

Abstract
It has become increasingly evident that chemotherapy regimens used to condition patients prior to bone marrow transplantation damage the hematopoietic microenvironment as dose-escalation reveals problems with hematopoietic recovery or engraftment. We have previously demonstrated that bone marrow stromal cells (BMSCs) exposed to dose escalated etoposide (VP-16) have reduced support of CXCR4+ cell chemotaxis and diminished stromal cell derived factor-1 (CXCL12) in the supernatants. Based on the identification of CXCL12 as a matrix metalloproteinase-2 (MMP-2) substrate, we investigated potential dysregulation of MMP-2 expression or activity in chemotherapy-treated BMSCs. BMSC exposure to VP-16 resulted in an immediate, but transient, increase in MMP-2 followed by reduced MMP-2 protein expression correlated with diminished CXCL12 protein and reduced chemotactic support. Consistent with these observations, BMSCs derived from MMP-2 knockout mice had significantly less chemotactic support of CXCR4+ cells than wild-type controls. Inhibition of BMSC MMP-2 activity by the specific inhibitor, OA-Hy, also reduced chemotactic support and CXCL12 protein detected in supernatants. VP-16-induced reduction of BMSC support of hematopoietic cell migration was restored by supplementing cultures with physiological levels of recombinant MMP-2 protein. These data suggest that MMP-2 is sensitive to chemotherapy-induced stress, and may regulate BMSC support of hematopoietic cell chemotaxis through diverse mechanisms. Increased MMP-2 expression during the acute phase of chemotherapy potentially mediates inactivation of CXCL12. Subsequently, chronic exposure to chemotherapy, with the associated downregulation of MMP-2, interrupts CXCL12 release from the extracellular matrix.
Introduction
Chemotaxis of hematopoietic progenitor or stem cells to the bone marrow microenvironment is essential for efficient hematopoietic recovery following bone marrow transplantation1,2. CXCL12 is the primary chemokine released by BMSCs that promotes chemotaxis of transplanted progenitors to the bone marrow microenvironment 3,4. Following migration to the bone marrow, hematopoietic progenitors interact with BMSC, which provide support to developing hematopoietic cells through the production of soluble cytokines and chemokines, and adhesion molecules that facilitate physical interaction 5,6. In addition, BMSCs cells deposit extracellular matrix that provides structural support and stabilizes hematopoietic growth factors in concentrated niches 7-10. CXCL12 is concentrated and stabilized in the bone marrow microenvironment on heparin sulfated proteoglycans produced by BMSCs 11.
Preparative regimens used prior to transplantation are aggressive, and have the potential to damage the hematopoietic microenvironment 12-15. One chemotherapeutic agent, etoposide (VP-16), has previously been shown to negatively influence BMSC support of hematopoiesis, in part, by diminishing vacsular cell adhesion molecule (VCAM-1) protein 16. Studies from our own laboratory have shown that VP-16 exposure also results in reduced ability of BMSCs to support pro-B cell chemotaxis 17. Together, these observations highlight the vulnerability of BMSCs to chemotherapy damage, and prompted our investigation of the mechanisms by which chemotherapy reduces efficiency of pro-B cell chemotaxis.
MMPs have traditionally been considered in the context of extracellular matrix regulation, however additional roles have been identified, including 18-20 regulation of hematopoiesis. Based on studies by others that documented the ability of MMP-2 to cleave and inactivate CXCL12 21, we determined whether VP-16 exposure increased MMP-2 activity or expression in BMSCs, contributing to reduced chemotactic support. An immediate increase in MMP-2 activity following initiation of VP-16 exposure was observed, that was pronounced and transient. BMSCs exposed to greater than 5 hours of chemotherapy expressed less MMP-2 protein than control BMSCs. Coincident with reduced MMP-2 expression is a reduction of CXCL12 protein and chemotactic support capacity. These observations position MMP-2 as a factor in the bone marrow microenvironment that can respond to external stresses, including chemotherapy, and influence support of hematopoietic reconstitution through regulation of the CXCL12 gradient.
Materials and Methods
Reagents
VP-16 (etoposide, Bristol Laboratories, Princeton, NJ) was stored at a stock concentration of 33.98 mM at -20°C and was diluted in α-Modification of Eagles Medium (α-MEM, GIBCO, Grand Island, NY) immediately prior to use. MMP-2 Inhibitor I (Cis-9-Octadeconyl-N-hydroxylamide, OA-Hy, Calbiochem, San Diego, CA) was reconstituted in DMSO at 10mM immediately prior to use. Recombinant human MMP-2 (Biomol International L.P., Plymouth Meeting, PA) and recombinant murine MMP-2 (R&D Systems Inc., Minneapolis, MN) were diluted in media at the indicated concentrations. Mouse anti-human MMP-2 (Ab 3) monoclonal antibody was obtained from Calbiochem, Boston, MA.
Cell lines and culture conditions
Stromal cell cultures were initiated from human bone marrow from consenting donors, with approval by the West Virginia University Institutional Review Board, as previously described 15. All primary BMSC cultures were initiated from donors with no previous chemotherapy exposure. BMSCs were maintained in α-MEM supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), 1% L-glutamine (GIBCO, Grand Island, NY), 1% Penicillin/Streptomycin (Sigma, St. Louis, MO), and 0.1% 2-beta-mercapthanol (Sigma, St. Louis, MO).
Murine BMSC line S10 was provided by Dr. Kenneth Dorshkind (University of California Los, Angeles). Characterization and maintenance of S10 has been previously described in detail 22. S10 BMSCs were grown to confluence in α-MEM supplemented with 2.5% fetal bovine serum, 1% L-glutamine (GIBCO, Grand Island, NY), 1% Penicillin/Streptomycin (Sigma, St. Louis, MO), and 0.1% 2-beta-mercapthanol (Sigma, St. Louis, MO).
Murine pro-B cell clone C1.92 was provided by Dr. Kenneth S. Landreth (West Virginia University). Derivation of C1.92 has been previously described 23. C1.92 was maintained in the presence of the BMSC line S-10 and 50U/mL recombinant murine IL-7 (mIL-7, Biosource International, Westlake Village, CA).
MMP-2-/- and wild type (WT) BMSCs were initiated from femurs isolated from C57BL/6 WT and C57BL/6 MMP-2 knockout mice (kindly provided by Dr. Farrah Kheradmand; Baylor College of Medicine) 24. WT and MMP-2-/- BMSCs were cultured in α-MEM supplemented with 2.5% fetal bovine serum, 1% L-glutamine (GIBCO, Grand Island, NY), 1% Penicillin/Streptomycin (Sigma, St. Louis, MO), and 0.1% 2-beta-mercapthanol (Sigma, St. Louis, MO).
Gelatin Zymography
BMSC supernatants were collected following 2 hours treatment in serum free α-MEM (GIBCO, Grand Island, NY) with 100μM VP-16 (Bristol Laboratories, Princeton, NJ). Supernatants were concentrated 10X using Amiconfi Ultra-15 Centrifugal Filters (Millipore, Billerica, MA). Samples were resolved in SDS-PAGE gels containing 1mg/mL gelatin (Sigma, St. Louis, MO). Following electrophoresis, gels were incubated for 30 minutes in 2.5% Triton-X-100 (Mallinckrodt, Inc., Paris, KY) and subsequently incubated overnight at 37°C in 1X developing buffer (1.2% Tris Base, 6.3% Tris HCl, 11.7% NaCl, 0.7% CaCl, 0.2% Brij 35). Gels were then stained with 0.5% Coomassie Blue R-250 (Bio-Rad Laboratories, Richmond, CA) for 30 minutes at room temperature and then destained (50% Methanol, 10% acetic acid, 40% dH20) until clear bands were detected indicative of MMP-2 and/or MMP-9.
Western blot analysis
Confluent BMSCs were treated with 25-100μM VP-16 for 24 hours. To determine protein stability, confluent BMSCs were set up in duplicate and treated with both 100μM VP-16 and 25:g/mL cycloheximide or VP-16 alone for 2 to 24 hours. Supernatants were collected following treatment and concentrated 10x using Amicon® Ultra-15 Centrifugal Filter Devices (Millipore, Billerica, MA). Media was centrifuged at 3,000xg for 10 minutes at room temperature. Concentrated supernatants were resolved on SDS-PAGE gels and transferred to nitrocellulose membranes (Schleicher & Schuell bioscience, Inc., Keene, NH). Membranes were blocked in TBS/5% nonfat dry milk/0.1% Tween-20 at room temperature for 1 hour and probed with mouse anti-human MMP-2 monoclonal antibody. Proteins were detected by incubation with horseradish peroxidase-conjugated secondary antibody and visualized with ECL reagents (Amersham, Pharmacia Biotech, Piscataway, NJ).
Confocal Microscopy
BMSCs were grown to confluence on glass coverslips and treated with 100μM VP-16 for 24 hours. Following treatment, BMSCs were rinsed in autoclaved 1X PBS and fixed in 1:1 methanol: acetone for 20 minutes. Non-specific antibody binding was blocked by incubation of BMSCs for 15 minutes in autoclaved 1X PBS/5% BSA. Intracellular MMP-2 was evaluated by incubation of BMSCs with MMP-2 monoclonal antibody for 1 hr. PE conjugated secondary antibody was then added to BMSCs for 60 minutes and coverslips were inverted on slides and evaluated by confocal microscopy (Zeiss LSM 510, Thornwood, NY)
RNA Isolation
Total RNA was isolated from BMSCs using the Micro-to-Midi Total RNA Isolation kit following the recommendations of the manufacturer (Invitrogen, Carlsbad, CA). Pelleted BMSCs were lysed by centrifugation through QIA shredder Spin Columns (QIAGEN Inc, Santa Clarita, CA). RNA was treated with 1U DNAse for 30 minutes at 37°C and samples were quantitated at 260nm (GENESYS-10UV, Spectronic Unicam, Rochester, NY).
PCR
To evaluate MMP-2 and β-actin RNA levels, semi-quantitative “One-Step” RT-PCR (QIAGEN Inc., Valencia, CA) was completed using 0.1μg RNA isolated from untreated or VP-16 treated BMSCs. Reverse transcription was completed by incubation of samples at 42°C for 90 minutes and amplification initiated by a hot start at 95°C for 5 minutes, followed by 35 cycles of 94°-1 minute, 55°-1 minute, and 72°-1 minute (Perkin-Elmer GeneAmp PCR System 9600). Actin and MMP-2 primer (0.1μg/sample) were added to each reaction. Actin specific primers included 5’TGACGGGGTCACCCACACTGTGCCCATCTA-3’ and 5’TAGAAGCATTTGCGGTGGACGATGGAGGG-3’ (Stratagene, La Jolla, CA) to generate an amplicon of 661 base pairs. MMP-2 primers were 5’-GGCCCTGTCACTCCTGAGAT3’ and 5’-GGCATCCAGGTTATCGGGGA-3’ (Biosource International, Camarillo, CA) to generate an amplicon of 474 base pairs. MMP-2 to actin ratios were quantitated by EagleSight Version 3.21 (Stratagene, La Jolla, CA) densitometric analysis. The linear range of amplification was determined for each primer set prior to use.
RNAse protection assay
Confluent BMSC layers were treated with 25, 50 or 100μM VP-16 for 24 hours. RNAse protection assays were performed using an RPAIII kit according to the protocol of the manufacturer (Ambion Inc., Austin, TX). 10μg of RNA from each sample was hybridized to 32P-labeled MMP-2, GAPDH, and L-32 specific probes. Anti-sense 32P-RNA probes were generated using T7 RNA polymerase-directed synthesis from RiboQuant DNA templates (PharMingen, San Diego, CA). Nucleic acids were treated with RNAse A and T1 to digest unhybridized sequences. Protected RNA fragments that corresponded to MMP-2 and GAPDH were visualized by exposure to Phospho Imager cassettes (Molecular Dynamics, Sunnyvale, CA). MMP-2 band intensities were normalized to GAPDH or L-32 controls in each treatment group.
CXCL12 and MMP-2 ELISA
Confluent BMSCs were treated with 1μM OA-Hy or 100μM VP-16 for 24 hours. 100μL of 24-hour BMSC conditioned supernatant was collected from each well to evaluate CXCL12 or MMP-2 protein by ELISA, following the recommendations of the manufacturer (R&D Systems, Inc, Minneapolis, MN). All samples were evaluated in triplicate. Colorimetric values were read on a plate reader (Biotek Instruments, Inc., Winoski, VT) and analyzed by KC Junior software with reference wavelengths set at 450nM and correction wavelengths set at 540nM.
Intracellular CXCL12 Staining
BMSCs were grown to confluence and exposed to 100μM VP-16, 1μM OA-Hy or DMSO for 24 hours. Following treatment, BMSCs were trypsinized, collected, and fixed in 10% formaldehyde for 30 minutes. BMSCs were rinsed in 1X PBS and permeabolized in 70% EtOH for 30 minutes on ice. To prevent non specific antibody binding, BMSCs were blocked in PBS/5% BSA for 15 minutes and subsequently incubated with 2ug of CXCL12 specific antibody or isotype control for 20 minutes. PE conjugated secondary antibody was added to BMSCs for 20 minutes. BMSCs were rinsed, evaluated by flow cytometry and data were analyzed using CellQuest (Becton Dickinson, San Jose, CA).
Chemotaxis assay
Confluent BMSC layers, grown in 24-well tissue culture plates (Becton Dickinson, Franklin Lakes, NJ), were treated with 100μM VP-16, 1μM OA-Hy, or left untreated for 24 hours. BMSCs were then rinsed with fresh medium and 350μL of medium, or medium supplemented with 10ng/mL recombinant murine MMP-2 or 25ng/mL recombinant human MMP-2 was placed in the bottom chamber of each well for 2 hours. In addition, C57BL/6 WT or MMP-2-/- BMSCs were grown in 350μL α-MEM in 24-well tissue culture plates for 24 hours. Following incubation, transwells with 5μ pores (Corning Inc., Corning, NY) containing 1.5×105 C1.92 were placed into each well. Following incubation at 37° C for 4 hours, cells migrating to the lower chamber were enumerated by flow cytometry (number of events/30-second collection). Recombinant CXCL12 (100ng/mL R&D Systems Inc., Minneapolis, MN), and medium alone served as the positive and negative control respectively. All samples were evaluated in triplicate.
Statistical Analysis
Statistical analysis was performed using Student’s T-test to detect differences among means (SigmaStat Version 4.0 software, SPSS Inc., Chicago, IL). All statistical comparisons represent treated samples compared to control levels. Statistically significant differences are indicated by and asterisk on appropriate graphs.
Results
MMP-2 expression is regulated distinctly during acute and chronic VP-16 exposure.
To evaluate alterations in MMP-2 protein, we performed gelatin zymography on supernatants from BMSCs that were exposed to VP-16. Following treatment, BMSC MMP-2 levels increased at 30 minutes and subsequently began to diminish following 5 hours of VP-16 exposure (Figure 1A).
Figure 1.
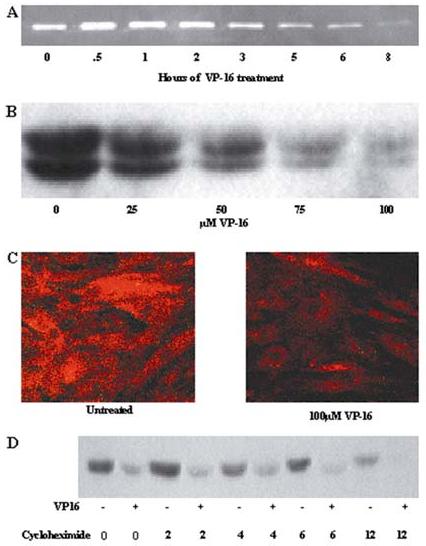
MMP-2 protein is dysregulated in BMSCs exposed to VP-16. (A) BMSC supernatants were conditioned in serum-free medium for 8 hours. At each time point (30 minutes OE 8 hours) 100:M VP-16 was added to the conditioned BMSCs media. After 8 hours the supernatants were collected and gelatin zymography performed to detect MMP-2. (B) Supernatants were collected from VP-16 treated BMSCs and concentrated as described in Materials and Methods. MMP-2 monoclonal antibody was used to detect MMP-2 protein in VP-16 treated groups compared to supernatants collected from untreated control BMSCs. Data are representative of three independent experiments. (C) BMSCs treated with 100μM VP-16 for 24 hours were fixed and stained with MMP-2 monoclonal antibody and subsequently incubated with PE-tagged secondary antibody. Fluorescence was detected by confocal microscopy. (D) Confluent BMSCs were treated with either VP-16 or VP-16 and cycloheximide for 2-24 hours. At each time point the supernatants were collected, concentrated 10X, and subjected to western blot with antibody specific to MMP-2.
To determine whether VP-16 alters BMSC production of MMP-2 during long-term exposure, we evaluated several primary human BMSC lines by ELISA following 24 hours of exposure to VP-16. MMP-2 protein was diminished in supernatants of BMSCs to approximately 12% to 58% of that in untreated controls (Data not shown). MMP-2 protein was also evaluated by western blot and the amount of active and latent MMP-2 protein in concentrated supernatants was determined to be reduced by VP-16 exposure compared to untreated controls (Figure 1B). MMP-2 protein was not altered in BMSCs treated with VP-16 solvent control (Data not shown). Reduction of MMP-2 protein in BMSCs exposed to VP-16 for 24 hours was not due to intracellular accumulation (Figure 1C) or reduced protein stability (Figure 1D).
In contrast to MMP-2 protein, MMP-2 mRNA was not reduced during VP-16 exposure. Semi-quantitative RT-PCR (Figure 2A) illustrates that BMSCs treated with 100μM VP-16 do not have diminished MMP-2 mRNA. This result was confirmed by RNAse protection (Figure 2B).
Figure 2.
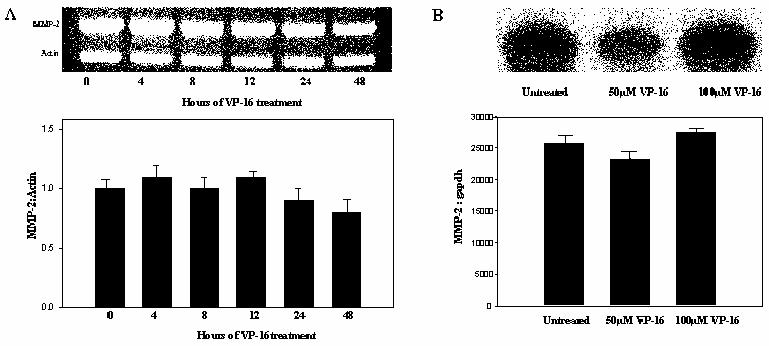
MMP-2 mRNA is not diminished following VP-16 exposure. (A) MMP-2 mRNA is not altered by VP-16 treatment of BMSCs. BMSCs were treated for the indicated times with 100uM VP-16. Semi-quantitative RT-PCR was performed to estimate the amount of MMP-2 message in each sample relative to β-actin. Representative data from three independent experiments are shown. (B) BMSCs were treated with 50 or 100μM VP-16 for 24 hours, RNA was extracted, and RNAse protection assay was completed with probes specific for MMP-2 and GAPDH sequences.
MMP-2 protein is necessary for optimal stromal cell support of chemotaxis.
To determine if MMP-2 protein is necessary for BMSC support of pro-B cell chemotaxis we compared control BMSCs with BMSCs treated with OA-Hy or BMSCs derived from WT and MMP-2-/- mice. Addition of OA-Hy to BMSCs resulted in approximately 50% reduction in the ability of human primary (P151) and murine S-10 BMSCs to support C1.92 pro-B cell chemotaxis (Figure 3A). Direct addition of OA-Hy to CXCL12 did not reduce support of chemotaxis (Figure 3A). MMP-2-/- BMSCs also had diminished ability to support chemotaxis compared to WT control BMSCs (Figure 3B). Addition of recombinant CXCL12 restored MMP-2-/- BMSC support of chemotaxis to approximately 93% of control BMSCs. (Figure 3B).
Figure 3.
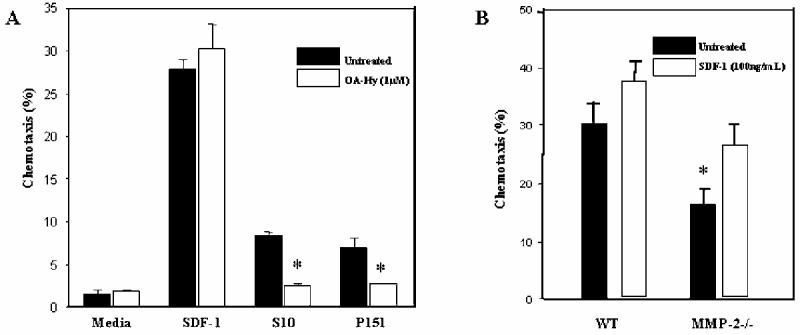
MMP-2 is necessary for optimal BMSC support of pro-B cell chemotaxis. (A) Murine S10 or P151 primary human BMSCs were left untreated or treated with 1μM OA-Hy for 24 hours in the bottom chamber of a transwell plate. Following 4 hours of chemotaxis towards BMSCs, media, or rCXCL12, the number of C1.92 cells that migrated to the bottom chamber was evaluated by flow cytometry. (B) C57BL/6 WT and C57BL/6 MMP-2-/- BMSCs were evaluated for their ability to support chemotaxis of C1.92 cells. Following 4 hours of incubation, C1.92 cells were collected from lower wells that contained either adherent WT or MMP-2-/- BMSC layers. C1.92 cells that migrated to the bottom chamber were enumerated by flow cytometry (p<.05).
CXCL12 protein is diminished in supernatants following MMP-2 inhibition.
To determine whether MMP-2 is required for release of CXCL12 protein into supernatants of adherent BMSCs, BMSCs were treated with 1μM OA-Hy for 24 hours. CXCL12 protein was diminished in OA-Hy treated BMSC supernatants compared to DMSO solvent control treated BMSCs (Figure 4A). The reduction of CXCL12 in supernatants was not due to intracellular accumulation of CXCL12 protein (Figure 4B). CXCL12 mRNA was not altered by OA-Hy treatment, determined by RT-PCR (Data not shown).
Figure 4.
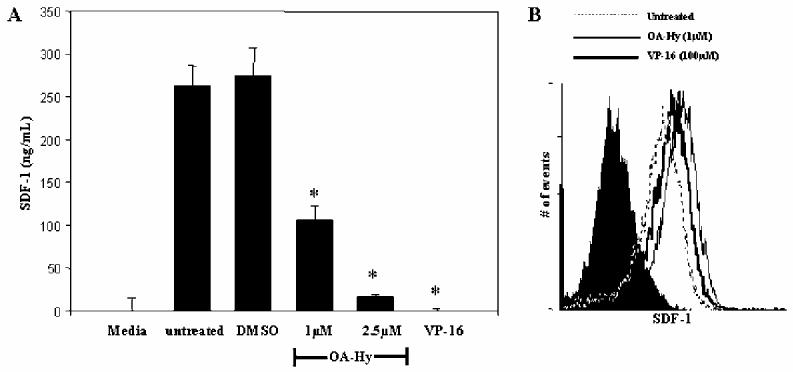
CXCL12 protein is diminished in the supernatants of BMSCs with diminished MMP-2 protein levels. (A) BMSCs were either untreated, or treated with 1μM OA-Hy, DMSO, or 100μM VP-16 for 24 hours. Supernatants were evaluated by a CXCL12 specific ELISA (p<.01). (B) CXCL12 intracellular staining was performed on BMSCs exposed to 1μM OA-Hy or DMSO for 24 hours. All samples were compared to isotype controls.
Recombinant MMP-2 protein restores VP-16 treated BMSC support of chemotaxis.
To determine whether addition of MMP-2 protein could restore VP-16 treated BMSC support of chemotaxis; we first determined the amount of MMP-2 protein that primary human BMSC line (P156) and C57BL/6 BMSCs produced at steady state (Data not shown). Based on our ELISA results, we added 25ng/mL rhMMP-2 to VP-16-treated P156 BMSCs or 10ng/mL rmMMP-2 to C57BL/6 MMP-2-/- BMSCs to approximate physiological levels. Addition of MMP-2 restored VP-16-treated and MMP-2-/- BMSC support of chemotaxis to greater than 100% of untreated human P156 BMSCs (Figure 5A) and 85% of that supported by WT murine BMSCs (Figure 5B).
Figure 5.

Recombinant MMP-2 protein restores VP-16 treated BMSC support of chemotaxis. P156 primary BMSCs were treated with 100μM VP-16 for 24 hours and C57BL/6 MMP-2-/- BMSCs were cultured for 24 hours prior to the addition of CXCR4+ cells to the top chamber. P156 BMSCs were then rinsed and either 10 mg/mL murine or 25 ng/mL human recombinant MMP-2 was added in 350μL of media to C57BL/6 MMP-2-/- or P156 BMSCs respectively for 2 hours. 5μm transwells were placed on top of the cells and 1×106 C1.92 were evaluated for their ability to migrate into the bottom chamber over a 4-hour period (p<.05).
Discussion
In the current study we found that BMSC MMP-2 is affected differentially by acute and chronic exposure to VP-16. Increased MMP-2 expression was transient in VP-16 treated BMSCs. Subsequent to the acute response, MMP-2 protein was reduced when BMSCs were exposed to VP-16 for longer periods of time. The consequence of chronic exposure of BMSCs to VP-16 is the main focus of the current study. Our data suggest that chemotherapy down regulates MMP-2 protein expression and disrupts CXCL12 supported chemotaxis, potentially, by inhibiting CXCL12 release from the BMSC surface. This novel role for MMP-2 in CXCL12 regulation broadens the context in which MMPs may influence hematopoiesis. Further, it contributes to our understanding of factors that may impact on chemotactic support by the bone marrow microenvironment following aggressive chemotherapy.
We have previously shown that VP-16 induces many alterations in BMSCs that reduce BMSC support of hematopoiesis 16,17. In the current report, we show that VP-16 treatment (100μM) increases, and then subsequently reduces, the amount of MMP-2 protein detected in BMSC supernatants (Figure 1). VP-16 exposure rapidly increases BMSC ROS generation, potentially allowing for immediate activation of MMP-2 through conformation changes resulting in auto-catalytic cleavage of MMP-2’s pro-domain (unpublished data). The mechanism of diminished MMP-2 protein during chronic VP-16 exposure is not due to reduced MMP-2 mRNA or intracellular accumulation of protein (Figure 1, 2). Further, the stability of MMP-2 protein is not reduced by VP-16 (Figure 1).
Based on this study, future investigations will focus, in part, on disruption of translation efficiency of MMP-2 transcripts in VP-16 treated BMSCs. In other models VP-16 has been shown to blunt phosphorylation of Eukaryotic Initiation Factor 4α (EIF4α)25 which is necessary for MMP-2 translational initiation to occur 26. Additionally, VP-16 treatment of Swiss 3T3 fibroblast cells increased association of cap binding protein eIF-4E with its inhibitory binding protein 4E-BP 27. Unsequestered eIF-4E is also necessary for efficient translation of MMP-2 message 26. These observations suggest that disrupted translation may be one consequence of VP-16-induced damage, resulting in potentially diverse effects on BMSC function.
Initial observations that preceded this study indicated that the addition of OA-Hy to established BMSC pro-B cell co-cultures resulted in diminished adhesion of pro-B cells to BMSCs, a subsequent accumulation of pro-B cells in G0/G1 phase of cell cycle, and increased apoptosis (Data not shown). Clearly, the effects of OA-Hy on the co-culture may be due to a direct effect of MMP-2 inhibition on BMSC function, pro-B cell proliferation or survival, or a combination of effects on both cell types. The current study was aimed at isolating the effects of diminished MMP-2 on BMSCs influence of hematopoietic support capacity.
B lymphopoiesis has not been evaluated in MMP-2-/- mice. However, MMP-2-/- mice have been used to study antibody induced asthma, arthritis, and Experimental Autoimmune Encephalomyelitis (EAE) 28-30. Increased incidence of EAE in MMP-2-/- mice is due to an increase in T-cell MMP-9 expression. B cells were not evaluated in this model so no conclusion can be drawn regarding B lymphopoiesis in the absence of MMP-2 in vivo 28. However, it has become increasingly evident that MMPs influence hematopoietic cell support in the bone marrow microenvironment from other studies. One report indicates that MMP-9 is required to release soluble Kit-ligand within the bone marrow microenvironment, which regulates stem cells movement from quiescent to proliferative niches 31. This study is just one that provides precedent for MMPs function within the bone marrow microenvironment as a regulator of growth-factor availability.
To determine whether diminished MMP-2 expression in BMSCs exposed to VP-16 contributes to reduced support of chemotaxis, we treated BMSCs with OA-Hy, and quantitated the ability of treated BMSCs to support pro-B cell chemotaxis. Consistent with VP-16 exposure, MMP-2 inhibition by OA-Hy diminished BMSCs support of chemotaxis (Figure 3A). Because the use of chemical inhibitors has the limitation of non-specific effects, we chose to further investigate MMP-2 in a more specific manner. To do so, we established BMSCs from MMP-2 knockout mice. MMP-2-/- BMSCs used in this study were established from the only MMP-2-/- mice currently available to our laboratory (femurs generously provided by Dr. Farrah Kheradmand). This MMP-2-/- was generated on the C57BL/6 background. We have previously noted that C57BL/6 BMSCs are very resistant to chemotherapy and display enhanced support of pro-B cells when compared to human or Balb/c derived BMSCs (unpublished data). MMP-2-/- BMSCs were less able to support pro-B cell chemotaxis than stromal cells established from wild-type control mice, further supporting a role for MMP-2 protein in CXCL12 directed migration of pro-B cells (Figure 3B).
OA-Hy treated BMSCs secrete less CXCL12 protein than controls (Figure 4A), however, we have confirmed that this is not due to intracellular accumulation of the CXCL12 protein (Figure 4B). This suggests that MMP-2 may regulate release of CXCL12 from the BMSC surface. Potentially, when MMP-2 protein is below physiological levels, as observed following VP-16 exposure or OA-Hy treatment, proteoglycan bound CXCL12 is not efficiently released and an optimal chemotactic gradient is not established. Consistent with a role for MMPs in regulating chemokine gradients, MMP-2-/- mice used in an antibody-induced asthma model have inflammatory cells sequestered in lung parenchyma resulting in asphyxiation. Notably, MMP-2 protein is required for release of eotaxin chemokine (CCL11) 24 which is necessary for directed migration of inflammatory cells out of the lung parenchyma.
Restoration of diminished BMSC chemotactic support by VP-16 treated BMSCs occurred only when recombinant MMP-2 protein was added back at physiological levels (Figure 5B). Levels that exceeded baseline decreased chemotactic support of BMSCS. A previous report indicates that MMP-2 can cleave and inactivate CXCL12, resulting in reduced chemotactic support 21. Our data suggests that inactivation of CXCL12 by MMP-2 may occur when active MMP-2 is elevated during the acute response to chemotherapy. This may reduce the concentration of active CXCL12 in the bone marrow microenvironment, contributing to diminished recruitment of CXCR4+ pro-B cells. Consistent with the report noted above, we found that increased MMP-2 diminished CXCL12 supported chemotaxis in a dose responsive manner (Data not shown). Our data suggest that at physiological levels BMSC MMP-2 may release proteoglycan bound CXCL12 establishing a chemotactic gradient, while inappropriately high levels observed during tissue damage inactivate CXCL12 protein. Potentially, dysregulation of MMP-2 that occurs during VP-16 exposure may contribute to diminished BMSC chemotactic support by this combination of effects on CXCL12 activity and availability.
Footnotes
Correspondance Laura F. Gibson, PhD Health Sciences Center Dept. of Pediatrics PO Box 9214 Morgantown, WV 26506 Email: lgibson@hsc.wvu.edu
This work was supported, in part, by NIH ROI HL056888 (LFG) and NRSA Institutional Training Grant in Environmental Science ES010953 (SDC).
Reference List
- 1.Hendrikx PJ, Martens CM, Hagenbeek A, Keij JF, Visser JW. Homing of fluorescently labeled murine hematopoietic stem cells. Exp.Hematol. 1996;24:129–140. [PubMed] [Google Scholar]
- 2.Papayannopoulou T, Craddock C. Homing and trafficking of hemopoietic progenitor cells. Acta Haematol. 1997;97:97–104. doi: 10.1159/000203665. [DOI] [PubMed] [Google Scholar]
- 3.Jo DY, Rafii S, Hamada T, Moore MA. Chemotaxis of primitive hematopoietic cells in response to stromal cell-derived factor-1. J.Clin.Invest. 2000;105:101–111. doi: 10.1172/JCI7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.D'Apuzzo M, Rolink A, Loetscher M, Hoxie JA, Clark-Lewis I, Melchers F, Baggiolini M, Moser B. The chemokine CXCL12, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur.J.Immunol. 1997;27:1788–1793. doi: 10.1002/eji.1830270729. [DOI] [PubMed] [Google Scholar]
- 5.Janczewska S, Ziolkowska A, Durlik M, Cybulska E, Olszewski WL, Lukomska B. Requirement of stromal cells in the bone marrow transplant for rapid lymphoid replenishment. Transplant.Proc. 1999;31:696–699. doi: 10.1016/s0041-1345(98)01614-5. [DOI] [PubMed] [Google Scholar]
- 6.Strobel ES, Mobest D, von Kleist S, Dangel M, Ries S, Mertelsmann R, Henschler R. Adhesion and migration are differentially regulated in hematopoietic progenitor cells by cytokines and extracellular matrix. Blood. 1997;90:3524–3532. [PubMed] [Google Scholar]
- 7.Morris AJ, Turnbull JE, Riley GP, Gordon MY, Gallagher JT. Production of heparan sulphate proteoglycans by human bone marrow stromal cells. J.Cell Sci. 1991;99(Pt 1):149–156. doi: 10.1242/jcs.99.1.149. [DOI] [PubMed] [Google Scholar]
- 8.Gallagher JT, Spooncer E, Dexter TM. Role of the cellular matrix in haemopoiesis. I. Synthesis of glycosaminoglycans by mouse bone marrow cell cultures. J.Cell Sci. 1983;63:155–171. doi: 10.1242/jcs.63.1.155. [DOI] [PubMed] [Google Scholar]
- 9.Drzeniek Z, Siebertz B, Stocker G, Just U, Ostertag W, Greiling H, Haubeck HD. Proteoglycan synthesis in haematopoietic cells: isolation and characterization of heparan sulphate proteoglycans expressed by the bone-marrow stromal cell line MS-5. Biochem.J. 1997;327(Pt 2):473–480. doi: 10.1042/bj3270473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts R, Gallagher J, Spooncer E, Allen TD, Bloomfield F, Dexter TM. Heparan sulphate bound growth factors: a mechanism for stromal cell mediated haemopoiesis. Nature. 1988;332:376–378. doi: 10.1038/332376a0. [DOI] [PubMed] [Google Scholar]
- 11.Amara A, Lorthioir O, Valenzuela A, Magerus A, Thelen M, Montes M, Virelizier JL, Delepierre M, Baleux F, Lortat-Jacob H, Arenzana-Seisdedos F. Stromal cell-derived factor-1alpha associates with heparan sulfates through the first beta-strand of the chemokine. J.Biol.Chem. 1999;274:23916–23925. doi: 10.1074/jbc.274.34.23916. [DOI] [PubMed] [Google Scholar]
- 12.Verdrengh M, Tarkowski A. Impact of topoisomerase II inhibition on cytokine and chemokine production. Inflamm.Res. 2003;52:148–153. doi: 10.1007/s000110300065. [DOI] [PubMed] [Google Scholar]
- 13.Galotto M, Berisso G, Delfino L, Podesta M, Ottaggio L, Dallorso S, Dufour C, Ferrara GB, Abbondandolo A, Dini G, Bacigalupo A, Cancedda R, Quarto R. Stromal damage as consequence of high-dose chemo/radiotherapy in bone marrow transplant recipients. Exp.Hematol. 1999;27:1460–1466. doi: 10.1016/s0301-472x(99)00076-4. [DOI] [PubMed] [Google Scholar]
- 14.Chamberlin W, Barone J, Kedo A, Fried W. Lack of recovery of murine hematopoietic stromal cells after irradiation-induced damage. Blood. 1974;44:385–392. [PubMed] [Google Scholar]
- 15.Gibson LF, Fortney J, Landreth KS, Piktel D, Ericson SG, Lynch JP. Disruption of bone marrow stromal cell function by etoposide. Biol.Blood Marrow Transplant. 1997;3:122–132. [PubMed] [Google Scholar]
- 16.Hall BM, Fortney JE, Gibson LF. Alteration of nuclear factor-kappaB (NF-6B) expression in bone marrow stromal cells treated with etoposide. Biochem.Pharmacol. 2001;61:1243–1252. doi: 10.1016/s0006-2952(01)00602-5. [DOI] [PubMed] [Google Scholar]
- 17.Hall BM. Human Bone Marrow Stromal Cell CXCL12 Production Is Reduced Following Exposure To Topoisosmerase II Inhibitors, Etoposide Or Doxorubicin. Fortney, J. E. and Gibson, L. F. Analytical Pharmacology. 2003;4(2):21–29. [Google Scholar]
- 18.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu.Rev.Cell Dev.Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marquez-Curtis LA, Dobrowsky A, Montano J, Turner AR, Ratajczak J, Ratajczak MZ, Janowska-Wieczorek A. Matrix metalloproteinase and tissue inhibitors of metalloproteinase secretion by haematopoietic and stromal precursors and their production in normal and leukaemic long-term marrow cultures. Br.J.Haematol. 2001;115:595–604. doi: 10.1046/j.1365-2141.2001.03160.x. [DOI] [PubMed] [Google Scholar]
- 20.Janowska-Wieczorek A, Matsuzaki A, Marquez A. The Hematopoietic Microenvironment: Matrix Metalloproteinases in the Hematopoietic Microenvironment. Hematol. 2000;4:515–527. [PubMed] [Google Scholar]
- 21.McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, Overall CM. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J.Biol.Chem. 2001;276:43503–43508. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- 22.Collins LS, Dorshkind K. A stromal cell line from myeloid long-term bone marrow cultures can support myelopoiesis and B lymphopoiesis. J.Immunol. 1987;138:1082–1087. [PubMed] [Google Scholar]
- 23.Gibson LF, Piktel D, Landreth KS. Insulin-like growth factor-1 potentiates expansion of interleukin-7-dependent pro-B cells. Blood. 1993;82:3005–3011. [PubMed] [Google Scholar]
- 24.Corry DB, Rishi K, Kanellis J, Kiss A, Song Lz LZ, Xu J, Feng L, Werb Z, Kheradmand F. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat.Immunol. 2002;3:347–353. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeffrey IW, Bushell M, Tilleray VJ, Morley S, Clemens MJ. Inhibition of protein synthesis in apoptosis: differential requirements by the tumor necrosis factor alpha family and a DNA-damaging agent for caspases and the double-stranded RNA-dependent protein kinase. Cancer Res. 2002;62:2272–2280. [PubMed] [Google Scholar]
- 26.Bradley JM, Kelley MJ, Rose A, Acott TS. Signaling pathways used in trabecular matrix metalloproteinase response to mechanical stretch. Invest Ophthalmol.Vis.Sci. 2003;44:5174–5181. doi: 10.1167/iovs.03-0213. [DOI] [PubMed] [Google Scholar]
- 27.Tee AR, Proud CG. DNA-damaging agents cause inactivation of translational regulators linked to mTOR signalling. Oncogene. 2000;19:3021–3031. doi: 10.1038/sj.onc.1203622. [DOI] [PubMed] [Google Scholar]
- 28.Esparza J, Kruse M, Lee J, Michaud M, Madri JA. MMP-2 null mice exhibit an early onset and severe experimental autoimmune encephalomyelitis due to an increase in MMP-9 expression and activity. FASEB J. 2004;18:1682–1691. doi: 10.1096/fj.04-2445com. [DOI] [PubMed] [Google Scholar]
- 29.Corry DB, Kiss A, Song LZ, Song L, Xu J, Lee SH, Werb Z, Kheradmand F. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004;18:995–997. doi: 10.1096/fj.03-1412fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itoh T, Matsuda H, Tanioka M, Kuwabara K, Itohara S, Suzuki R. The role of matrix metalloproteinase-2 and matrix metalloproteinase-9 in antibody-induced arthritis. J.Immunol. 2002;169:2643–2647. doi: 10.4049/jimmunol.169.5.2643. [DOI] [PubMed] [Google Scholar]
- 31.Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MA, Werb Z, Rafii S. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]