

There is growing evidence that the efficacy of anti-tumour necrosis factor α (TNF-α) therapies in Crohn’s disease (CD) may critically depend on the binding of the transmembrane precursor of TNF-α (mTNF-α), thus eliciting complex intracellular signalling events, a process described as “reverse signalling”.1–3 In their recent paper (Gut 2004;53:70–7), Di Sabatino et al showed that infliximab reverted defective peripheral and lamina propria lymphocyte apoptosis in CD patients via a caspase dependent mechanism, further corroborating the findings of previous studies.1–4 It has also been suggested that failure of another TNF binding agent, etanercept (Enbrel; a recombinant TNFR2:Fc fusion protein), to induce peripheral and lamina propria lymphocyte apoptosis,4 provides a possible molecular explanation for the lack of efficacy of etanercept in a randomised placebo controlled trial in active CD.5
However, the authors do not discuss other signalling pathways that are activated via ligation of transmembrane TNF-α by infliximab (for example, we have shown that infliximab also transiently activates p38 mitogen activated protein kinase (MAPK) in monocytes in vitro and in the lamina propria of CD patients in vivo3). Responders and non-responders to infliximab differ in the pattern of mucosal p38MAPK target phosphorylation, but not caspase-3 activation, further emphasising the complex modulation of intracellular signalling pathways beyond mere neutralisation of sTNF-α.6 To show if these signalling pathways are also activated in primary T cells, we analysed the influence of infliximab and etanercept on p38MAPK activation and apoptosis in an established model of non-transformed in situ activated T lymphocytes.7,8
According to the findings of van den Brande et al,4 we observed PARP cleavage as a molecular hallmark of apoptosis in cultures grown with infliximab (fig 1A▶) but not in the presence of etanercept. Whereas no increase in phosphorylated p38MAPK could be detected after etanercept stimulation, significant activation (that is, dual phosphorylation) of p38MAPK 24 hours after infliximab treatment was observed in 4/5 of the cell lines derived from CD patients (fig 1A▶).
Figure 1.
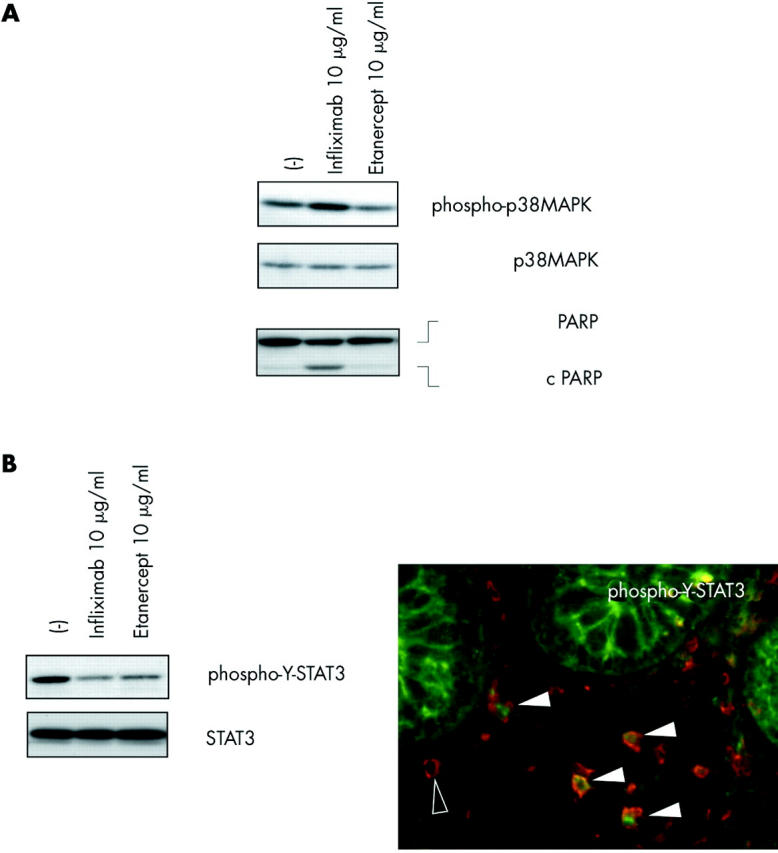
Intestinal CD4+ T cells were obtained from colonic biopsies from five patients with Crohn’s disease (median age 34 years (range 18–49)) with an established diagnosis of Crohn’s disease based on histopathological and endoscopic criteria. All patients had active disease at inclusion and were treated with 5-ASA (2–4 g/day, five patients), prednisone (20 mg/day, one patient), and azathioprine (150–200 mg/day, four patients). None of the patients had received anti-TNF-α treatment. Neither antigen nor feeder cells were added to the intestinal T cells cultures, which consisted of more than 97% CD3+/CD4+ T cells. (A) Infliximab, but not etanercept, activated p38α and induced apoptosis in intestinal T lymphocytes from Crohn’s disease patients. Levels of (phosphorylated) p38 mitogen activated protein kinase (MAPK) and PARP in in situ activated intestinal T lymphocytes were investigated by western blot 24 hours after stimulation with the respective TNF-α binding agent, as described previously.3 Data are representative of experiments performed in all patients, n = 5. (B) Signal transducer and activator of transcription 3 (STAT3) was activated in CD4+ cells in the intestinal mucosa in Crohn’s disease patients in vivo and downregulated by both infliximab and etanercept in intestinal T cells from Crohn’s disease patients in vitro. Western blot and immunofluorescence staining were performed as described previously.3 Filled arrowheads, cells positive for phospho-Y-STAT3 (anti-phospho-Y-STAT3, 1/100; Cell Signaling Technology) and CD4 (anti-CD4, 1/500; BD PharMingen, San Diego, California, USA); open arrowhead, CD4 immunoreactivity only (n = 5 for immunofluorescence and western blot; representative result for all experiments).
We have demonstrated previously that constitutive tyrosine phosphorylation of the transcription factor signal transducer and activator of transcription 3 (STAT3) may represent a specific feature of intestinal T cells from Crohn’s disease.8 Tyrosine phosphorylated STAT3 can be found in CD4+ cells in the inflamed mucosa, as shown by immunofluorescence analysis (fig 1B▶). Surprisingly, infliximab and etanercept were able to reduce STAT3 phosphorylation in T cells from CD patients (cultured with interleukin (IL)-2 and IL-4) to a similar extent (fig 1B▶). It is tempting to speculate that the previously reported downregulation of interferon γ/granulocyte macrophage-colony stimulating factor by both infliximab and etanercept is also involved in the decrease in STAT3 tyrosine phosphorylation as both cytokines are potent inducers of STAT3 activation via Janus kinases. Mechanistically, this common action of infliximab and etanercept might be due to neutralisation of an autocrine loop of constitutively released sTNF-α. Although the findings suggest that inhibition of cytokine dependent inducible STAT3 phosphorylation could be dispensable for therapeutic efficacy in CD, aberrant constitutive STAT phosphorylation in T lymphocytes may still have an important modulatory role in chronic intestinal inflammation.8
These observations corroborate the hypothesis that TNF-α binding agents may exert distinct functions on lymphocyte activation and survival, either by “reverse signalling” via binding of mTNF-α resulting in p38MAPK activation and apoptosis or by neutralisation of sTNF-α, which in this model may serve to inhibit STAT3 signalling. The individual properties of TNF-α blockers to induce either of these complex molecular actions may be differentially responsible for therapeutic success or failure in chronic inflammatory disorders such as rheumatoid arthritis, psoriasis, or CD. In CD, ligation of mTNF-α, subsequent apoptotic processes, and MAPK signalling seem to be critically required. Molecular dissection of mTNF-α apoptotic and non-apoptotic signalling may have important implications for the design of future therapeutic strategies in inflammatory bowel disease.
Conflict of interest: None declared.
References
- 1.ten Hove T , van Montfrans C, Peppelenbosch MP, et al. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn’s disease. Gut 2002;50:206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lugering A , Schmidt M, Lugering N, et al. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn’s disease by using a caspase-dependent pathway. Gastroenterology 2001;121:1145–57. [DOI] [PubMed] [Google Scholar]
- 3.Waetzig GH, Seegert D, Rosenstiel P, et al. p38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J Immunol 2002;168:5342–51. [DOI] [PubMed] [Google Scholar]
- 4.Van den Brande JM, Braat H, van den Brink GR, et al. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn’s disease. Gastroenterology 2003;124:1774–85. [DOI] [PubMed] [Google Scholar]
- 5.Sandborn WJ, Hanauer SB, Katz S, et al. Etanercept for active Crohn’s disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology 2001;121:1088–94. [DOI] [PubMed] [Google Scholar]
- 6.Waetzig GH, Rosenstiel P, Nikolaus S, et al. Differential p38 mitogen-activated protein kinase target phosphorylation in responders and nonresponders to infliximab. Gastroenterology 2003;125:633–4. [DOI] [PubMed] [Google Scholar]
- 7.Agnholt J , Dahlerup JF, Kaltoft K. The effect of etanercept and infliximab on the production of tumour necrosis factor alpha, interferon-gamma and GM-CSF in in vivo activated intestinal T lymphocyte cultures. Cytokine 2003;23:76–85. [DOI] [PubMed] [Google Scholar]
- 8.Lovato P , Brender C, Agnholt J, et al. Constitutive STAT3 activation in intestinal T cells from patients with Crohn’s disease. J Biol Chem 2003;278:16777–81. [DOI] [PubMed] [Google Scholar]