

Abstract
Background: Angiotensin converting enzyme (ACE) 2 is a recently identified homologue of ACE that may counterregulate the actions of angiotensin (Ang) II by facilitating its breakdown to Ang 1–7. The renin-angiotensin system (RAS) has been implicated in the pathogenesis of cirrhosis but the role of ACE2 in liver disease is not known.
Aims: This study examined the effects of liver injury on ACE2 expression and activity in experimental hepatic fibrosis and human cirrhosis, and the effects of Ang 1–7 on vascular tone in cirrhotic rat aorta.
Methods: In sham operated and bile duct ligated (BDL) rats, quantitative reverse transcriptase-polymerase chain reaction was used to assess hepatic ACE2 mRNA, and western blotting and immunohistochemistry to quantify and localise ACE2 protein. ACE2 activity was quantified by quenched fluorescent substrate assay. Similar studies were performed in normal human liver and in hepatitis C cirrhosis.
Results: ACE2 mRNA was detectable at low levels in rat liver and increased following BDL (363-fold; p<0.01). ACE2 protein increased after BDL (23.5-fold; p<0.05) as did ACE2 activity (fourfold; p<0.05). In human cirrhotic liver, gene (>30-fold), protein expression (97-fold), and activity of ACE2 (2.4 fold) were increased compared with controls (all p<0.01). In healthy livers, ACE2 was confined to endothelial cells, occasional bile ducts, and perivenular hepatocytes but in both BDL and human cirrhosis there was widespread parenchymal expression of ACE2 protein. Exposure of cultured human hepatocytes to hypoxia led to increased ACE2 expression. In preconstricted rat aorta, Ang 1–7 alone did not affect vascular tone but it significantly enhanced acetylcholine mediated vasodilatation in cirrhotic vessels.
Conclusions: ACE2 expression is significantly increased in liver injury in both humans and rat, possibly in response to increasing hepatocellular hypoxia, and may modulate RAS activity in cirrhosis.
Keywords: liver, fibrosis, renin-angiotensin system, angiotensin converting enzyme
The main effector of the renin angiotensin system (RAS), angiotensin (Ang) II, is a potent systemic vasoconstrictor that plays a central role in cardiovascular homeostasis. However, there is evidence from studies in a wide range of organs that, in addition to its systemic effects, locally produced Ang II contributes to tissue injury and fibrosis in many chronic diseases. A number of studies have provided evidence that the RAS also contributes to the pathogenesis of chronic liver disease. There is marked upregulation of intrahepatic RAS components in experimental injury, and RAS inhibitors have been shown to inhibit hepatic fibrosis in several animal models.1–4 In addition, in advanced cirrhosis there is activation of the systemic RAS in response to mesenteric and systemic vasodilatation. Although vasoconstriction responses to Ang II are significantly impaired in patients with end stage liver disease,5 this response of the systemic RAS plays a central role in maintaining blood pressure and renal perfusion.5,6
Angiotensin converting enzyme (ACE), a dipeptidyl carboxypeptidase, is a key enzyme in the RAS and converts Ang I to the potent vasoconstrictor Ang II.7 ACE2 is a recently identified homologue of ACE8,9 which unlike ACE, has only one active enzymatic site, degrades Ang II to Ang 1–7, and cleaves a single residue from Ang I to generate angiotensin 1–9.10 Ang 1–9 has no known effects but is converted to Ang 1–7 by ACE (fig 1 ▶).11 There is evidence that Ang 1–7 may modulate the effects of RAS activation via several mechanisms, including inhibition of ACE, blockade of the AT1 receptor, and by its direct effects on vascular tone.12 Thus ACE2 may play an important counterregulatory role to the RAS, protecting tissues from Ang II mediated injury by both degrading Ang II and inhibiting its production, and by increasing production of Ang 1–7.12
Figure 1.

Schematic diagram of the renin angiotensin system (RAS) showing the main pathway for angiotensin (Ang) II generation from Ang I via angiotensin converting enzyme (ACE), and the role of ACE2 to degrade Ang I to Ang 1–9, and Ang II to the vasodilator Ang 1–7.
ACE2 was described in the endothelium of heart, kidney, and testis and is also detected in the normal large and small intestine.13 Evidence for a biological role for ACE2 is provided by studies in ACE2 knockout mice. These animals have impaired cardiac contractility and upregulation of hypoxia induced genes in the heart associated with impaired degradation of Ang II,14 suggesting ACE2 is an important regulator of cardiac function and a possible link between ACE2 and tissue oxygenation. In experimental models of hypertension and diabetes, renal expression of this enzyme is reduced and this may contribute to Ang II mediated tissue injury.14,15 ACE2 is also a functional receptor for the severe acute respiratory syndrome (SARS) coronavirus, suggesting that it may play a role in SARS infection.16,17
Although expression of ACE2 in normal human liver tissue is low,9 the possibility that ACE2 is modulated in liver disease has not been explored. This study examined hepatic expression of ACE2 in normal rats and in the bile duct ligated (BDL) rat, a model of hepatic fibrosis, as well as in normal human livers and hepatitis C cirrhotic livers removed at liver transplantation. We also examined the possible vascular effects of Ang1–7 in cirrhosis by studying the effects of Ang 1–7 in preconstricted aortic rings from healthy and cirrhotic rats.
METHODS
Experimental procedures were approved by the Animal and Human Research Ethics Committees of Austin Health and performed according to the NHMRC of Australia Guidelines for animal experimentation and the principles of the Helsinki declaration. Sprague-Dawley rats (200–250 g) were housed in a 12:12 h light-dark cycle, with ad libitum food containing 0.4–0.6% NaCl (Norco, Lismore, NSW, Australia) and water.
Animal model
Double bile duct ligation and transection were performed in rats (n = 9), as previously described.18 Sham operated (control) rats underwent the same surgical procedure but the bile duct was not ligated (n = 9). At day 21, animals were sacrificed by excision of the liver following lethal anaesthesia. Liver tissue was fixed in 4% paraformaldehyde and paraffin embedded for immunohistochemical studies. Tissue was frozen for quantitative reverse transcriptase-polymerase chain reaction (QRT-PCR) analysis, western blotting and measurement of ACE2 activity.
In a second group of experiments, rats were sacrificed at 7, 14, and 21 days post BDL and 21 days after sham operation (n = 4 in each group) in order to determine the location of ACE2 protein expression by immunohistochemistry during the evolution of liver injury and its possible relationship to cellular hypoxia, as assessed by immunostaining of pimonidazole adducts. For these experiments, pimonidazole hydrochloride (Hypoxyprobe-1) was obtained from Chemicon Australia Pty Ltd. (part No 90201, Victoria, Australia) in the form of sealed vials containing 1.0 g of pimonidazole hydrochloride. Prior to sacrifice, rats were anaesthetised with an intraperitoneal injection of pentobarbitone sodium (Nembutal), and a bolus injection of pimonidazole hydrochloride (80 mg/kg) dissolved in sterile 0.9% saline was injected into the jugular vein. One hour after injection, rats were anaesthetised again, the livers removed, fixed in 4% paraformaldehyde phosphate buffered saline (PBS) solution, and then embedded in paraffin.
Human liver tissue
Explanted liver tissue from patients who had undergone liver transplantation for hepatitis C induced cirrhosis (n = 9) was obtained at the time of transplantation and frozen for real time QRT-PCR analysis, western blotting, and measurement of ACE2 activity. Control livers included one donor liver, one explant from a patient transplanted for hypercholesterolaemia, and one specimen from resection of a colorectal metastasis taken well clear of the tumour margin. All three liver samples were histologically normal and were stored in the same manner as cirrhotic tissues. Tissue from three of the hepatitis C cirrhotic livers and three normal livers was fixed in 10% buffered formalin for immunohistochemical and in situ hybridisation studies.
Isolation of total RNA and synthesis of cDNA
Total RNA was isolated from liver tissue suspensions by homogenising with an Ultra-Turrax (Janke and Kunkel IKA, Labortechnik, Germany) in Trizol (Invitrogen Life Technologies, Melbourne, Victoria, Australia).15,19 cDNA was synthesised with a reverse transcriptase reaction carried out with the use of standard techniques (Superscript First Strand Synthesis System for QRT-PCR; Life Technologies Inc) with random hexamers, dNTPs, and total RNA extracted from control and diseased livers. An aliquot of the resulting single strand cDNA was used in the real time PCR (QRT-PCR) experiments, as described below. To assess genomic DNA contamination, controls without reverse transcriptase were included.
Quantitative real time RT-PCR (QRT-PCR)
QRT-PCR is a fully quantitative method for determination of the amounts of mRNA. Briefly, gene specific 5′-oligonucleotide corresponding to the human ACE (5′-TGC CTA TCC TTC CTA TAT CAG TCC AA), ACE 3′-oligonucleotide primer (5′-TTG TCC AAA ATC TAC CCC ACA TAT C), and ACE probe (FAM 5′-ATG CCT CCC TGC TCA TTT GCT TGG T-TAMRA) for rat ACE2 gene specific 5′-oligonucleotide (5′-GCC AGG AGA TGA CCG GAA A), an ACE2 3′-oligonucleotide primer (5′-CTG AAG TCT CCA TGT CCC AGA TC), and ACE2 probe (FAM5′-TTG TCT GCC ACC CCA CA-MGBNFQ) were designed using the software program, Primer Express (PE Applied Biosystems, Foster City, California, USA).
Generation of amplicons was defined by the point during cycling when amplification of the PCR product was first detected. The QRT-PCR reaction took place with 500 nmol/l of forward and reverse primer and 50 nM of FAM/TAMRA ACE/ACE2 probe and VIC/TAMRA 18S ribosomal probe, in 1× Taqman universal PCR master mix (PE Biosystems). Each sample was run and analysed in duplicate. Samples from sham livers were used as the calibrator with a given value of 1 and the BDL groups compared with this calibrator.15,19
Western blotting
Western blotting for ACE2 was performed as previously described.15 Liver tissue from rat and human tissue was minced, resuspended in buffer containing 10 mM HEPES, 150 mM NaCl, 1 mM EGTA, 5 mM MgCl2, and 0.02% NaN3, 5% Triton ×100, pH 7.4, to which 0.5 μg/ml pepstatin, 0.25 mg/ml leupeptin (Sigma, St Louis, Missouri, USA), 0.1 mg/ml benzamidine, and 0.1 mg/ml bacitracin were added, and homogenised at 13 000 rpm with the Ultra-Turrax (Janke and Kunkel IKA) and centrifuged at 1000 g at 4°C for 30 minutes. The resultant supernatant was harvested and stored in aliquots at −80°C. Samples (100 µg protein) were loaded and run on a 10% sodium dodecyl sulphate denaturing gel system and transblotted onto nitrocellulose filters (Hybond P, Amersham-Pharmacia Biotech, Buckinghamshire, UK) using a transfer tank at 15 V for 30 minutes. At the end of the transfer, filters were blocked with 10% non-fat skim milk powder in Tris buffered saline and 0.1% Tween (TBS/Tween) for one hour at room temperature. The primary ACE2 antibody diluted to a concentration of 1/5000 with 10% skim milk powder in TBS/Tween was incubated overnight at room temperature. The membrane was then washed thoroughly three times in wash solution (TBS/Tween). Positive bands were developed using the western blotting analysis system (Amersham-Pharmacia Biotech, Buckinghamshire, UK) in which horseradish peroxidase labelled secondary sheep antirabbit antibody (Chemicon, Temecula, California, USA) was diluted at 1/2000 and incubated for one hour at room temperature. Exposed Biomax film of bands representing ACE2 protein were quantified on an Automated Imaging System (Imaging Research Inc., St Catherines, Ontario, Canada).
Quenched fluorescent substrate assay of ACE2 activity
Liver homogenate (10 µl) was incubated with an ACE2 specific quenched fluorescent substrate (QFS: (7-methoxycoumarin-4-yl)-acetyl-Ala-Pro-Lys(2, 4-dintirophenyl)); Auspep, Parkville, Victoria, Australia), as previously described,20 in duplicate. Assays were performed in black 96 well microtitre plates with 50 µM QFS in a final volume of 200 µl per well ACE2 assay buffer (100 mM Tris, 1 M NaCl, pH 6.5). Final concentration of DMSO (used to solubilise QFS) was 0.7%. Reactions proceeded at 37°C for 30 minutes within a thermostat fMax fluorescence microplate reader (Molecular Devices, Sunnyvale, California, USA), prior to reading the liberated fluorescence (λex = 320 nm, λem = 420 nm). Cleavage of QFS was attributed to ACE2 by the use of a specific inhibitor (S,S) 2-(1-carboxy-2-[3-(3,5-dichloro-benzyl)-3H-imidazol-4-yl]-ethylamino)-4-methyl-pentanoic acid (MLN-4760; a generous gift from Dr Natalie Dales, Millennium Pharmaceuticals, Cambridge, Massachusetts, USA.) at 1 µM final concentration.21 As QFS can be cleaved by prolyl endopeptidase, a specific inhibitor of this enzyme, Z-Pro-prolinal (10 µM), was included in all wells. The specific activity of the preparations was expressed as units of fluorescence/mg membrane protein/h.
Immunohistochemistry
Immunohistochemical studies for ACE2 were performed as previously described on 4 µm sections of paraffin embedded liver (human and rat) tissue mounted on silane coated slides.15 Slides were dewaxed in histolene and hydrated in graded ethanol, and endogenous peroxidase activity was removed by treating sections with 3% hydrogen peroxide in PBS for 20 minutes. The primary antibody to ACE2 (donated by Dr S Acton, Millennium Pharmaceuticals, Cambridge, Boston, Massachusetts, USA) was applied at room temperature for one hour to slides15 with a dilution of 1:500 for rat sections and 1:800 for human sections. Specific staining in human sections was detected using the standard ABC (avidin-biotin complex) method. Slides were incubated for 20 minutes with the secondary antibody (biotin conjugated goat antirabbit IgG; Dako, Copenhagen, Denmark) at a concentration of 1/250. The avidin-biotin Vectastain ABC system (Vector Laboratories, Burlingame, California, USA) was applied for 20 minutes. For rat sections, specific staining was detected using Zymed SuperPicTure polymer detection kit. Slides were incubated with antirabbit horseradish peroxidase polymer for 15 minutes. The final detection step for all slides was carried out using 3,3′-diaminobenzidine (Sigma) as the chromogen. Sections were counterstained with haematoxylin. Three types of control tests were performed to determine the specificity of the antibody. Firstly, control sections were incubated with anti-ACE2 antibody solutions which had been preabsorbed with the synthetic peptide to which the antibody was raised (peptide sequence: NTNITEENVQNMNNAGDKW aa 51–69); secondly, sections were incubated with rabbit non-immune serum; and thirdly, sections were incubated with PBS without the primary antibodies. These control sections did not reveal any staining (see fig 4A ▶).
Figure 4.
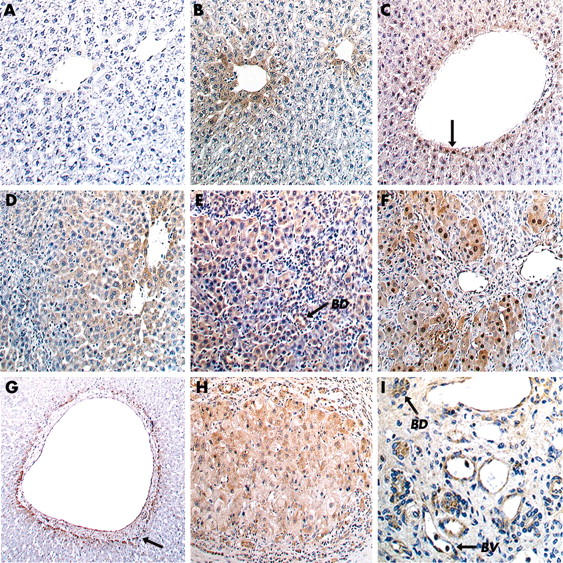
Immunohistochemical staining of angiotensin converting enzyme 2 (ACE2) protein in rat and human liver. In rat liver, pimonidazole immunostaining was used as a marker of cellular hypoxia. Normal rat liver is shown in the top row (A–C). There was no hepatic ACE2 staining detectable following ACE2 peptide preabsorption (A), confirming the specificity of the antibody used to detect ACE2. In normal livers. ACE2 immunolabelling (B) and staining of pimonidazole adducts (C, arrow) were restricted to perivenular hepatocytes. Bile duct ligated (BDL) rat livers are shown in the second row (D–F). In rat livers at seven days post BDL (D), ACE2 expression was more heterogenous and widespread than in normal livers. At 21 days post BDL, immunostaining of both ACE2 (E) and pimonidazole adducts (F) was detected in more than 80% of hepatocytes, with ACE2 also detected in bile duct cells and endothelial cells. (G–I) Human liver. In normal liver, ACE2 was detected in hepatocytes surrounding occasional larger central veins (G, arrow). In cirrhotic liver, there was widespread ACE2 expression throughout cirrhotic nodules (H) and in bile duct cells (BD) and endothelial cells lining small blood vessels (BV) (I). Original magnifications ×850, except (4G) and (4H) × 450, and (4I) ×1700.
Immunostaining for pimonidazole binding was performed on sections (4 μm) from formalin fixed paraffin embedded liver samples following the manufacturer’s instructions (Chemicon International). Briefly, sections were deparaffinised, rehydrated, and incubated with 3% aqueous hydrogen peroxide for five minutes at room temperature to inactivate endogenous peroxidase. After antigen retrieval by Pronase digestion for 40 minutes at 40°C, the primary monoclonal antibody (Hypoxyprobe-1Mab1; Chemicon Australia Pty Ltd, part No 90204) for pimonidazole adducts at a dilution of 1:50 was applied for 40 minutes at room temperature. Biotin-conjugated F(c) secondary goat antimouse antibody (Sigma-Aldrich Pty Ltd, Sydney, Australia), at a dilution of 1:500, was then applied for 10 minutes at room temperature followed by incubation with the avidin-biotin Vectastain ABC system (Vector Laboratories) for 10 minutes at room temperature. Positive signals were detected by incubation of the tissue sections with DAB chromogen for 10 minutes at room temperature. Sections were counterstained with haematoxylin for 15 seconds and mounted. As a negative control, normal goat serum was used instead of primary antibody.
Effects of hypoxia on ACE2 expression in cultured hepatocytes
The human hepatoma cell line HepG2 was obtained from the ATCC. Cell culture media and supplements were purchased from Invitrogen (Carlsbad, California, USA). Cells were seeded at a density of approximately 105 cells per 10 cm diameter culture dish in 10 ml MEM supplemented with 10% heat inactivated fetal bovine serum, 100 U/ml penicillin, and 0.1 mg/ml streptomycin, and grown to confluence at 37°C in a humidified atmosphere of 95% air and 5% carbon dioxide.
When cultures reached confluence, the culture medium was removed and replaced with fresh medium. Cells were then incubated in a sealed container (AnaeroPack, Mitsubishi Gas Chemical Co., Japan) under either normal (room air, n = 5 replicate experiments) or hypoxic conditions using the AnaeroGen system (AnaeroGen, Oxoid, Hants, UK) as previously described.22 After 2, 4, or 6 hours of hypoxia (n = 6 replicate experiments in each group), the culture medium was removed from replicate cultures and cell monolayers rinsed briefly with PBS before storage at −70°C for later analysis.
Vascular responses to Ang 1–7
These studies were performed in vessels from Sprague-Dawley rats, four weeks after undergoing bile duct ligation, and in age and sex matched controls using previously described techniques.23 Briefly, animals were sacrificed and the thoracic aorta removed and cleaned of connective tissue and fat. Rings were cut into 3 mm segments and placed between two metal hooks in an organ bath chamber (six rings). The chambers were kept at a constant 37°C and continuously bubbled with carbogen. Rings were left to equilibrate for one hour with no tension, and then the tension was progressively increased to 1 g. After this, rings were allowed to equilibrate for 30 minutes and then the maximum contraction was obtained by incubating the vessels with a high potassium solution (K+ = 124 mM). After the contraction reached a plateau, vessels were rinsed with Krebs three times (10 minute intervals) and allowed to rest for one hour. Vessels were then contracted to 30–50% of maximum with phenylephrine.
In an initial study, vessels were then exposed to increasing concentrations of Ang 1–7 (10−8–10−5) at one minute intervals over 10 minutes. In a second study, after vessels reached a plateau, 10−6 Ang(1–7) was added to every second bath. After one minute, increasing concentrations (10−8–10−5) of acetylcholine (Ach) were added to all baths, at half log concentrations only after each vasorelaxation had reached a plateau. Per cent relaxation was obtained by measuring the change in tension from the initial contraction to phenylephrine.
Statistics
All data are shown as mean (SEM) unless otherwise specified. Data were analysed by the Mann-Whitney U test. A p value of less than 0.05 was considered statistically significant.
RESULTS
Hepatic fibrosis in the BDL model
Light microscopy of haematoxylin and eosin stained liver sections from 21 day BDL animals showed extensive fibrosis, bile duct proliferation, and nodular transformation without cirrhosis.
Human liver histology
All livers had established hepatitis C related cirrhosis with varying degrees of inflammatory activity and no evidence of other chronic liver diseases.
ACE2 expression and activity in rat tissue
Only low levels of ACE2 mRNA expression were detected in control rat liver tissue but these were increased 363-fold following BDL (p<0.01) (fig 2A ▶). Western analysis revealed low levels of ACE2 protein in control livers which increased 23.5-fold following bile duct ligation (p<0.05) (fig 2B, 2C ▶ ▶). The increase in ACE2 expression was associated with a fourfold increase in ACE2 enzyme activity in BDL livers compared with controls (p<0.05) (fig 2D ▶).
Figure 2.
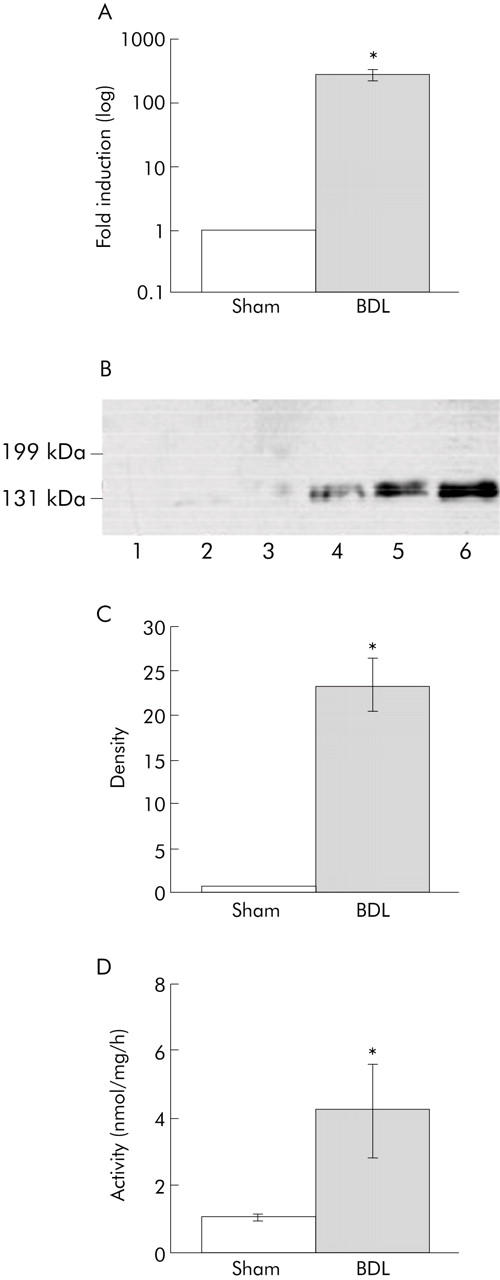
Angiotensin converting enzyme 2 (ACE2) expression and activity in sham operated control and bile duct ligated (BDL) rat liver. (A) Quantification of ACE2 mRNA by quantitative reverse transcriptase-polymerase chain reaction, with control values arbitrarily standardised to 1. Values are mean (SEM), n = 6 in each group; *p<0.01, BDL versus control. (B) Western blot of ACE2 protein in control and BDL livers. (C) Quantification of western blots. Values are mean (SEM); *p<0.05, BDL versus control. (D) ACE2 activity in sham and BDL. Values are mean (SEM), n = 9 in each group; *p<0.05, BDL versus control.
ACE2 protein and activity in human liver
As in rat liver, ACE2 mRNA was detectable at low levels in healthy livers. ACE2 mRNA levels were upregulated 34-fold in hepatitis C induced cirrhotic liver tissue compared with controls (p<0.01) (fig 3A ▶). This was associated with a 97-fold increase in ACE2 protein by western blotting (p<0.01) (fig 3B ▶, C) while ACE2 activity was increased 2.4-fold compared with controls (p<0.01) (fig 3D ▶).
Figure 3.

Angiotensin converting enzyme 2 (ACE2) expression and activity in normal and cirrhotic (HepC) human liver. (A) Quantification of ACE2 mRNA by quantitative reverse transcriptase-polymerase chain reaction of normal human (n = 3) and cirrhotic livers (n = 20) with control values arbitrarily standardised to 1. Values are mean (SEM); *p<0.01, cirrhosis versus control. (B) Western blotting of ACE2 protein in normal and cirrhotic liver. (C) Quantitation of western blotting in normal and cirrhotic liver. Values are mean (SEM), n = 3 and 4 per group, respectively; *p<0.01, cirrhotics versus normal. (D) ACE2 activity in normal and cirrhotic liver. Values are mean (SEM), n = 3 and n = 9, respectively; *p<0.01, cirrhotics versus normal.
Localisation of ACE2 expression and pimonidazole staining
Immunohistochemistry demonstrated that in livers from sham operated control rats, only a small percentage of cells expressed ACE2 protein and this was almost entirely confined to perivenular hepatocytes (fig 4B ▶). In these livers, hypoxia was detected by the presence of pimonidazole adduct staining in a perivenular pattern affecting the last row of perivenular hepatocytes, similar to that seen for ACE2 immunolabelling (fig 4C ▶). In livers from animals that were studied at seven days post BDL (fig 4D ▶), ACE2 expression was markedly increased, involving 30–40% of cells in each lobule. However, the strongest labelling was still seen in perivenular cells (fig 4D ▶). At 14 days post BDL there was a further increase in ACE2 staining. At 21 days there was very widespread ACE2 expression, with staining identified in more than 80% of hepatocytes as well as bile duct epithelial and endothelial cells (fig 4E ▶). In these livers, pimonidazole adducts were detected in most hepatocytes in keeping with the pattern of ACE2 expression (fig 4E ▶, F).
In normal human liver, ACE2 staining was minimal and confined to occasional bile duct cells, vascular endothelium, and perivenular hepatocytes (fig 4G ▶). In contrast, in cirrhotic human liver, ACE2 was detected in most hepatocytes within cirrhotic nodules, as well as bile duct cells and vascular endothelial cells (fig 4H ▶, I).
Effects of hypoxia on ACE2 expression in hepatocytes
Expression of ACE2 mRNA in HepG2 cells cultured under normoxic (room air) and hypoxic conditions was assessed by QRT-PCR. ACE2 expression was significantly higher in cells incubated under hypoxic conditions and increased with duration of hypoxia (1.69-fold increase at two hours, 1.81-fold at four hours, and 2.61-fold at six hours (p = 0.01, 0.04, and 0.01, respectively).
Vascular responses to Ang 1–7
Addition of Ang 1–7 alone to preconstricted aortic rings produced no significant effects on vascular tone. Ach produced significant vasodilatation in both healthy and cirrhotic rat vessels (fig 5 ▶). However, while Ang 1–7 had no effect on the response to Ach in normal vessels, it enhanced Ach mediated vasodilatation in cirrhotic vessels (p<0.05).
Figure 5.

Effects of angiotensin (Ang) 1–7 on responses to acetylcholine (Ach) in aortic rings from normal (Con) and bile duct ligated (BDL) rats. Addition of Ang 1–7 to the organ bath enhanced Ach mediated vasodilatation in vessels from BDL rats. **p<0.01, ***p<0.001 versus control; †p = 0.043 versus BDL.
DISCUSSION
Although ACE2 expression is low in normal liver tissue,9,13 the results of this study show that in rat and human liver disease there is major upregulation of hepatic ACE2 at the gene and protein levels, accompanied by significant increases in ACE2 activity. These findings demonstrate that chronic liver injury causes a major increase in the hepatic expression of ACE2 and raise the possibility that it may play a role in disease pathogenesis.
Evidence to date has indicated that in normal physiology expression of ACE2 is more restricted than ACE, and is localised to the heart, kidney,9 testis,8 and large and small intestine.13 With the use of highly sensitive QRT-PCR techniques, low levels of ACE2 mRNA have been detected in the normal human liver13 and this is consistent with our own findings in control rodent and human liver. We have previously shown in the BDL liver that ACE protein was abundant in proliferating bile duct epithelial cells, consistent with studies in the kidney which identified epithelial cells as a major site of both ACE and ACE2 expression.15 However, the current study has demonstrated that ACE2 is also strongly expressed in hepatocytes in this model of chronic liver injury. Whether these patterns of upregulation of ACE and ACE2 are specific to the BDL model is unknown, and expression of the RAS in other models of experimental hepatic fibrosis deserves study. However, our findings in human hepatitis C induced cirrhotic liver suggest that a widespread increase in ACE2 expression may be a general feature of liver injury in both rats and humans.
To date, there are few reports addressing ACE2 expression in other disease states. The first by Crackower et al showed that ACE2 mRNA and protein levels were downregulated in rat models of hypertension.14 Furthermore, in the same study, the finding that the ACE2 knockout mouse developed impaired cardiac function lends credence to the hypothesis that this enzyme is a critical regulator of cardiac function. A second study demonstrated that ACE2 gene and protein expression are reduced in the kidney in experimental diabetes and that this paralleled changes in ACE expression.15 It has been postulated that ACE2 may function in conjunction with ACE to regulate the generation and degradation of angiotensin peptides, including Ang I, Ang II, Ang 1–9, and Ang 1–7.12 This is supported by the finding that tissue Ang II levels are significantly increased in ACE2 knockout mice,14 and suggests that ACE and ACE2 have counterregulatory functions.
The finding that in the hearts of ACE2 knockout mice elevation of Ang II levels is associated with upregulation of hypoxia inducible genes14 indicates that an important role of ACE2 may be to maintain local tissue perfusion and oxygenation, and to reduce Ang II mediated vasoconstriction. The concept that ACE2 may be upregulated in response to tissue hypoxia is supported by our recent finding that ACE2 expression is increased in response to myocardial ischaemia.19 Our findings in the liver are also consistent with this idea, as ACE2 expression in normal livers was largely restricted to perivenular hepatocytes, the cells of the lobule exposed to the lowest oxygen tension. As has been previously shown,22 the presence of hypoxia in these cells was confirmed by pimonidazole staining. There is considerable evidence that hepatic fibrogenesis is associated with progressive impairment of hepatocellular oxygenation, with the likely causes including sinusoidal capillarisation, intrahepatic shunting, vasoconstriction, and disruption of sinusoidal flow. As has been previously demonstrated, in our study there was extensive staining of pimonidazole adducts in fibrotic livers, indicating the development of widespread hepatocellular hypoxia that may be a stimulus for increased ACE2 production. Consistent with this hypothesis, we found that in HepG2 cells, ACE2 mRNA expression progressively rose in response to increasing periods of hypoxia. Using a similar experimental approach, Corpechot et al demonstrated a similar degree of upregulation of the hypoxia inducible growth factor vascular endothelial growth factor in hypoxic isolated rat hepatocytes.22
However, in the BDL liver, as with other models of tissue injury,7 there is a general increase in expression of ACE and other RAS components.2 Upregulation of ACE2 and its widespread expression throughout the liver in BDL may therefore represent a more general counterregulatory response to increased ACE levels and the multiple deleterious effects of increased local Ang II production. ACE2 upregulation may also contribute to the vasodilatation of cirrhosis by degrading Ang II, and this may help explain why vasoconstrictor responses to Ang II are impaired5 and vasodilatation persists in patients with advanced liver disease despite activation of the systemic RAS.5,6 In addition, ACE2 generates Ang 1–7, and this peptide may reduce Ang II production by inhibiting ACE and reduce Ang II mediated vasoconstriction by blocking AT1 receptors.24 It has also been shown that, in some tissues, Ang 1–7 may act as a potent vasodilator.12 Our study has confirmed that in both rat and human liver there is increased ACE2 expression and enzyme activity that would be expected to facilitate the degradation of Ang II and the formation of Ang 1–7. We also found that while in the rat aorta Ang 1–7 had no measurable effects on vascular tone, it enhanced Ach mediated vasodilatation in vessels from cirrhotic rats. Possible mechanisms for this effect include enhanced prostanoid release from vascular smooth muscle cells and direct stimulation of nitric oxide synthesis.25–27 These vascular effects of Ang 1–7 suggest a further mechanism by which ACE2 might contribute to altered vascular tone in cirrhosis.
ACE2 is also a functional receptor for the SARS coronavirus and plays an important role in SARS infection. Liver impairment is common in patients with SARS, and it has been recently shown that the SARS coronavirus causes a distinctive hepatitis characterised by lobular inflammation and hepatocyte apoptosis.28 Our finding that the major cellular receptor for SARS is present in the liver and is increased in liver injury provides a potential explanation for the finding that SARS infects the liver.
Recent research has been directed towards the importance of the RAS in the development of hepatic fibrosis4 and the potential benefits of modulation of this system either by ACE inhibition or Ang receptor antagonism.2,3 The finding of major upregulation of ACE2 in the BDL rat and cirrhotic human liver provides further insight into the complexities of the RAS and its regulation in liver injury. In the future, the development of specific modulators of ACE2 activity and function will help determine the role of ACE2 in the pathophysiology of liver disease.
Acknowledgments
Dr Paizis received a National Health and Medical Research Council (NHMRC) Medical Postgraduate Research Scholarship. The study was funded by grants from the NHMRC, the Sir Edward Dunlop Foundation, the Australian Brewer’s Association, and the Austin Hospital Medical Research Foundation.
Abbreviations
ACE, angiotensin converting enzyme
RAS, renin-angiotensin system
Ang, angiotensin
QRT-PCR, quantitative reverse transcriptase-polymerase chain reaction
SARS, severe acute respiratory syndrome
BDL, bile duct ligated
QFS, quenched fluorescent substrate
PBS, phosphate buffered saline
ABC, avidin-biotin complex
Ach, acetylcholine
Published online first 15 September 2005
Conflict of interest: None declared.
REFERENCES
- 1.Paizis G, Gilbert RE, Cooper ME, et al. Effect of angiotensin II type 1 receptor blockade on experimental hepatic fibrogenesis. J Hepatol 2001;35:376–85. [DOI] [PubMed] [Google Scholar]
- 2.Paizis G, Cooper ME, Schembri JM, et al. Up-regulation of components of the renin-angiotensin system in the bile duct-ligated rat liver. Gastroenterology 2002;123:1667–76. [DOI] [PubMed] [Google Scholar]
- 3.Jonsson JR, Clouston AD, Ando Y, et al. Angiotensin-converting enzyme inhibition attenuates the progression of rat hepatic fibrosis. Gastroenterology 2001;121:148–55. [DOI] [PubMed] [Google Scholar]
- 4.Bataller R, Sancho-Bru P, Gines P, et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 2003;125:117–25. [DOI] [PubMed] [Google Scholar]
- 5.Helmy A, Jalan R, Newby DE, et al. Role of angiotensin II in regulation of basal and sympathetically stimulated vascular tone in early and advanced cirrhosis. Gastroenterology 2000;118:565–72. [DOI] [PubMed] [Google Scholar]
- 6.Newby DE, Jalan R, Masumori S, et al. Peripheral vascular tone in patients with cirrhosis: role of the renin-angiotensin and sympathetic nervous systems. Cardiovasc Res 1998;38:221–8. [DOI] [PubMed] [Google Scholar]
- 7.Johnston CI. Tissue angiotensin converting enzyme in cardiac and vascular hypertrophy, repair, and remodeling. Hypertension 1994;23:258–68. [DOI] [PubMed] [Google Scholar]
- 8.Tipnis SR, Hooper NM, Hyde R, et al. A human homolog of angiotensin-converting enzyme—Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 2000;275:33238–43. [DOI] [PubMed] [Google Scholar]
- 9.Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 2000;87:E1–9. [DOI] [PubMed] [Google Scholar]
- 10.Ferrario CM, Chappell MC, Tallant EA, et al. Counterregulatory actions of angiotensin-(1–7). Hypertension 1997;30:535–41. [DOI] [PubMed] [Google Scholar]
- 11.Burrell LM, Johnston CI, Tikellis C, et al. ACE2, a new regulator of the renin angiotensin system. Trends Endocrinol Metab 2004;15:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yagil Y, Yagil C. Hypothesis: ACE2 modulates blood pressure in the mammalian organism. Hypertension 2003;41:871–3. [DOI] [PubMed] [Google Scholar]
- 13.Harmer D, Gilbert M, Borman R, et al. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett 2002;532:107–10. [DOI] [PubMed] [Google Scholar]
- 14.Crackower MA, Sarao R, Oudit GY, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002;417:822–8. [DOI] [PubMed] [Google Scholar]
- 15.Tikellis C, Johnston CI, Forbes J, et al. Characterisation of renal angiotensin converting enzyme 2 in diabetic nephropthy. Hypertension 2003;41:392–7. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003;426:450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dimitrov DS. The secret life of ACE2 as a receptor for the SARS virus. Cell 2003;115:652–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kountouras J, Billing BH, Scheuer PJ. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br J Exp Pathol 1984;65:305–11. [PMC free article] [PubMed] [Google Scholar]
- 19.Burrell LM, Risvanis J, Kubota E, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J 2005;26:369–75. [DOI] [PubMed] [Google Scholar]
- 20.Vickers C, Hales P, Kaushik V, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem 2002;277:14838–43. [DOI] [PubMed] [Google Scholar]
- 21.Dales NA, Gould AE, Brown JA, et al. Substrate-based design of the first class of angiotensin-converting enzyme-related carboxypeptidase (ACE2) inhibitors. J Am Chem Soc 2002;124:11852–3. [DOI] [PubMed] [Google Scholar]
- 22.Corpechot C, Barbu V, Wendum D, et al. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology 2002;35:1010–21. [DOI] [PubMed] [Google Scholar]
- 23.Zulli A, Widdop RE, Hare DL, et al. High methionine and cholesterol diet abolishes endothelial relaxation. Arterioscler Thromb Vasc Biol 2003;23:1358–63. [DOI] [PubMed] [Google Scholar]
- 24.Roks AJ, van Geel PP, Pinto YM, et al. Angiotensin-(1–7) is a modulator of the human renin-angiotensin system. Hypertension 1999;34:296–301. [DOI] [PubMed] [Google Scholar]
- 25.Porsti I, Bara AT, Busse R, et al. Release of nitric oxide by angiotensin-(1–7) from porcine coronary endothelium: implications for a novel angiotensin receptor. Br J Pharmacol 1994;111:652–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muthalif MM, Benter IF, Uddin MR, et al. Signal transduction mechanisms involved in angiotensin-(1–7)-stimulated arachidonic acid release and prostanoid synthesis in rabbit aortic smooth muscle cells. J Pharmacol Exp Ther 1998;284:388–98. [PubMed] [Google Scholar]
- 27.Brosnihan KB, Li P, Ferrario CM. Angiotensin-(1–7) dilates canine coronary arteries through kinins and nitric oxide. Hypertension 1996;27:523–8. [DOI] [PubMed] [Google Scholar]
- 28.Chau TN, Lee KC, Yao H, et al. SARS-associated viral hepatitis caused by a novel coronavirus: report of three cases. Hepatology 2004;39:302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]