT CAC AGA AGG AGT GGC TAA G -3′ and 5′-TCT GAC CAC AGT GAG GAA TGT CCA C -3′, annealing temperature at 62°C, 4 mM MgCl2.
IL-10: 5′-TCC TTA ATG CAG GAC TTT AAG GGT TAC TTG-3′ and 5′’-GAC ACC TTG GTC TTG GAG CTT ATT AAA ATC-3′, annealing temperature at 62°C, 3 mM MgCl2.
All primers were purchased from MWG-Biotech AG (Ebersberg, Germany). Cytokine mRNA was quantified for each individual mouse.
BrdU labelling and immunohistochemistry
Animals were injected intraperitoneally with 1 mg/mouse BrdU (Sigma Aldrich, München, Germany) for three consecutive days before the mice were sacrificed. Immunohistochemical staining of BrdU in frozen sections of mesenteric lymph nodes was performed following the manufacturer’s instructions using the BrdU in situ detection kit (BD Pharmingen).
Statistics
Statistical analysis was performed using the Student’s t test (cytokine levels), the Mann-Whitney rank sum test (histological score), or the general linear model (daily weight loss). In all experiments including more than two groups, a statistically significant difference according to the Kruskal-Wallis analysis of variance on ranks among the treatment groups was confirmed, before comparing two groups using the Mann-Whitney rank sum test. Error bars represent the standard error of the mean (SEM). Statistically significant differences were accepted when p<0.05.
RESULTS
Effects of CpG-ODN pretreatment of donor mice on weight loss and intestinal inflammation in the SCID transfer model of colitis
To evaluate whether CD4+ T cells are involved in the previously described protective effects of CpG-ODN administration in different colitis models,26,27 we used the SCID transfer model in which colitis is induced in SCID mice by transfer of splenic CD4+CD62L+ cells. Donor mice were treated with either CpG-ODN or control GpG-ODN over five days or left untreated before CD4+CD62L+ splenic T cells were isolated and transferred into recipient animals. As shown in fig 1A ▶, SCID mice, reconstituted with lymphocytes from CpG-ODN treated donors, gained weight over time (week 8: +14 (5)%) and developed no signs of colitis comparable with animals that did not receive any T cells (week 8: +28 (4)%). In contrast, mice transferred with CD4+CD62L+ cells from untreated donors or donors pretreated with control GpG-ODN lost weight over time (week 8: −8 (5)% and −5 (2)%) and developed colitis, as expected.
Figure 1.

Effects of cytosin-guanosin containing oligodeoxynucleotide (CpG-ODN) treatment of donor animals in the SCID transfer model of colitis on colitis development. Donor animals were treated with either CpG-ODN or GpG-ODN (each 10 µg/day over five days) or left untreated, and CD4+CD62L+ cells were transferred to SCID recipients. As a negative control, SCID mice were injected with phosphate buffered saline (PBS). (A) Weight change after transfer. (B) Histological score in the different groups was examined at the end of the experiments (eight and 12 weeks after transfer). (C) Representative colonic haematoxylin-eosin sections of non-transferred mice and mice transferred with CD4+CD62L+ cells from CpG-ODN or GpG-ODN treated (control) or untreated donors (control) are shown (magnification 50-fold). (D) Toll-like receptor 9 (TLR9) deficient or wild-type (Wt) littermate controls were treated with CpG-ODN (10 µg/day over five days), CD4+CD62L+ cells were transferred to SCID recipients, and the histological score was examined at the end of the experiment. Data presented in (A–C) were derived from 5–8 mice per group and are representative of five independent experiments. Values are mean (SEM). *Significantly different from both groups transferred with CD4+CD62L+ cells from either GpG-ODN or untreated donor mice. Data presented in (B) (12 week data) and (D) were derived from 5–8 mice per group and are representative of two independent experiments. Values are mean (SEM). *Significantly different.
Differences in the course of weight loss were reflected by histological signs of mucosal inflammation. Hardly any inflammation was observed within the intestinal mucosa of SCID mice reconstituted with lymphocytes from CpG-ODN treated donors (histological score: 0.4 (0.4)) whereas animals that received T cells from GpG-ODN or untreated donor mice displayed fully established colitis with no significant difference between the two groups (histological score: transfer from GpG-ODN treated donors 2.2 (0.3); transfer from untreated donors 1.8 (0.4)) (fig 1B ▶).
To check whether CpG-ODN treatment of donor mice results in a delayed onset of intestinal inflammation or in a sustained reduced colitis, we extended the observation period to 12 weeks. As shown in fig 1B ▶, we found a significant protective effect of CpG-ODN donor treatment even 12 weeks after the transfer of cells (CpG-ODN treated donors 1.1 (0.3); untreated donors 3.8 (0.2); GpG-ODN treated control donors 3.0 (0.4); p<0.001). The protective CpG-ODN effects were recently shown to be TLR9 dependent in other colitis models.30 To verify this in the SCID transfer model of colitis, we treated both TLR9 deficient donor mice and wild-type littermates with CpG-ODN. In contrast with wild-type littermates, there was no protective effect of CpG-ODN treatment of TLR9 deficient donor mice, indicating that the protective effects of CpG-ODN are TLR9 dependent (fig 1D ▶).
Effects of CpG-ODN pretreatment of donor mice on cytokine production in the SCID transfer model of colitis
To verify whether amelioration of intestinal inflammation was accompanied by modulation of cytokine production, we measured cytokine secretion from mesenteric lymph node cells (fig 2A ▶) and mRNA in the colon (fig 2B ▶).
Figure 2.

Effects of cytosin-guanosin containing oligodeoxynucleotide (CpG-ODN) treatment of donor animals in the SCID transfer model of colitis on cytokine production. Donor animals were treated with either CpG-ODN or GpG-ODN (each 10 µg/day daily over five days) or left untreated, and CD4+CD62L+ cells were transferred to SCID recipients. As a negative control, SCID mice were injected with phosphate buffered saline (PBS). (A) Mesenteric lymph node cells were isolated at the end of the experiment and spontaneous cytokine secretion was measured in overnight cultures by ELISA. IL, interleukin; IFN-γ, interferon γ. (B) mRNA from colonic tissue of individual mice was isolated and expression of different cytokines was quantified by real time polymerase chain reaction, as described in materials and methods. Data presented were derived from five independent experiments with at least 20 mice per group. Values are mean (SEM). *Significantly different from both the group transferred with CD4+CD62L+ cells from untreated and GpG-ODN treated donor mice if not indicated otherwise.
Interestingly, recipients of CD4+CD62L+ donor cells from CpG pretreated mice showed no increase in IFN-γ or IL-6 secretion compared with non-transferred animals whereas recipients of cells from untreated mice showed significantly upregulated synthesis of both proinflammatory cytokines (IFN-γ 700-fold; IL-6 sevenfold) (fig 2A ▶). In contrast, IL-10 secretion significantly increased (fourfold) in mice transferred with cells from CpG-ODN treated donors compared with controls. Pretreatment of donor mice with GpG-ODN also affected cytokine production but at a significantly lower level. Gene expression of proinflammatory cytokines in colonic tissue, as measured by RT-PCR, was also significantly lower (IL-6 6.7-fold; IFN-γ 20-fold) whereas IL-10 expression was upregulated by approximately 3.5-fold by transfer of CpG-ODN pretreated donor cells compared with untreated or GpG treated donors cells (fig 2B ▶).
Transfer of CD4+CD62L+ cells from TLR9 deficient mice results in increased intestinal inflammation in SCID recipients
In the experiments described above, donor mice were exogenously exposed to a substantial amount of CpG-ODN over five days, finally leading to CD4+CD62L+ T cells with a significantly reduced inflammatory potential. To investigate whether “physiological” steady state TLR9/CpG DNA interaction in donor mice influences the colitis stimulating potential of CD4+CD62L+ cells, we used TLR9 deficient mice as donor animals in which this interaction is missing. As demonstrated in fig 3A ▶, SCID recipients showed significantly increased weight loss when transferred with cells from TLR9−/− donors compared with mice transferred with cells from control littermates (transferred from TLR9−/− donors −17%; transferred from wild-type controls −8%). Increased weight loss was paralleled by a significantly increased histological score (transfer from TLR9−/− donors 3.2 (0.2); transfer from wild-type donors 1.3 (0.5)) and significantly higher IFN-γ (7.9-fold), IL-5 (7.6-fold), and IL-6 (1.8-fold) production from mesenteric lymph node cells whereas IL-10 secretion was decreased by 9.7-fold (fig 3C ▶). Taken together, this suggests that CD4+CD62L+ cells from TLR9 deficient mice display an increased inflammatory potential. To exclude the possibility that genetic differences between donors and recipients qualitatively influenced the increased inflammation observed in recipients transferred with TLR9−/− cells, we also used C57/Bl6 RAG1−/− recipient mice and obtained qualitatively the same results (data not shown).
Figure 3.
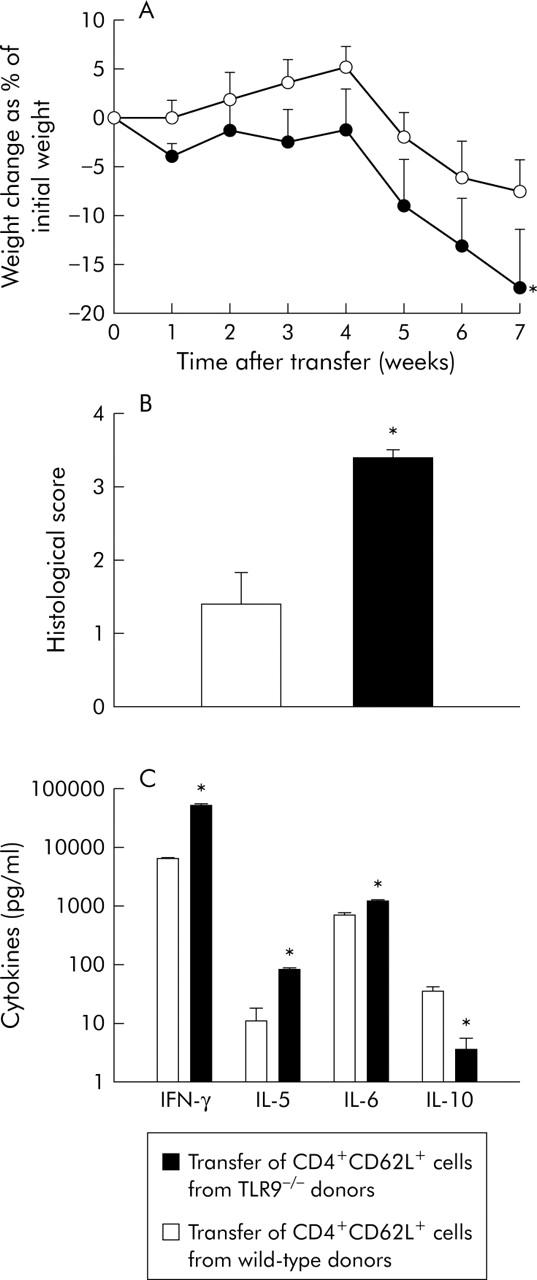
Transfer of CD4+CD62L+ cells from toll-like receptor 9 (TLR9) deficient mice versus wild-type mice. CD4+CD62L+ cells from TLR9 deficient donors or wild-type control littermates were isolated and transferred to SCID recipients. (A) Weight change after transfer. (B) Histological score of the different groups was examined at the end of the experiment (seven weeks after transfer). (C) Mesenteric lymph node cells were isolated at the end of the experiment and cytokine secretion was measured after 24 hours in supernatants by ELISA. IL, interleukin; IFN-γ, interferon γ. *Significantly different from the group transferred with CD4+CD62L+ cells from wild-type littermates. Data presented were derived from five mice per group and are representative of two independent experiments.
Cotransfer of CD4+CD62L+ cells from CpG-ODN and untreated donors leads to reduced intestinal inflammation in the SCID transfer model of colitis
To determine whether CD4+CD62L+ cells from CpG-ODN treated donors lost their capacity to induce intestinal inflammation or whether they obtained regulatory capabilities, we performed cotransfer experiments. Figure 4A ▶ demonstrates that cotransfer of cells from CpG-ODN treated and untreated donor mice prevented colitis development in SCID hosts. Compared with mice only transferred with CD4+CD62L+ cells from untreated donors, the histological score was reduced 2.2-fold and IFN-γ production was significantly lower (5.7-fold) (fig 4B ▶) Furthermore, we found a clearly reduced number of proliferating cells (BrdU staining, brown colour in fig 5 ▶) in mesenteric lymph nodes from SCID mice transferred with CD4+CD62L+ cells from CpG-ODN pretreated donors and also in cotransferred hosts compared with SCID mice transferred with cells from untreated donors. Taken together, these data suggest that CpG-ODN treatment induced the appearance of regulatory activity among CD4+CD62L+ cells.
Figure 4.

Cotransfer of CD4+CD62L+ cells from cytosin-guanosin containing oligodeoxynucleotide (CpG-ODN) treated and untreated donor animals in SCID mice. Donor animals were treated with CpG-ODN (10 µg/day over five days) or left untreated and either CD4+CD62L+ cells from untreated or CpG-DNA treated donors or both were transferred to SCID recipients. As a negative control, SCID mice were injected with phosphate buffered saline (PBS). Histological score (A) and interferon γ (IFN-γ) secretion of isolated mesenteric lymph node cells (B) at the end of the experiment (eight weeks after transfer) are shown. *Significantly different from the group transferred with CD4+CD62L+ cells from untreated donor mice. Data presented were derived from three independent experiments, with 10–15 mice per group. ND, not detected.
Figure 5.

Cotransfer of CD4+CD62L+ cells from cytosin-guanosin containing oligodeoxynucleotide (CpG-ODN) treated and untreated donor animals in SCID mice. Donor animals were treated with CpG-ODN (10 µg/day over five days) or left untreated and either CD4+CD62L+ cells from untreated or CpG-DNA treated donors or both were transferred to SCID recipients. In vivo proliferation of cells in mesenteric lymph nodes was detected by BrdU incorporation.
Characterisation of CD4+CD62L+ cells from CpG-ODN treated donors
To further investigate the changes induced by CpG-ODN exposition in CD4+CD62L+ cells, we compared anti-CD3 induced cytokine secretion and proliferation, and FACS analysis was performed to detect markers described as being preferentially expressed on regulatory/inhibitory T cells (table 2 ▶). CpG-ODN pretreatment of mice decreased secretion of IFN-γ (3.9-fold) and IL-5 (5.2-fold) by CD4+CD62L+ cells whereas no significant difference was found for IL-10 or transforming growth factor β (TGF-β) secretion (fig 6A ▶). Furthermore, their proliferation on CD3 ligation was significantly reduced (approximately twofold; fig 6B ▶). There was no marked difference in CD25, GITR, CTLA-4, or αEβ7 surface display in these cells compared with controls (table 2 ▶).
Table 2.
Expression of surface markers on CD4+CD62L+ cells detected by flow cytometric analysis
| Treatment of mice | None | CpG-ODN | GpG-ODN (control) |
| CD25 | 13.8 | 14.6 | 15.5 |
| αEβ7 | 9.8 | 5.7 | 6.6 |
| GITR | 5.8 | 5.1 | 7.3 |
| CTLA-4 | 0.4 | 0.6 | 0.4 |
Results are given as positive cells (in per cent) and are representative of three independent experiments.
CpG-ODN, cytosin-guanosin containing oligodeoxynucleotide.
Figure 6.

Cytokine secretion and proliferation of anti-CD3 stimulated CD4+CD62L+ cells isolated from cytosin-guanosin containing oligodeoxynucleotide (CpG-ODN) treated donor mice. Mice were treated with either CpG-ODN or GpG-ODN (each 10 µg/day over five days) or left untreated, and CD4+CD62L+ cells were isolated and incubated in anti-CD3 coated wells, as described in materials and methods, over 24 hours for cytokine detection (A) or 72 hours for 3H-thymidine incorporation (B). PBS, phosphate buffered saline; IL, interleukin; IFN-γ, interferon γ; TGF-β, transforming growth factor β. *Significantly different from both GpG-ODN or untreated mice. Incubations were performed in quadruplicate and results are representative of three independent experiments.
Characterisation of CD4+CD62L+ cells from TLR9 deficient mice
Anti-CD3-stimulated CD4+CD62L+ cells from TLR9 deficient mice were compared with cells from wild-type littermate controls. IFN-γ (2.3-fold), IL-5 (10-fold), and IL-6 (2.4-fold) secretion was significantly increased and IL-10 secretion was significantly decreased (1.6-fold) (fig 7A ▶). Furthermore, we found a significant increase in proliferation after 48 hours in unstimulated cells (44%) compared with wild-type controls. In contrast, there was no significantly different proliferation in anti-CD3 activated cells (115% of wild-type controls) (fig 7B ▶).
Figure 7.

Cytokine secretion and proliferation of anti-CD3 stimulated CD4+CD62L+ cells isolated from toll-like receptor 9 (TLR9) deficient mice. CD4+CD62L+ cells were isolated from TLR9 deficient mice or wild-type littermate controls and incubated in anti-CD3 coated wells, as described in materials and methods, over 24 hours for cytokine detection (A) or 72 hours for 3H-thymidine incorporation (both unstimulated and anti-CD3 stimulated) (B). IL, interleukin; IFN-γ, interferon γ. *Significantly different from littermate controls. Incubations were performed in quadruplicate and results are representative of at least two experiments.
DISCUSSION
Recently, it was demonstrated by us and others that prophylactic CpG-ODN treatment ameliorates colitis in various animal models of intestinal inflammation.26,27 However, the mechanism of this protective effect remained unclear. Our data provide evidence that CpG induced protection is mediated via a T cell dependent mechanism and further suggest that naturally occurring CpG/TLR9 interactions in healthy mice may contribute to the regulatory potential of T cells.
Our previous results in the acute dextran sodium sulphate model of colitis demonstrated reduced IFN-γ and increased IL-10 secretion in CpG-ODN pretreated mice at the end of colitis induction.26 We therefore assumed that T cells were involved in the protective CpG-ODN effects. To test this hypothesis we used the SCID transfer model of colitis. This model is widely used to investigate the role of specific T cells in induction of intestinal inflammation because colitis is mediated by transfer of “pathogenic” CD4+ lymphocytes into immune deficient mice. We found that T lymphocytes from untreated or control GpG-ODN injected donor mice induced severe colitis in SCID mice whereas cells transferred from CpG-ODN pretreated animals failed to mediate significant intestinal inflammation.
This effect was surprising because the most prominent activity of CpG-ODN is their strong Th1 inducing capacity.31,32 However, it is also known that CpG motifs not only induce secretion of proinflammatory cytokines by antigen presenting cells but also secretion of the anti-inflammatory cytokine IL-10. Additionally, other reports described a protective role of CpG-ODN in several models of Th2 mediated pathologies.23,33,34 In these models, the beneficial effect was attributed to induction of a strong Th1 response that was thought to counteract the Th2 mediated pathomechanisms. Interestingly, it was also shown previously that the positive effects of prophylactic immunisation with CpG-DNA before schistosome egg application were not due to induction of a Th1 immune response but mainly IL-10 and cell/cell contact dependent. This suggested that induction of a regulatory immune response by CpG motifs limits pathology in this model.35 Furthermore, prophylactic CpG-ODN application was also able to prevent Th1 driven autoimmune diabetes in NOD mice.36 Again, the protective effect correlated with increased IL-10 production. These results, together with our finding that mesenteric lymph node cells from SCID mice that received T cells from CpG-ODN pretreated donors secreted more IL-10 than control mice, prompted us to speculate that CD4+CD62L+ cells isolated from CpG-ODN injected animals not only lose their potential to induce colitis but, in contrast, obtain regulatory capacities. This hypothesis was confirmed by cotransfer experiments in which CD4+CD62L+ cells from CpG-ODN pretreated donors abolished the colitis inducing capacity of CD4+CD62L+ cells from untreated donors.
Furthermore, IFN-γ production was significantly reduced and lymphocyte proliferation was not detectable in mesenteric lymph node cells from the cotransferred recipients. This indicated that CD4+CD62L+ cells (or a subpopulation of these cells) from CpG-ODN pretreated donors suppressed intestinal inflammation, presumably by inhibition of proinflammatory cytokine production and suppression of T cell proliferation. This potential induction of regulatory T cells by CpG-ODN treatment would also explain why prophylactic CpG-ODN administration suppresses colitis in Th1 as well as in Th2 mediated models of intestinal inflammation, as shown by Rachmilewitz and colleagues.27
Over the past few years many new insights into the process of immune response regulation have been gained that demonstrated the importance of regulatory T cells for maintenance of immunological steady states. To date, different subsets of CD4+ cells with a suppressive function were characterised and the existence of at least two basic subsets of CD4+ regulatory T cells was proposed which differ in terms of development, specificity, mechanism of action, and dependence on T cell receptor and costimulatory signalling.37 The best characterised subset consists of the so-called naturally occurring CD4+CD25+ regulatory cells that develop in the thymus. These CD25+ cells are characterised by expression of glucocorticoid induced TNF receptor related protein GITR and the transcription factor Foxp3.38–40 We did not find increased amounts of CD25, GITR, or Foxp3 mRNA (preliminary results derived from semiquantitative PCR) in CD4+CD62L+ cells from CpG-ODN pretreated animals.
We therefore favour the adaptive or “induced” regulatory T cell population.37 It is thought that these cells are induced under certain conditions of antigenic stimulation involving cytokines such as IL-10 and type I interferons, and also corticosteroids, vitamin D3, and/or cell/cell contact.41,42 This hypothesis is supported by a recent report that demonstrated that CD8α+ CpG-ODN matured dendritic cells support the induction of regulatory CD4+ cells.43 The mechanism by which these adaptive T cells exert their regulatory function is not yet identified. In some in vivo models, IL-10 and/or TGF-β secretion are thought to play an important role.44–46 However, similar to natural regulatory cells, cell-cell contact dependent mechanisms are suggested to mediate the inhibitory functions of adaptive regulatory T lymphocytes.37 Our results indicate that in vivo, CpG-ODN pretreatment inhibited the secretion of both Th1 (IFN-γ) and Th2 (IL-5) cytokines from CD4+CD62L+ cells while the production of cytokines which are attributed to regulatory cells (IL-10, TGF-β) was unaffected, indicating a CpG-ODN induced shift towards a regulatory pattern of cytokine secretion among CD4+CD62L+ cells.
Another quality of regulatory T cells is their reduced capacity to proliferate. CD4+CD62L+ cells from CpG-ODN treated mice exhibited a markedly reduced proliferation which became evident after only 72 hours of in vitro culture. In vivo, the antiproliferative effect appeared to be even stronger as we found hardly any proliferating cells in mesenteric lymph nodes of SCID mice transferred with lymphocytes from CpG treated donors in contrast with controls transferred with cells from untreated donors. Taken together, these results argue for a regulatory type of CD4+ T cell which results from in vivo CPG-ODN exposition and which is part of the CD4+CD62L+ lymphocyte preparation capable of preventing the development of colitis.
Based on these results and on a recent report which indicated that the prophylactic CpG-ODN effect on experimental colitis is in fact TLR9 dependent,30 we raised the important question of whether, in a similar way, exposition to “endogenous” bacterial DNA, possibly derived from the normal luminal bacterial flora, also modulates the potential of CD4+CD62L+ cells to induce colitis. CD4+CD62L+ cells isolated from TLR9 deficient mice, which cannot be stimulated by bacterial DNA, and their wild-type littermates were compared after transfer to SCID or RAG-1 deficient mice. Concerning all measured parameters of colitis, TLR9 deficient cells displayed a significantly greater inflammatory potential. Thus constant exposure of a healthy organism to bacterial DNA may contribute to the steady state homeostasis of the intestinal immune system regulating the delicate balance between effector and regulatory mechanisms. Interestingly, similar findings have been observed recently in human T cells indicating that CpG motifs are potent inducers of regulatory qualities.47 In this respect, our data are of special interest with regard to results from epidemiological studies which suggest that the incidence of IBD positively correlates with the increase in hygienic standards in developed countries, leading to less contact of an organism with bacteria and bacterial products.10,48 It is therefore tempting to speculate that reduced exposure to microbial components (such as bacterial DNA) contributes to an increased risk of developing a deregulated intestinal immune response due to a reduction in regulatory T lymphocyte activities, eventually leading to IBD in genetically susceptible hosts.
Acknowledgments
This work was supported by a grant from the EU to WF (QLG1-CT-1999-00549), a grant from the DFG to UGS, and a research grant from the University of Regensburg, Germany, as part of the ReForM programme to FO.
Abbreviations
IBD, inflammatory bowel disease
CpG, cytosin-guanosin dinucleotide
CpG-ODN, CpG containing oligodeoxynucleotide
TLR, toll-like receptor
IL, interleukin
TNF, tumour necrosis factor
IFN-γ, interferon γ
TGF-β, transforming growth factor β
PBS, phosphate buffered saline
RT-PCR, reverse transcription-polymerase chain reaction
Conflict of interest: None declared.
Published online first 5 May 2005
REFERENCES
- 1.Andus T, Gross V. Etiology and pathophysiology of inflammatory bowel disease—environmental factors. Hepatogastroenterology 2000;47:29–43. [PubMed] [Google Scholar]
- 2.Sartor RB. Pathogenesis and immune mechanisms of chronic inflammatory bowel diseases. Am J Gastroenterol 1997;92 (12 suppl) :5–11S. [PubMed] [Google Scholar]
- 3.Brimnes J, Reimann J, Nissen M, et al. Enteric bacterial antigens activate CD4(+) T cells from scid mice with inflammatory bowel disease. Eur J Immunol 2001;31:23–31. [DOI] [PubMed] [Google Scholar]
- 4.Dianda L, Hanby AM, Wright NA, et al. T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a microbial environment. Am J Pathol 1997;150:91–7. [PMC free article] [PubMed] [Google Scholar]
- 5.Madsen KL, Malfair D, Gray D, et al. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis 1999;5:262–70. [DOI] [PubMed] [Google Scholar]
- 6.Schultz M, Tonkonogy SL, Sellon RK, et al. IL-2-deficient mice raised under germfree conditions develop delayed mild focal intestinal inflammation. Am J Physiol 1999;276:G1461–72. [DOI] [PubMed] [Google Scholar]
- 7.Veltkamp C, Tonkonogy SL, De Jong YP, et al. Continuous stimulation by normal luminal bacteria is essential for the development and perpetuation of colitis in Tg(epsilon26) mice. Gastroenterology 2001;120:900–13. [DOI] [PubMed] [Google Scholar]
- 8.Sartor RB. Review article: Role of the enteric microflora in the pathogenesis of intestinal inflammation and arthritis. Aliment Pharmacol Ther 1997;11:17–22. [DOI] [PubMed] [Google Scholar]
- 9.Sartor RB. The influence of normal microbial flora on the development of chronic mucosal inflammation. Res Immunol 1997;148:567–76. [DOI] [PubMed] [Google Scholar]
- 10.Duggan AE, Usmani I, Neal KR, et al. Appendicectomy, childhood hygiene, Helicobacter pylori status, and risk of inflammatory bowel disease: a case control study. Gut 1998;43:494–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001;411:603–6. [DOI] [PubMed] [Google Scholar]
- 12.Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 2003;278:8869–72. [DOI] [PubMed] [Google Scholar]
- 13.Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem 2003;278:5509–12. [DOI] [PubMed] [Google Scholar]
- 14.Ogura Y, Inohara N, Benito A, et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem 2001;276:4812–18. [DOI] [PubMed] [Google Scholar]
- 15.Krieg AM. Now I know my CpGs. Trends Microbiol 2001;9:249–52. [DOI] [PubMed] [Google Scholar]
- 16.Krieg AM, Yi AK, Matson S, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995;374:546–9. [DOI] [PubMed] [Google Scholar]
- 17.Krieg AM. Immune effects and mechanisms of action of CpG motifs. Vaccine 2000;19:618–22. [DOI] [PubMed] [Google Scholar]
- 18.Burge C, Campbell AM, Karlin S. Over- and under-representation of short oligonucleotides in DNA sequences. Proc Natl Acad Sci U S A 1992;89:1358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stacey KJ, Young GR, Clark F, et al. The molecular basis for the lack of immunostimulatory activity of vertebrate DNA. J Immunol 2003;170:3614–20. [DOI] [PubMed] [Google Scholar]
- 20.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000;408:740–5. [DOI] [PubMed] [Google Scholar]
- 21.Bauer S, Wagner H. Bacterial CpG-DNA licenses TLR9. Curr Top Microbiol Immunol 2002;270:145–54. [DOI] [PubMed] [Google Scholar]
- 22.O’Hagan DT, MacKichan ML, Singh M. Recent developments in adjuvants for vaccines against infectious diseases. Biomol Eng 2001;18:69–85. [DOI] [PubMed] [Google Scholar]
- 23.Krieg AM. From bugs to drugs: therapeutic immunomodulation with oligodeoxynucleotides containing CpG sequences from bacterial DNA. Antisense Nucleic Acid Drug Dev 2001;11:181–8. [DOI] [PubMed] [Google Scholar]
- 24.Metzger WJ, Nyce JW. Oligonucleotide therapy of allergic asthma. J Allergy Clin Immunol 1999;104:260–6. [DOI] [PubMed] [Google Scholar]
- 25.Katakura K, Sato Y, Sato N, et al. CpG motifs in plasmid DNA exacerbate inflammation in experimental colitis. Gastroenterology 2001;120:A518. [Google Scholar]
- 26.Obermeier F, Dunger N, Strauch UG, et al. Contrasting activity of cytosin-guanosin dinucleotide oligonucleotides in mice with experimental colitis. Clin Exp Immunol 2003;134:217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rachmilewitz D, Karmeli F, Takabayashi K, et al. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology 2002;122:1428–41. [DOI] [PubMed] [Google Scholar]
- 28.Sparwasser T, Koch ES, Vabulas RM, et al. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur J Immunol 1998;28:2045–54. [DOI] [PubMed] [Google Scholar]
- 29.Neurath MF, Weigmann B, Finotto S, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J Exp Med 2002;195:1129–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rachmilewitz D, Katakura K, Karmeli F, et al. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology 2004;126:520–8. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi H, Horner AA, Takabayashi K, et al. Immunostimulatory DNA pre-priming: a novel approach for prolonged Th1-biased immunity. Cell Immunol 1999;198:69–75. [DOI] [PubMed] [Google Scholar]
- 32.Krieg AM. The role of CpG motifs in innate immunity. Curr Opin Immunol 2000;12:35–43. [DOI] [PubMed] [Google Scholar]
- 33.Broide D, Schwarze J, Tighe H, et al. Immunostimulatory DNA sequences inhibit IL-5, eosinophilic inflammation, and airway hyperresponsiveness in mice. J Immunol 1998;161:7054–62. [PubMed] [Google Scholar]
- 34.Schwartz DA, Wohlford-Lenane CL, Quinn TJ, et al. Bacterial DNA or oligonucleotides containing unmethylated CpG motifs can minimize lipopolysaccharide-induced inflammation in the lower respiratory tract through an IL-12-dependent pathway. J Immunol 1999;163:224–31. [PubMed] [Google Scholar]
- 35.Chiaramonte MG, Hesse M, Cheever AW, et al. CpG oligonucleotides can prophylactically immunize against Th2-mediated schistosome egg-induced pathology by an IL-12-independent mechanism. J Immunol 2000;164:973–85. [DOI] [PubMed] [Google Scholar]
- 36.Quintana FJ, Rotem A, Carmi P, et al. Vaccination with empty plasmid DNA or CpG oligonucleotide inhibits diabetes in nonobese diabetic mice: modulation of spontaneous 60-kDa heat shock protein autoimmunity. J Immunol 2000;165:6148–55. [DOI] [PubMed] [Google Scholar]
- 37.Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol 2003;3:253–7. [DOI] [PubMed] [Google Scholar]
- 38.Khattri R, Kasprowicz D, Cox T, et al. The amount of scurfin protein determines peripheral T cell number and responsiveness. J Immunol 2001;167:6312–20. [DOI] [PubMed] [Google Scholar]
- 39.Khattri R, Cox T, Yasayko SA, et al. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol 2003;4:337–42. [DOI] [PubMed] [Google Scholar]
- 40.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003;4:330–6. [DOI] [PubMed] [Google Scholar]
- 41.Levings MK, Bacchetta R, Schulz U, et al. The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells. Int Arch Allergy Immunol 2002;129:263–76. [DOI] [PubMed] [Google Scholar]
- 42.Barrat FJ, Cua DJ, Boonstra A, et al. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med 2002;195:603–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bilsborough J, George TC, Norment A, et al. Mucosal CD8alpha+ DC, with a plasmacytoid phenotype, induce differentiation and support function of T cells with regulatory properties. Immunology 2003;108:481–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med 2001;194:629–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Powrie F, Carlino J, Leach MW, et al. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J Exp Med 1996;183:2669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Groux H, O’Garra A, Bigler M, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997;389:737–42. [DOI] [PubMed] [Google Scholar]
- 47.Moseman EA, Liang X, Dawson AJ, et al. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. J Immunol 2004;173:4433–42. [DOI] [PubMed] [Google Scholar]
- 48.Gent AE, Hellier MD, Grace RH, et al. Inflammatory bowel disease and domestic hygiene in infancy. Lancet 1994;343:766–7. [DOI] [PubMed] [Google Scholar]