

Abstract
Background: Oxidative stress, including the generation of reactive oxygen species (ROS), is involved in hepatofibrogenesis. The authors’ previous studies have shown that oestradiol suppresses hepatic fibrosis in animal models and attenuates the activation of cultured rat hepatic stellate cells (HSCs), which possess oestrogen receptor subtype β and are also activated by ROS.
Aims: To define the mechanisms by which female sex hormones play an antifibrogenic role in activated HSCs, the effects of oestradiol and progesterone on ROS generation processes and intracellular pathways, leading to the activation of HSCs undergoing oxidative stress, was examined.
Methods: HSCs, isolated from rats, were cultured for 7 days with oestradiol or progesterone for 24 hours as pretreatment, and oxidative stress was then induced by exposure to low doses of hydrogen peroxide for another 24 hours.
Results: Oestradiol inhibited ROS generation and antioxidant enzyme loss via the suppression of NADH/NADPH oxidase activity, and attenuated hydrogen peroxide induced transforming growth factor-β1 (TGF-β1) expression, HSC proliferation and transformation, and the activation of mitogen activated protein kinase (MAPK) pathways and transcription factors. Progesterone exerted a stimulatory effect through the progesterone receptor on the induction of ROS generation processes and intracellular pathways, resulting in TGF-β1 expression and HSC activation, and fibrogenic effects were inhibited by oestradiol.
Conclusion: These findings show for the first time that oestradiol inhibits the activation of transcription factors by suppressing ROS generation processes and the MAPK pathways, and inactivates the downstream transcription processes involved in TGF-β1 expression and HSC activation, whereas progesterone acts in opposition to the favourable effects of oestradiol and its effects are blocked by oestradiol.
Keywords: oestradiol, progesterone, hepatic stellate cells, ROS, TGF-β
Parenchymal cell membrane damage could produce reactive oxygen species (ROS) derived from lipid peroxidative processes, which represent the general feature of sustained inflammatory response and liver injury, and play a causative role in hepatic fibrosis development.1 Cells are well equipped to neutralise the effects of ROS, using a series of antioxidant protective systems, including enzymatic defence molecules such as superoxide dismutase (SOD), glutathione peroxidase, and catalase. Oestradiol is a potent endogenous antioxidant, which reduces lipid peroxidation and increases levels of SOD and glutathione peroxidase activity in the liver of a hepatic fibrosis model,2 and inhibits ROS generation in cultured rat hepatocytes in a state of prooxidant induced oxidative stress.3,4 We have already reported that oestradiol treatment results in the dose dependent suppression of hepatic fibrosis, accompanied by reduced collagen production in the liver of rat models of hepatic fibrosis,5,6 and attenuated iron induced lipid peroxidation in rat liver mitochondria.7 Hepatitis C virus infections are recognised as a major causative factor in the development of hepatic fibrosis to cirrhosis. Epidemiological studies support the view that cirrhosis development is more common in men than women with the exception of classical autoimmune diseases. Using multivariate analysis with chronic hepatitis C patients, it has been reported that male gender is associated with advanced hepatic fibrosis, independent of age at the time of hepatitis C virus infection and of alcohol consumption, and that hepatic fibrosis begins to progress at 50 years of age, irrespective of the duration of virus infection.8 These findings implicate female sex hormones in a protective role against the development of hepatic fibrosis.9
In the injured liver, hepatic stellate cells (HSCs) in the space of Disse are regarded as the primary target cells for inflammatory stimuli, and undergo proliferation and transformation into α smooth muscle actin (αSMA) positive myofibroblast-like cells. These HSCs are activated cells and are responsible for much of the collagen synthesis observed during hepatic fibrosis development to cirrhosis, although liver myofibroblasts, which are located in the portal field and around the pericentral area, have also been observed in hepatic fibrogenesis.10,11 It has been reported that paracrine stimuli derived from hepatocytes undergoing oxidative stress induce HSC proliferation and collagen synthesis.12 HSCs are activated by the generation of ROS with Fe2+/ascorbate13 and by malondialdehyde (MDA)14 and 4-hydroxynonenal,15 end products of lipid peroxidation. In studies with cultured rat HSCs we have also shown that HSCs possess functional oestrogen receptor β, but not oestrogen receptor α of oestrogen receptor subtypes, which respond directly to oestradiol,14 and that oestradiol suppresses HSC activation with decreased proliferation and collagen production.5,6 However, there is little information about the possible role of progesterone, another female sex steroid, in hepatic fibrosis. Although in vitro studies showed that progesterone inhibited arterial smooth muscle cell proliferation,16 systemic hormone replacement therapy with oestrogens and progestins increases the dermal content of collagen.17 In contrast, progesterone combined with oestradiol attenuates the type I procollagen synthesis and fibroblast proliferation of the anterior cruciate ligament in a dose dependent manner.18 This discrepancy might result from differences in the sex steroid compounds, the dosage combinations, and the experimental procedures employed. It remains to be elucidated whether progesterone can modulate HSC activation.
A primary source of ROS production in non-phagocytic cells such as HSCs is NADH/NADPH oxidase. Compared with the other ROS species, hydrogen peroxide is more stable and membrane permeable leading to the hypothesis that it acts as a second messenger to regulate signaling events including mitogen activated protein kinase (MAPK) activation. The MAPK family includes three major subgroups, extracellular signal regulated kinase (ERK), p38 MAPK (p38), and c-Jun N-terminal kinase/stress activated protein kinase (JNK). ERK and JNK lie upstream of activator protein-1 (AP-1).19 JNK and p38 activation are more important in stress responses such as inflammation, which can also activate nuclear factor κB (NF-κB).20 AP-1 and NF-κB are key transcription factors that induce multiple genes involved in inflammation and fibrogenesis, including cytokines and growth factors such as platelet derived growth factor (PDGF) and transforming growth factor-β1 (TGF-β1). TGF-β1 is a major fibrogenic cytokine, acting as a paracrine and autocrine (from HSCs) mediator, which triggers and activates the proliferation and transformation of HSCs in vivo.
To define the mechanisms by which female sex hormones might play an antifibrogenic role in activated HSCs, the effects of oestradiol and progesterone on NADH/NADPH oxidase activity, ROS generation, lipid peroxidation, and levels of SOD, catalase and glutathione peroxidase activity, as well as the activation of MAPK pathways and transcription factors of AP-1 and NF-κB, and TGF-β1 expression were examined in cultured rat HSCs undergoing oxidative stress.
MATERIALS AND METHODS
Isolation and culture of rat HSCs
HSCs were isolated from the livers of male and female Wistar rats and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) on uncoated 35 mm diameter plastic dishes, as described previously.21 On day six of culture, the culture medium was removed, and cells were incubated in serum free DMEM with and without 17β-oestradiol (Sigma, St Louis, MO, USA) or progesterone (Wako, Osaka, Japan) for 24 hours. Oxidative stress was then induced by incubation with hydrogen peroxide for an additional 24 hours. In some experiments, cells were treated with ICI 182,780 (Tocris Cookson, Ballwin, MO, USA), a high affinity oestrogen receptor antagonist, the progesterone receptor antagonist RU486 (Wako), or diphenyleneiodonium (DPI, Sigma), a specific inhibitor of NADH and NADPH oxidases, in the presence and absence of oestradiol or progesterone for 24 hours. Steroid sex hormones and receptor antagonists were initially prepared as an ethanolic stock solution (10−2 mol/l) and then diluted with culture medium to the appropriate working solution concentration.
For the immunohistochemical examination of progesterone receptor, cells were prepared as previously described,14 and incubated overnight at 4°C with a 1:200 dilution of a polyclonal antibody against human progesterone receptor (C-20) (Santa Cruz, Santa Cruz, CA, USA). The samples were then washed and incubated with biotin conjugated goat antirabbit IgG F(ab’)2. Following incubation with the avidin-biotin complex (Vectastain R ABC reagent, Vector Laboratories, Burlingame, CA, USA), the antigen-antibody complexes were visualised with diaminobenzidine, and photographed using a differential interference contrast microscope. As a negative control, normal mouse IgG was substituted for the specific antibody at the same dilution. Paraffin sections of a normal rat uterus were used as a positive control.
Measurement of NADH and NADPH oxidase activities
HSCs were washed five times, scraped in ice cold phosphate buffered saline (PBS), homogenised, and added to a cuvette containing lucigenin (500 μmol/l) as the electron acceptor and either NADH (100 μmol/l) or NADPH (100 μmol/l) as the substrate in 50 mmol/l phosphate buffer (pH 7.0).22 NADH or NADPH oxidase activity was measured by a luminescence assay, as previously described.22 All measurements were expressed as nanomoles of substrate/minute per milligram of cellular protein. Protein concentrations were determined by the Lowry method.
Measurement of intracellular ROS and lipid peroxidation
Intracellular ROS was monitored using the 2′,7′-dichlorofluorescein diacetate (H2DCF-DA) assay, as previously reported.23 H2DCF-DA, dissolved in ethanol, was added at a final concentration of 10 μmol/l. 2′,7′-Dichlorofluorescein fluorescence was measured using a flow cytometer (EPICS XL, Coulter, Hialeah, FL, USA) with excitation at 488 nm and emission at 525 nm.
Lipid peroxidation in the cells was determined by measuring MDA levels, using the thiobarbiturate method, as described previously.21
Antioxidant enzyme assays
Protein levels of a predominantly cytosolic copper, zinc dependent SOD (CuZn-SOD) were detected using an enzyme linked immunosorbent assay (ELISA) system kit (Amersham, Little Chalfont, UK), and glutathione peroxidase activities were determined using a Cellular Glutathione Peroxidase Assay kit (Calbiochem, San Diego, CA, USA), as described previously.3 The results are expressed as nanograms of immunoreactive protein levels for CuZn-SOD and the units for glutathione peroxidase per milligram of cellular protein. Catalase proteins were detected immunologically as described below.
Western blot analysis
Cell lysates were obtained with a sodium dodecyl sulfate (SDS) loading buffer as described previously.21 Ten micrograms of cell associated proteins were electrophoresed on 12% SDS-polyacrylamide gels, and the proteins were then transferred onto Hybond-ECL membranes (Amersham, Arlington Heights, IL, USA). Membranes were incubated in blocking buffer containing antibodies against catalase (Sigma), αSMA (DAKO, Glostrup, Denmark), IκB-α (inhibitory subunit of NF-κB; New England Biolabs, Beverly, MA, USA) and intracellular signaling proteins of ERK (Zymed, San Francisco, CA, USA), phosphorylated ERK (p-ERK) (Santa Cruz), p38 (Santa Cruz), phosphorylated p38 (p-p38) (Santa Cruz), JNK (Santa Cruz), and phosphorylated JNK (p-JNK) (Santa Cruz). A rabbit antibody against β-actin (Biomedical Technologies, Stoughton, MA, USA) was used to control protein loading. All antibodies were used at a dilution of 1:1000. Immunoreactive bands were visualised with an ECL western blotting detection system (chemiluminescence) kit (Amersham) as described previously,21 scanned with Gel Doc 2000 (Bio-Rad, Hercules, CA, USA) and analysed using Quantity One software (Bio-Rad).
Cell proliferation
Using a Biotrak cell proliferation ELISA system (Amersham),24 DNA synthesis was measured by incubating cells in 96-well plates with 10 μmol/l of the pyrimidine analogue, bromodeoxyuridine (BrdU) for 24 hours. After removing the culture medium, the cells were fixed and the incorporated BrdU was detected according to the manufacturer’s instructions, then compared with the effects of a positive control, PDGF-BB (5 ng/ml, Calbiochem).
ELISA analysis of TGF-β1 protein
To determine TGF-β1 protein, cells were rinsed with cold PBS and removed by scraping as described previously.25 Rat TGF-β1 in the cells was measured using an ELISA kit (R&D Systems) according to the manufacturer’s instructions.
Reverse transcription-polymerase chain reaction
Cells were collected using ISOGEN (Nippon gene, Tokyo, Japan) and total RNA was prepared according to the manufacture’s instructions. Reverse transcription-polymerase chain reaction (RT-PCR) was performed using a one-step RNA PCR kit (AMV) (Takara, Kyoto, Japan). Briefly, 1 μg of total RNA for each sample was added to the PCR reaction. For the gene expression of the progesterone receptor, after reverse transcription (30 minutes at 50°C; 2 minutes at 94°C), 30 cycles of PCR were performed for 30 seconds at 94°C, 45 seconds at 61°C, and 1 minute at 72°C, and the final elongation time was 7 minutes at 72°C. The set of primers was described previously.25,26 The resultant fragment of the progesterone receptor was 320 bp in size. Rat uterus was used as a positive control for the progesterone receptor, and yeast transfer RNA was used as a negative control.
For TGF-β1 gene expression, real-time PCR was performed using a SYBR Green PCR Core Reagents kit (Biosystems, Warrington, UK) according to the manufacturer’s protocol. The primer sequences were designed using Primer Express Software (Applied Biosystems, Foster City, CA, USA): 5′-TGGCGTTACCTTGGTAACC-3′ for the forward primer and 5′-GGTGTTGAGCCCTTTCCAG-3′ for the reverse primer. The primers for 18 S rRNA were purchased form a commercial vender. Quantitative PCR was performed on the ABI PRISM 7000 Sequence Detection System (Applied Biosystems) according to the manufacturer’s protocol as follows: 2 minutes at 50°C, 10 minutes at 95°C, and then 40 cycles with denaturation at 95°C for 15 seconds, and annealing at 60°C for 1 minute. The specificity of the produced amplification products was confirmed by examination of the dissociation reaction plots.27,28 A distinct single peak indicated that a single DNA sequence was amplified during PCR.
Electrophoretic mobility shift assay
Cellular extract proteins were prepared as described elsewhere.7 For the electrophoretic mobility shift assay (EMSA), an oligonucleotide corresponding to the DNA binding consensus site for AP-1 or NF-κB was end-labeled with [γ-32P]ATP and T4 polynucleotide kinase using an AP-1 family (T-cell activation) Gelshift kit (Geneka Biotechnology, Quebec, Canada) or an NF-κB family (T-cell activation) Gelshift kit (Geneka Biotechnology) as previously reported.3 The bandshift was visualised by autoradiography. Competition was performed by adding specific unlabeled double stranded oligonucleotide to the reaction mixture in a 20- or 100-fold molar excess.
Statistical analysis
Data are presented as the mean (standard deviation), unless otherwise indicated. Means were compared between two groups using Wilcoxon’s signed rank test and the Mann-Whitney U test. All p values are two tailed. A p value of less than 0.05 was considered to be statistically significant.
RESULTS
Protein and gene expressions of progesterone receptor in cultured rat HSCs with and without hydrogen peroxide exposure
We performed western blot analysis of extracts from the rat uterus using the antiprogesterone receptor antibody (C-20). When lysates of seven day cultured HSCs (fig 1A ▶) obtained from male and female rats were analysed by western blotting in the same manner, we found 94 kD and 114 kD bands for the progesterone receptor.29 The progesterone receptor expression was not significantly different when cultured with and without hydrogen peroxide, or between genders (fig 1A ▶). Immunohistochemistry for the progesterone receptor showed a strong nuclear positivity in HSCs obtained from male (fig 1B ▶) and female (data not shown) animals. No significant difference was found in the cellular localisation of the progesterone receptor between treatments with and without hydrogen peroxide or genders. In the negative control, the reaction of samples with normal IgG at the same dilution instead of the specific antibody showed no staining above background (data not shown).
Figure 1.
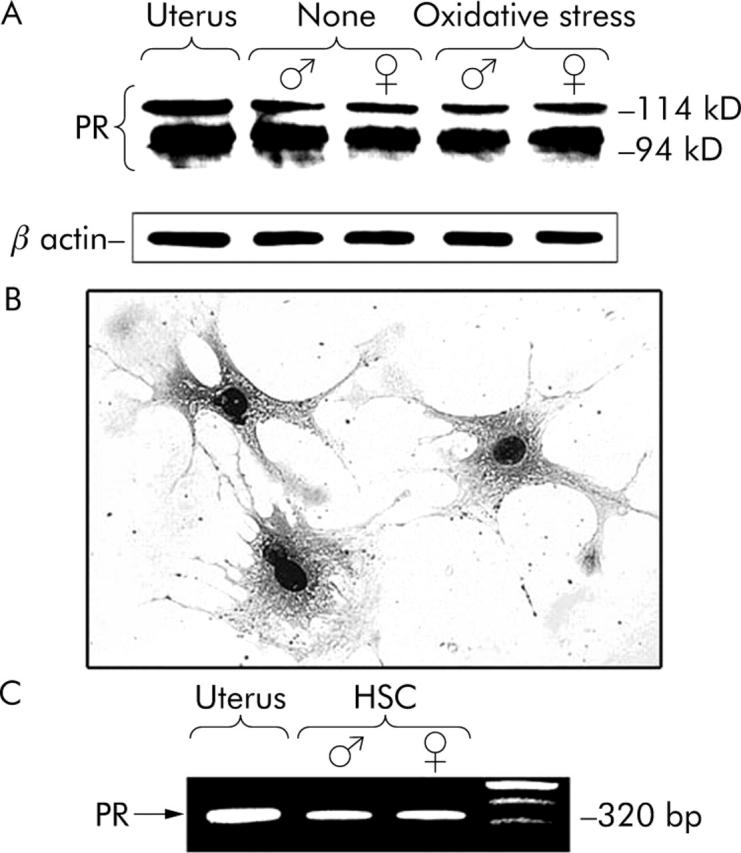
Western blot, immunohistochemical, and RT-PCR analyses of progesterone receptor in cultured HSCs from male and female rats. (A) On culture day seven, HSCs, preincubated in serum-free DMEM for 24 hours, were then incubated with (oxidative stress) and without (none) 10−5 mol/l hydrogen peroxide for another 24 hours. Cell lysates were subjected to 12% SDS-polyacrylamide gel electrophoresis and transferred onto nylon membranes, and progesterone receptor was detected immunologically. The graphs represent typical results of five independent experiments. (B) Reaction of cultured HSCs from males with an antibody against progesterone receptor (C-20). Original magnification ×200. (C) RT-PCR showed 320 bp transcripts for progesterone receptor in cultured HSCs from males as well as the uterus as a positive control. One of four similar studies is shown. PR, progesterone receptor.
The occurrence of the progesterone receptor in cultured HSCs from both genders was also investigated by RT-PCR. A transcript of 320 bp for the progesterone receptor was detected in the rat uterus (positive control) (fig 1C ▶), but not in yeast transfer RNA (negative control) (data not shown). The message for the progesterone receptor was detected in RNA samples of HSCs from males and females (fig 1C ▶).
Effects of oestradiol and progesterone on the activation of MAPK pathways, transcription factors, and TGF-β1 expression in cultured rat HSCs with and without hydrogen peroxide exposure
On culture day seven, exposure to low doses of hydrogen peroxide (10−7–10−5 mol/l) for 24 hours in HSCs preincubated in serum-free DMEM for 24 hours was observed to stimulate αSMA expression, a marker of HSC transformation, and cell proliferation in a dose dependent manner (data not shown). DNA synthesis in HSCs was measured by evaluating BrdU incorporation. Subsequent studies used a dose of 10−5 mol/l of hydrogen peroxide for further activation of cultured HSCs. Exposure to hydrogen peroxide induced the time dependent and transient phosphorylation of ERK, p38, and JNK, peaking at 15–60 minutes, over a 24 hour period (fig 2A ▶). The levels of non-phosphorylated MAPKs were unaffected by hydrogen peroxide exposure. In hydrogen peroxide activated HSCs, the time dependent activation of AP-1 and NF-κB and the degradation of inhibitory protein of NF-κB, IκB-α were observed, as shown in figures 2B and C ▶. The specificity of AP-1 or NF-κB DNA binding induced by hydrogen peroxide was confirmed in competition experiments. Incubation with excess mutant oligonucleotide had no effect on AP-1 or NF-κB binding (data not shown). The activation of AP-1 and NF-κB as determined by EMSA and the degradation of IκB-α, as detected by western blot analysis were readily detected after 15 minutes and peaked after 1 hour (figs 2B and C ▶). The DNA synthesis and αSMA expressions peaked after 1 hour and 24 hours, respectively (fig 3A ▶) as did the intracellular levels of TGF-β1 mRNA and protein (figs 3B and C ▶). Subsequent studies used an incubation time of 30 minutes to activate MAPK pathways, 1 hour for the activation of AP-1 and NF-κB with degradation of IκB-α, DNA synthesis, and TGF-β1 mRNA level, and 24 hours for αSMA expression and TGF-β1 protein level, after hydrogen peroxide exposure.
Figure 2.
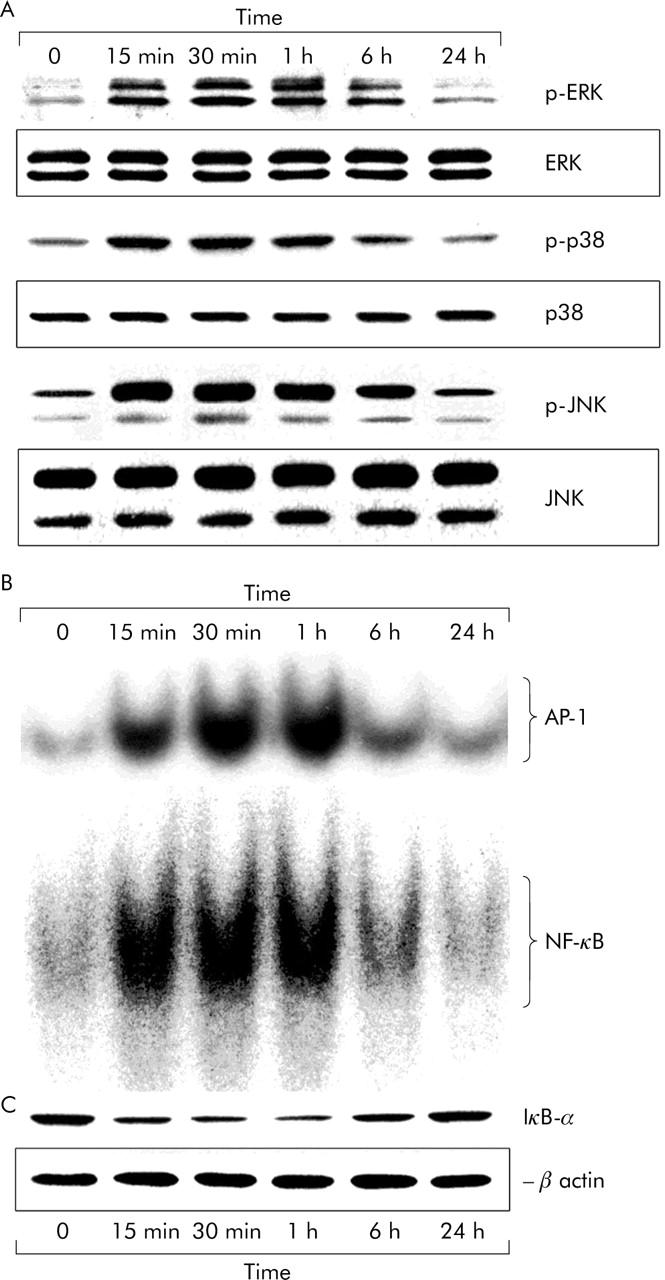
Activation of MAPK pathways and transcription factors of AP-1 and NF-κB after exposure to hydrogen peroxide in cultured rat HSCs. On culture day seven, HSCs, preincubated in serum-free DMEM for 24 hours, were then exposed to 10−5 mol/l hydrogen peroxide for another 24 hours. Following exposure to hydrogen peroxide, MAPK pathways of ERK, p38, and JNK (A) and transcription factors of AP-1 and NF-κB (B) were activated and IκB-α was degraded (C) in a time dependent manner. The micrographs represent typical results of three independent experiments.
Figure 3.
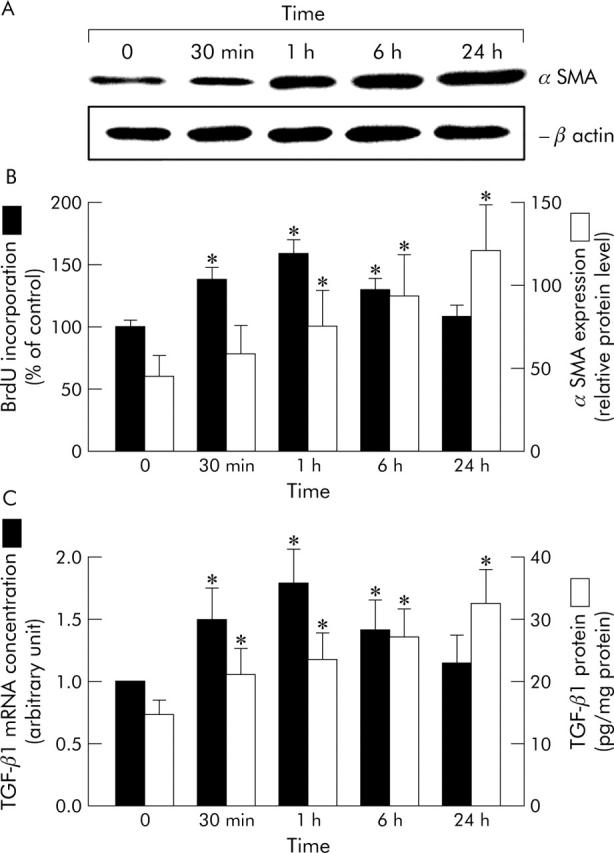
Stimulation of proliferation and αSMA expression and intracellular levels of TGF-β1 mRNA and protein after exposure to hydrogen peroxide in cultured rat HSCs. On culture day seven, HSCs, preincubated in serum-free DMEM for 24 hours, were then exposed to 10−5 mol/l hydrogen peroxide for another 24 hours. Following exposure to hydrogen peroxide, αSMA expression (A), DNA synthesis (B), and intracellular levels of TGF-β1 mRNA and protein (C) were increased after hydrogen peroxide exposure. DNA synthesis in the presence of PDGF-BB (5 ng/ml) for 1 hour, as a positive control, was 142 (SD 10)%. Results of densitometric analysis are presented as the mean percentages of β-actin signal intensity of for αSMA expression (B). The levels of TGF-β1 gene expression were quantitatively analysed by real-time PCR, and the results are expressed in arbitrary units (C). Values are means (SD) for six dishes. *p<0.05 compared with cultures before hydrogen peroxide exposure.
The hydrogen peroxide induced activation of MAPK pathways and transcription factors of AP-1 and NF-κB, the degradation of IκB-α, and stimulation of DNA synthesis, αSMA expression, and intracellular expression of TGF-β1 in the cultures were inhibited in a dose dependent manner by oestradiol (10−9–10−7 mol/l) (fig 4 ▶ and table 1 ▶). The inhibitory effects of oestradiol at a dose of 10−7 mol/l on the activation of MAPK pathways and transcription factors, the degradation of IκB-α, and the stimulation of proliferation, αSMA expression, and TGF-β1 expression were blocked by the specific ER antagonist ICI 182,780 in a dose dependent manner (data not shown), which was complete at 10−6 mol/l ICI 182,780 (fig 4 ▶ and table 1 ▶).
Figure 4.
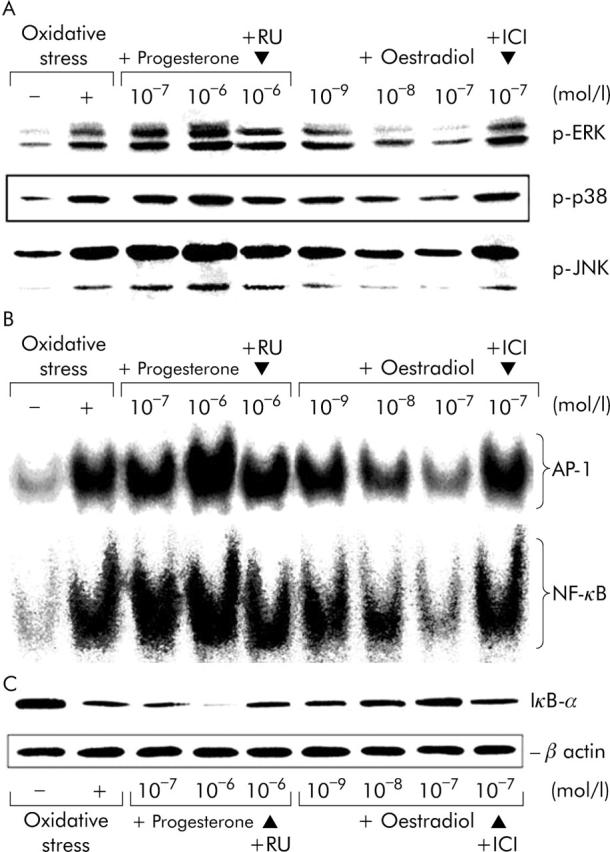
Effects of oestradiol and progesterone on the activation of MAPK pathways and transcription factors of AP-1 and NF-κB in cultured rat HSCs with and without hydrogen peroxide exposure. On culture day seven, oxidative stress was induced in HSCs by exposure to 10−5 mol/l hydrogen peroxide (oxidative stress) for 24 hours in pretreatment in serum-free DMEM with and without oestradiol (10−9–10−7 mol/l) or progesterone (10−7–10−6 mol/l) in the presence and absence of 10−6 mol/l ICI 182,780 (ICI) or 10−6 mol/l RU486 (RU) for 24 hours. After exposure to hydrogen peroxide, activation of the MAPK pathways of ERK, p38, and JNK (A) was evaluated at 30 minutes, activation of transcription factors of AP-1 and NF-κB (B) with the degradation of IκB-α (C) at 1 hour. The micrographs represent typical results of three independent experiments.
Table 1.
Effects of oestradiol and progesterone on the stimulation of proliferation and αSMA expression and intracellular levels of TGF-β1 mRNA and protein in cultured rat HSCs with and without hydrogen peroxide exposure
| BrdU incorporation (% of control) | αSMA (relative protein level) | TGF-β1 MRNA (arbitrary unit) | TGF-β1 protein (pg/mg protein) | |
| None | 100 (7)* | 55 (13)* | 0.58 (0.08)* | 15.1 (2.7)* |
| + Oxidative stress only | 147 (9) | 116 (20) | 1.00 | 31.3 (6.0) |
| + Oxidative stress | 184 (11)* | 146 (25)* | 1.25 (0.21)* | 39.8 (6.1)* |
| + Progesterone 10−7 mol/l | ||||
| + Oxidative stress | 205 (15)* | 168 (28)* | 1.47 (0.28)* | 46.5 (8.5)* |
| + Progesterone 10−6 mol/l | ||||
| + Oxidative stress | 151 (9) | 118 (21) | 1.02 (0.18) | 29.2 (6.9) |
| + Progesterone 10−6 mol/l + RU | ||||
| + Oxidative stress | 123 (9)* | 96 (13)* | 0.70 (0.09)* | 21.9 (5.3)* |
| + Oestradiol 10−9 mol/l | ||||
| + Oxidative stress | 106 (8)* | 75 (15)* | 0.61 (0.08)* | 17.4 (3.5)* |
| + Oestradiol 10−8 mol/l | ||||
| + Oxidative stress | 90 (7)* | 54 (10)* | 0.53 (0.07)* | 13.3 (3.1)* |
| + Oestradiol 10−7 mol/l | ||||
| + Oxidative stress | 146 (9) | 113 (21) | 0.95 (0.19) | 28.5 (5.1) |
| + Oestradiol 10−7 mol/l | ||||
| + ICI |
On culture day seven, oxidative stress was induced in HSCs by exposure to 10−5 mol/l hydrogen peroxide (oxidative stress) for 24 hours in pretreatment with serum-free DMEM with and without oestradiol (10−9–10−7 mol/l) or progesterone (10−7–10−6 mol/l) in the presence and absence of 10−6 mol/l ICI 182,780 (ICI) or 10−6 mol/l RU486 (RU) for 24 hours. After exposure to hydrogen peroxide, DNA synthesis and TGF-β1 mRNA level were evaluated at 1 hour, and αSMA expression and TGF-β1 protein level at 24 hours. The level of DNA synthesis in the presence of PDGF-BB (5 ng/ml), as a positive control, was 140 (SD 9)%. The results of densitometric analysis are presented as mean percentages of the signal intensity of beta-actin for αSMA expression. The levels of TGF-β1 gene expression were quantitatively analysed by real-time PCR, and results are expressed in arbitrary units. Values are means (SD) for six dishes. *p<0.05 compared with cultures after hydrogen peroxide exposure (oxidative stress).
In contrast to oestradiol, progesterone pretreatment for 24 hours resulted in the further activation of MAPK pathways and transcription factors and further stimulation of proliferation, αSMA expression and TGF-β1 expression in the hydrogen peroxide induced activation of cultured HSCs. The effect of progesterone (10−7–10−6 mol/l) was dose dependent and was blocked by the progesterone receptor antagonist RU486 at a dose of 10−6 mol/l (fig 4 ▶ and table 1 ▶). Treatment with ICI 182,780 or RU486 alone had no effect on any parameters examined herein (data not shown).
Effects of oestradiol and progesterone on NADH/NADPH oxidase activity, ROS generation, lipid peroxidation, and antioxidant enzyme levels in cultured rat HSCs without hydrogen peroxide exposure
The effect of oestradiol and progesterone in cultured HSCs on the activity of NADH/NADPH oxidase was evaluated by measuring the photon emission and the initial rate of NADH or NADPH oxidase activity. On culture day seven, HSCs, preincubated in serum-free DMEM for 24 hours, were then incubated for another 24 hours in the presence and absence of oestradiol (10−9–10−7 mol/l) without hydrogen peroxide exposure. The activities of both NADH and NADPH oxidases decreased in the presence of oestradiol in a dose dependent manner, compared with cultures in the absence of oestradiol without hydrogen peroxide exposure (none) (table 2 ▶). In addition, oestradiol treatment led to the dose dependent inhibition of intracellular ROS and MDA generation and the loss of antioxidant enzyme levels of CuZn-SOD, glutathione peroxidase, and catalase expression (table 2 ▶). The inhibitory effects of oestradiol were blocked by 10−6 mol/l ICI 182,780. These findings suggest that oestradiol is able to inhibit ROS generation, lipid peroxidation, and antioxidant enzyme loss via the suppression of NADH/NADPH oxidase activity through the oestrogen receptor. However, it cannot be deduced from the data whether oestradiol binding of the oestrogen receptor may affect the expression of antioxidant enzymes at the transcriptional level. In contrast, treatment with progesterone (10−7–10−6 mol/l) for 24 hours induced a dose dependent enhancement in ROS generation, lipid peroxidation, and antioxidant enzyme loss via NADH/NADPH oxidase activation (table 2 ▶). The stimulatory effects of progesterone were blocked by 10−6 mol/l RU486, suggesting that progesterone exerted its stimulatory effects through the progesterone receptor.
Table 2.
Effects of estradiol and progesterone on NADH/NADPH oxidase activities, ROS generation, lipid peroxidation, and antioxidant enzyme levels in cultured rat HSCs without hydrogen peroxide exposure
| NADH oxidase (mmol/min/mg protein) | NADPH oxidase (mmol/min/mg protein) | ROS (channel number/mg protein) | MDA (mmol/mg protein) | CuZn-SOD (ng/mg protein) | Glutathione peroxidase (U/mg protein) | Catalase (relative protein level) | |
| None | 29.0 (4.1) | 8.1 (1.3) | 7.5 (1.2) | 0.46 (0.07) | 3.2 (0.4) | 2.0 (0.3) | 65 (12) |
| + Progesterone 10−7 mol/l | 37.3 (6.6)* | 10.6 (2.1)* | 9.3 (1.5)* | 0.58 (0.09)* | 2.7 (0.4)* | 1.5 (0.3)* | 53 (10)* |
| + Progesterone 10−6 mol/l | 43.5 (9.4)* | 13.9 (2.9)* | 11.0 (2.2)* | 0.68 (0.10)* | 2.1 (0.5)* | 1.1 (0.2)* | 39 (10)* |
| + Progesterone 10−6 mol/l | 30.6 (3.4) | 8.6 (1.3) | 7.8 (1.0) | 0.48 (0.06) | 3.2 (0.5) | 2.0 (0.4) | 65 (11) |
| + RU | |||||||
| + Oestradiol 10−9 mol/l | 25.0 (3.3)* | 7.5 (1.0)* | 6.0 (1.0)* | 0.43 (0.06) | 4.5 (0.7)* | 2.6 (0.4)* | 82 (16)* |
| + Oestradiol 10−8 mol/l | 22.2 (3.2)* | 5.8 (0.8)* | 4.8 (0.7)* | 0.30 (0.05)* | 7.0 (1.3)* | 4.0 (0.9)* | 119 (22)* |
| + Oestradiol 10−7 mol/l | 16.9 (2.4)* | 4.8 (0.6)* | 3.8 (0.5)* | 0.22 (0.04)* | 8.1 (1.4)* | 4.8 (1.0)* | 140 (27)* |
| + Oestradiol 10−7 mol/l | 28.5 (4.1) | 7.9 (1.3) | 7.4 (1.1) | 0.46 (0.07) | 3.4 (0.6) | 2.0 (0.4) | 64 (15) |
| + ICI |
On culture day seven, HSCs, preincubated in serum-free DMEM for 24 hours, were then incubated with and without oestradiol (10−9–10−7 mol/l) or progesterone (10−7–10−6 mol/l) in the presence and absence of 10−6 mol/l ICI 182,780 (ICI) or 10−6 mol/l RU486 (RU) for another 24 hours without hydrogen peroxide exposure. NADH/NADPH oxidase activities, the generation of ROS and MDA, and the levels of CuZn-SOD, glutathione peroxidase, and catalase expression were then compared with cultures in the absence of steroid sex hormones and receptor antagonists without hydrogen peroxide exposure (none). The densitometric analysis results of catalase expression are presented as the mean percentages of β-actin signal intensity. Values are means (SD) for six dishes. *p<0.05 compared with cultures without any treatment (none).
Effects of oestradiol on progesterone induced NADH/NADPH oxidase activity, lipid peroxidation, proliferation, αSMA expression, and TGF-β1 expression in cultured rat HSCs with hydrogen peroxide exposure
Hydrogen peroxide exposure for 24 hours resulted in a further enhancement in NADH/NADPH oxidase activity, lipid peroxidation, and antioxidant enzyme loss in cultured HSCs compared with culture without hydrogen peroxide exposure. In the oxidative stress induced activation of cultured HSCs, progesterone induced a further increase in NADH/NADPH oxidase activity and intracellular ROS and MDA generation as well as proliferation, αSMA, and intracellular levels of TGF-β1 mRNA and protein (table 3 ▶) and the activation of MAPK pathways and transcription factors in a dose dependent manner (data not shown). The stimulatory effects of progesterone at a dose of 10−6 mol/l were inhibited by simultaneous coincubation with oestradiol (10−8–10−7 mol/l) in a dose dependent manner. Treatment with 10−6 mol/l ICI 182,780 led to a complete block of oestradiol mediated responses to progesterone exposure in cultured HSCs in a state of oxidative stress (table 3 ▶).
Table 3.
Effects of oestradiol on progesterone induced NADH/NADPH oxidase activities, ROS generation, lipid peroxidation, proliferation, αSMA expression, and intracellular levels of TGF-β1 mRNA and protein in cultured rat HSCs with hydrogen peroxide exposure
| NADH oxidase (mmol/min/mg protein) | NADPH oxidase (mmol/min/mg protein) | ROS (channel number/mg protein) | MDA (mmol/mg protein) | BrdU incorporation (% of control) | αSMA (relative protein level) | TGF-β1 mRNA (arbitrary unit) | TGF-β1 protein (pg/mg protein) | |
| None | 28.9 (5.5)*† | 8.1 (2.1)*† | 7.7 (1.1)*† | 0.52 (0.14)*† | 100 (6)*† | 63 (15)*† | 0.60 (0.10)*† | 15.6 (3.5)*† |
| + Oxidative stress only | 78.3 (14.8) | 27.2 (5.2) | 20.9 (4.0) | 1.44 (0.23) | 155 (10) | 129 (24) | 1.00 | 33.3 (6.7) |
| + Oxidative stress | 31.4 (5.4)*† | 9.0 (2.1)*† | 7.8 (1.1)*† | 0.55 (0.11)*† | 108 (7)*† | 64 (15)*† | 0.62 (0.11)*† | 16.3 (3.6)*† |
| + DPI 10−6 mol/l | ||||||||
| + Oxidative stress | 111.1 (17.3)* | 35.1 (6.2)* | 29.5 (4.5)* | 1.92 (0.32)* | 202 (20)* | 193 (37)* | 1.44 (0.21)* | 51.2 (10.1)* |
| + Progesterone 10−6 mol/l | ||||||||
| + Oxidative stress | 32.3 (5.6)*† | 9.4 (2.5)*† | 8.5 (1.2)*† | 0.58 (0.14)*† | 109 (7)*† | 66 (13)*† | 0.64 (0.13)*† | 16.5 (3.5)*† |
| + Progesterone 10−6 mol/l | ||||||||
| + DPI 10−6 mol/l | ||||||||
| + Oxidative stress | 73.6 (13.2)† | 22.4 (4.3)† | 19.0 (3.5)† | 1.24 (0.22)† | 165 (12)† | 130 (23)† | 1.09 (0.15)† | 40.4 (9.1)† |
| + Progesterone 10−6 mol/l | ||||||||
| + Estradiol 10−8 mol/l | ||||||||
| + Oxidative stress | 50.2 (13.3)*† | 14.5 (4.2)*† | 13.3 (2.8)*† | 0.81 (0.18)*† | 122 (11)*† | 84 (20)*† | 0.70 (0.15)*† | 26.7 (6.2)*† |
| + Progesterone 10−6 mol/l | ||||||||
| + Estradiol 10−7 mol/l | ||||||||
| + Oxidative stress | 110.5 (16.1)* | 35.7 (6.4)* | 28.4 (4.3)* | 1.87 (0.28)* | 222 (25)* | 186 (38)* | 1.59 (0.30)* | 52.6 (10.7)* |
| + Progesterone 10−6 mol/l | ||||||||
| + Oestradiol 10−7 mol/l | ||||||||
| + ICI | ||||||||
| + Oxidative stress | 33.8 (6.1)*† | 9.9 (2.5)*† | 8.3 (1.5)*† | 0.59 (0.15)*† | 110 (8)*† | 68 (15)*† | 0.65 (0.13)*† | 17.1 (3.7)*† |
| + Progesterone 10−6 mol/l | ||||||||
| + Oestradiol 10−7 mol/l | ||||||||
| + DPI 10−6 mol/l |
On culture day seven, oxidative stress was induced in HSCs by exposure to 10−5 mol/l hydrogen peroxide (oxidative stress) for 24 hours in the pretreatment in serum-free DMEM with and without progesterone (10−6 mol/l) and/or oestradiol (10−8–10−7 mol/l) in the presence and absence of 10−6 mol/l ICI 182,780 or 10−6 mol/l DPI for 24 hours. NADH/NADPH oxidase activities, the generation of ROS and MDA were then compared with cultures treated with progesterone alone after hydrogen peroxide exposure. DNA synthesis and TGF-β1 mRNA level were evaluated at 1 hour, and αSMA expression and TGF-β1 protein level at 24 hours. DNA synthesis in the presence of PDGF-BB (5 ng/ml), as a positive control, was 148 (SD 9)%. Results of densitometric analysis are presented as mean percentages of β-actin signal intensity of for αSMA expression. The levels of TGF-β1 gene expression were quantitatively analysed by real-time PCR, and the results are expressed in arbitrary units. Values are means (SD) for six dishes. *p<0.05 compared with cultures after hydrogen peroxide exposure (oxidative stress). †p<0.05 compared with cultures treated with progesterone alone after hydrogen peroxide exposure.
To assess the central or main role of the NADH/NADPH oxidase, a separate series of competition experiments was conducted with the NADH/NADPH oxidase inhibitor DPI (10−6 mol/l) in cultured HSCs (table 3 ▶). DPI treatment in HSCs cultured with hydrogen peroxide exposure induced a decrease in NADH/NADPH oxidase activity and the intracellular generation of ROS and MDA as well as the proliferation, αSMA and intracellular levels of TGF-β1 mRNA and protein. The stimulatory effects of progesterone were blocked by DPI, and the concomitant treatment of oestradiol with DPI resulted in further inhibition in the parameters examined herein (table 3 ▶).
DISCUSSION
This study indicates for the first time that there is a competing regulation between the actions of oestradiol and progesterone on oxidative stress induced ROS generation mainly originating from NADH/NADPH oxidase, the activation of MAPK pathways and transcription factors of AP-1 and NF-κB, TGF-β1 expression, proliferation, and transformation in cultured rat HSCs. Oestradiol was found to inhibit ROS generation, lipid peroxidation, and antioxidant enzyme loss via the suppression of NADH/NADPH oxidase activity and to attenuate the hydrogen peroxide induced activation of ERK, p38, and JNK and transcription factors of AP-1 and NF-κB with the degradation of IκB-α, TGF-β1 expression, and HSC activation through the oestrogen receptor in a dose dependent manner. In contrast, HSCs were observed to possess the progesterone receptor using immunohistochemistry, western blotting, and RT-PCR. Progesterone treatment induced the dose dependent enhancement of ROS generation, lipid peroxidation, and antioxidant enzyme loss via NADH/NADPH oxidase activation, the activation of MAPK pathways, and transcription factors, TGF-β1 expression, and HSC activation through the progesterone receptor in cultures with and without hydrogen peroxide exposure. The stimulatory actions of progesterone leading to the activation of HSCs were blocked by oestradiol at a one hundredth and one tenth dose of progesterone through the oestrogen receptor in a dose dependent manner. These findings suggest that oestradiol and progesterone may work together with the resulting suppression of hepatofibrogenesis in women.
MAPK pathways are comprised of key regulatory proteins that control the cellular response to both proliferation and stress signals, followed by the stimulation of at least two well defined transcription factors AP-1 and NF-κB. ERK and p38 can be activated by exposure to low doses of hydrogen peroxide (about 10−5 mol/l),19,30 such as might typically occur in cells.31 These findings agree with the data presented here, indicating that hydrogen peroxide at a dose of 10−5 mol/l stimulated the activation of AP-1 and NF-κB, acting through the MAPK pathways of ERK, p38, and JNK, followed by TGF-β1 expression and HSC activation. The hydrogen peroxide stimulated intracellular pathways and activation processes during HSC activation were inhibited by oestradiol in cultured HSCs. This occurs at physiological relevant concentrations (10−9–10−7 mol/l) of oestradiol, equivalent to the oestrogen receptor dissociation constant (about 10−9 mol/l), and to serum levels of oestradiol measured in women during their reproductive years (about 10−9–10−8 mol/l),32 and tissue levels of steroid sex hormones may actually be greater.33 Oestradiol and its derivatives (2-hydroxyestradiol) are strong endogenous antioxidants that reduce lipid peroxide levels in the liver and serum.4,34 This study confirms that oestradiol can inhibit the intracellular generation of ROS and MDA and the loss of antioxidant enzyme levels of SOD, glutathione peroxidase, and catalase expression via the suppressed activity of NADH and NADPH oxidases. The possible stimulatory effects of oestradiol binding the oestrogen receptor on the expression of antioxidant enzymes at the transcriptional level remain to be elucidated.
NADH and NADPH oxidases represent the most important source of ROS in both endothelial cells and vascular smooth muscle cells.35 Vascular smooth muscle cells are anatomically analogous to HSCs, and have been reported to express oestrogen receptor β at a higher level after vascular injury with no significant changes in oestrogen receptor α expression.36 Our earlier study showed that HSCs contain functional oestrogen receptor β but not oestrogen receptor α in both males and females.14 In this study, the specificity of the oestradiol mediated inactivation of HSCs with the suppressed induction of intracellular pathways and ROS generation processes through oestrogen receptor β was blocked by the pure oestrogen receptor antagonist ICI 182,780 in cultured HSCs with and without hydrogen peroxide exposure.
In contrast to oestradiol, the ability of progesterone to modulate HSC activation is much less clear. During the luteal phase of the menstrual cycle, the serum concentration of endogenous progesterone rises up to about 10−7 mol/l, ten to hundred times higher than oestradiol, and, during pregnancy, up to about 10−6 mol/l. These data clearly show that progesterone, at doses of 10−7–10−6 mol/l, acts in opposition to the favourable effects of oestradiol and exerts stimulatory effects on the induction of ROS generation processes and intracellular pathways, resulting in TGF-β1 expression and HSC activation in a dose dependent manner. The stimulatory effects of progesterone were blocked by the progesterone receptor antagonist RU486. In cultured vascular smooth muscle cells, although several contradictory reports exist concerning the effect of progesterone on cell proliferation,37,38 this is the first report to address the fibrogenic response of progesterone through the progesterone receptor in HSCs to low levels of oxidative stress, and the inhibitory effect of oestradiol on progesterone induced HSC activation. This suggests that the resultant inhibition of HSC activation and hepatic fibrosis induced by the combinations of oestradiol and progesterone at physiological relevant concentrations may help explain the preponderance of men suffering from chronic liver disease.
In the TGF-β1 response to oestradiol, it was reported that systemic hormone replacement therapy with oestrogens and progestins accelerated human cutaneous wound healing, associated with the upregulation of TGF-β1;39 however, our previous study showed that catalase treatment resulted in decreased ROS generation and TGF-β1 levels in cultured HSCs.25 It has been proposed that ROS, particularly hydrogen peroxide, acts as a signaling mediator for TGF-β1.40 De Bleser et al reported that exogenous TGF-β1 increased the production of hydrogen peroxide by HSCs, whereas the addition of hydrogen peroxide induced TGF-β1 production and secretion.41 This study revealed that hydrogen peroxide elicited ROS generation was inhibited by the NADH/NADPH oxidase inhibitor DPI in cultured HSCs. Concomitant DPI treatment led to further enhancement of the oestradiol mediated responses to progesterone exposure. Judging from these findings and these data, oestradiol can modify TGF-β1 expression, at least in part, via the suppression of NADH and NADPH oxidases activity.
Better understanding of the basic mechanisms underlying the gender associated differences observed in the development of hepatic fibrosis may open new avenues for the prevention and treatment of chronic liver disease.
Abbreviations
αSMA, α smooth muscle actin
AP-1, activator protein-1
BrdU, bromodeoxyuridine
CuZn-SOD, zinc dependent SOD
DMEM, Dulbecco’s modified Eagle’s medium
ELISA, enzyme linked immunosorbent assay
EMSA, electrophoretic mobility shift assay
ERK, extracellular signal regulated kinase
FBS, fetal bovine serum
H2DCF-DA, 2′,7′-dichlorofluorescein diacetate
HSC, hepatic stellate cell
JNK, c-Jun N-terminal kinase/stress activated protein kinase
MAPK, mitogen activated βprotein kinase
MDA, malondialdehyde
NF-κB, nuclear factor κB
p38, p38 MAPK
p-p38, phosphorylated p38
PBS, phosphate buffered saline
PDGF, platelet derived growth factor
p-ERK, phosphorylated ERK
p-JNK, phosphorylated JNK
RT-PCR, reverse transcription-polymerase chain reaction
ROS, reactive oxygen species
SDS, sodium dodecyl sulfate
SOD, superoxide dismutase
TGF-β1, transforming growth factor-β1
Published online first 13 July 2005
Competing interest: none declared.
REFERENCES
- 1.Shimizu I. Antifibrogenic therapies in chronic HCV infection. Curr Drug Targets Infect Disord 2001;1:227–40. [DOI] [PubMed] [Google Scholar]
- 2.Lu G, Shimizu I, Cui X, et al. Antioxidant and antiapoptotic activities of idoxifene and estradiol in hepatic fibrosis in rats. Life Sci 2004;74:897–907. [DOI] [PubMed] [Google Scholar]
- 3.Inoue H, Shimizu I, Lu G, et al. Idoxifene and estradiol enhance antiapoptotic activity through the estrogen receptor β in cultured rat hepatocytes. Dig Dis Sci 2003;48:570–80. [DOI] [PubMed] [Google Scholar]
- 4.Lacort M, Leal AM, Liza M, et al. Protective effect of estrogens and catecholestrogens against peroxidative membrane damage in vitro. Lipids 1995;30:141–6. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu I, Mizobuchi Y, Shiba M, et al. Inhibitory effect of oestradiol on activation of rat hepatic stellate cells in vivo and in vitro. Gut 1999;44:127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yasuda M, Shimizu I, Shiba M, et al. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats [see comments]. Hepatology 1999;29:719–27. [DOI] [PubMed] [Google Scholar]
- 7.Omoya T, Shimizu I, Zhou Y, et al. Effects of idoxifene and estradiol on NF-κB activation in cultured rat hepatocytes undergoing oxidative stress. Liver 2001;21:183–91. [DOI] [PubMed] [Google Scholar]
- 8.Poynard T, Ratziu V, Charlotte F, et al. Rates and risk factors of liver fibrosis progression in patients with chronic hepatitis C. J Hepatol 2001;34:730–9. [DOI] [PubMed] [Google Scholar]
- 9.Shimizu I. Impact of estrogens on the progression of liver disease. Liver Int 2003;23:63–9. [DOI] [PubMed] [Google Scholar]
- 10.Dudas J, Saile B, El Armouche H, et al. Endoreplication and polyploidy in primary culture of rat hepatic stellate cells. Cell Tissue Res 2003;313:301–11. [DOI] [PubMed] [Google Scholar]
- 11.Tateaki Y, Ogawa T, Kawada N, et al. Typing of hepatic nonparenchymal cells using fibulin-2 and cytoglobin/STAP as liver fibrogenesis-related markers. Histochem Cell Biol 2004;122:41–9. [DOI] [PubMed] [Google Scholar]
- 12.Baroni GS, D’Ambrosio L, Ferretti G, et al. Fibrogenic effect of oxidative stress on rat hepatic stellate cells. Hepatology 1998;27:720–6. [DOI] [PubMed] [Google Scholar]
- 13.Lee KS, Buck M, Houglum K, et al. Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. J Clin Invest 1995;96:2461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Y, Shimizu I, Lu G, et al. Hepatic stellate cells contain the functional estrogen receptor beta but not the estrogen receptor alpha in male and female rats. Biochem Biophys Res Commun 2001;286:1059–65. [DOI] [PubMed] [Google Scholar]
- 15.Parola M, Pinzani M, Casini A, et al. Stimulation of lipid peroxidation or 4-hydroxynonenal treatment increases procollagen (I) gene expression in human liver fat-storing cells. Biochem Biophys Res Commun 1993;194:1044–50. [DOI] [PubMed] [Google Scholar]
- 16.Lee WS, Harder JA, Yoshizumi M, et al. Progesterone inhibits arterial smooth muscle cell proliferation. Nat Med 1997;3:1005–8. [DOI] [PubMed] [Google Scholar]
- 17.Pierard GE, Letawe C, Dowlati A, et al. Effect of hormone replacement therapy for menopause on the mechanical properties of skin. J Am Geriatr Soc 1995;43:662–5. [DOI] [PubMed] [Google Scholar]
- 18.Yu WD, Panossian V, Hatch JD, et al. Combined effects of estrogen and progesterone on the anterior cruciate ligament. Clin Orthop 2001;383:268–81. [DOI] [PubMed] [Google Scholar]
- 19.Guyton KZ, Liu Y, Gorospe M, et al. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem 1996;271:4138–42. [DOI] [PubMed] [Google Scholar]
- 20.Xia Z, Dickens M, Raingeaud J, et al. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995;270:1326–31. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu I, Ma Y-R, Mizobuchi Y, et al. Effects of Sho-saiko-to, a Japanese herbal medicine, on hepatic fibrosis in rats [see comments]. Hepatology 1999;29:149–60. [DOI] [PubMed] [Google Scholar]
- 22.Thannickal VJ, Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. J Biol Chem 1995;270:30334–8. [DOI] [PubMed] [Google Scholar]
- 23.Maxwell DP, Wang Y, McIntosh L. The alternative oxidase lowers mitochondrial reactive oxygen production in plant cells. Proc Natl Acad Sci USA 1999;96:8271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muir D, Varon S, Manthorpe M. An enzyme-linked immunosorbent assay for bromodeoxyuridine incorporation using fixed microcultures. Anal Biochem 1990;185:377–82. [DOI] [PubMed] [Google Scholar]
- 25.Cui X, Shimizu I, Lu G, et al. Inhibitory effect of a soluble transforming growth factor b type II receptor on the activation of rat hepatic stellate cells in primary culture [see comments]. J Hepatol 2003;39:731–7. [DOI] [PubMed] [Google Scholar]
- 26.Hagihara K, Hirata S, Osada T, et al. Expression of progesterone receptor in the neonatal rat brain cortex: detection of its mRNA using reverse transcription-polymerase chain reaction. J Steroid Biochem Molec Biol 1992;41:637–40. [DOI] [PubMed] [Google Scholar]
- 27.Winer J, Jung CK, Shackel I, et al. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem 1999;270:41–9. [DOI] [PubMed] [Google Scholar]
- 28.Wickert L, Steinkruger S, Abiaka M, et al. Quantitative monitoring of the mRNA expression pattern of the TGF-beta-isoforms (beta 1, beta 2, beta 3) during transdifferentiation of hepatic stellate cells using a newly developed real-time SYBR Green PCR. Biochem Biophys Res Commun 2002;295:330–5. [DOI] [PubMed] [Google Scholar]
- 29.Conneely OM, Mulac-Jericevic B, DeMayo F, et al. Reproductive functions of progesterone receptors. Recent Prog Horm Res 2002;57:339–55. [DOI] [PubMed] [Google Scholar]
- 30.Kurata S. Selective activation of p38 MAPK cascade and mitotic arrest caused by low level oxidative stress. J Biol Chem 2000;275:23413–16. [DOI] [PubMed] [Google Scholar]
- 31.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J 1991;10:2247–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.White MM, Zamudio S, Stevens T, et al. Estrogen, progesterone, and vascular reactivity: potential cellular mechanisms. Endocr Rev 1995;16:739–51. [DOI] [PubMed] [Google Scholar]
- 33.Meizel S, Turner KO. Progesterone acts at the plasma membrane of human sperm. Mol Cell Endocrinol 1991;11:R1–R5. [DOI] [PubMed] [Google Scholar]
- 34.Yoshino K, Komura S, Watanabe I, et al. Effect of estrogens on serum and liver lipid peroxide levels in mice. J Clin Biochem Nutr 1987;3:233–9. [Google Scholar]
- 35.Rajagopalan S, Kurz S, Munzel T, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 1996;97:1916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindner V, Kim SK, Karas RH, et al. Increased expression of estrogen receptor-β mRNA in male blood vessels after vascular injury. Circ Res 1998;83:224–9. [DOI] [PubMed] [Google Scholar]
- 37.Akishita M, Ouchi Y, Miyoshi H, et al. Estrogen inhibits cuff-induced intimal thickening of rat femoral artery: effects on migration and proliferation of vascular smooth muscle cells. Atherosclerosis 1997;130:1–10. [DOI] [PubMed] [Google Scholar]
- 38.Morey AK, Pedram A, Razandi M, et al. Estrogen and progesterone inhibit vascular smooth muscle proliferation. Endocrinology 1997;138:3330–9. [DOI] [PubMed] [Google Scholar]
- 39.Ashcroft GS, Dodsworth J, van Boxtel E, et al. Estrogen accelerates cutaneous wound healing associated with an increase in TGF-beta1 levels. Nat Med 1997;3:1209–15. [DOI] [PubMed] [Google Scholar]
- 40.Ohba M, Shibanuma M, Kuroki T, et al. Production of hydrogen peroxide by transforming growth factor-beta 1 and its involvement in induction of egr-1 in mouse osteoblastic cells. J Cell Biol 1994;126:1079–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Bleser PJ, Xu G, Rombouts K, et al. Glutathione levels discriminate between oxidative stress and transforming growth factor-beta signaling in activated rat hepatic stellate cells. J Biol Chem 1999;274:33881–7. [DOI] [PubMed] [Google Scholar]