

SUMMARY
Toll-like receptors (TLR) and NOD2 are emerging as key mediators of innate host defence in the intestinal mucosa, crucially involved in maintaining mucosal as well as commensal homeostasis. Recent observations suggest new (patho-) physiological mechanisms of how functional versus dysfunctional TLRx/NOD2 pathways may oppose or favour inflammatory bowel disease (IBD). In health, TLRx signalling protects the intestinal epithelial barrier and confers commensal tolerance whereas NOD2 signalling exerts antimicrobial activity and prevents pathogenic invasion. In disease, aberrant TLRx and/or NOD2 signalling may stimulate diverse inflammatory responses leading to acute and chronic intestinal inflammation with many different clinical phenotypes.
INTRODUCTION
The intestinal mucosa must rapidly recognise detrimental pathogenic threats to the lumen to initiate controlled immune responses but maintain hyporesponsiveness to omnipresent harmless commensals. Charles Janeway Jr first suggested that so-called pattern recognition receptors (PRRs) may play an essential role in allowing innate immune cells to discriminate between “self” and microbial “non-self” based on the recognition of broadly conserved molecular patterns.1 Toll-like receptors (TLRs) which comprise a class of transmembrane PRRs play a key role in microbial recognition, induction of antimicrobial genes, and the control of adaptive immune responses. NODs (NOD1 and NOD2) are a structurally distinct family of intracellular PRRs which presumably in the context of microbial invasion subserve similar functions (fig 1 ▶). TLRs and NOD2 are widely expressed on various cell types of the gastrointestinal mucosa, participating in host defence against microbial pathogens in at least four ways:
Figure 1.
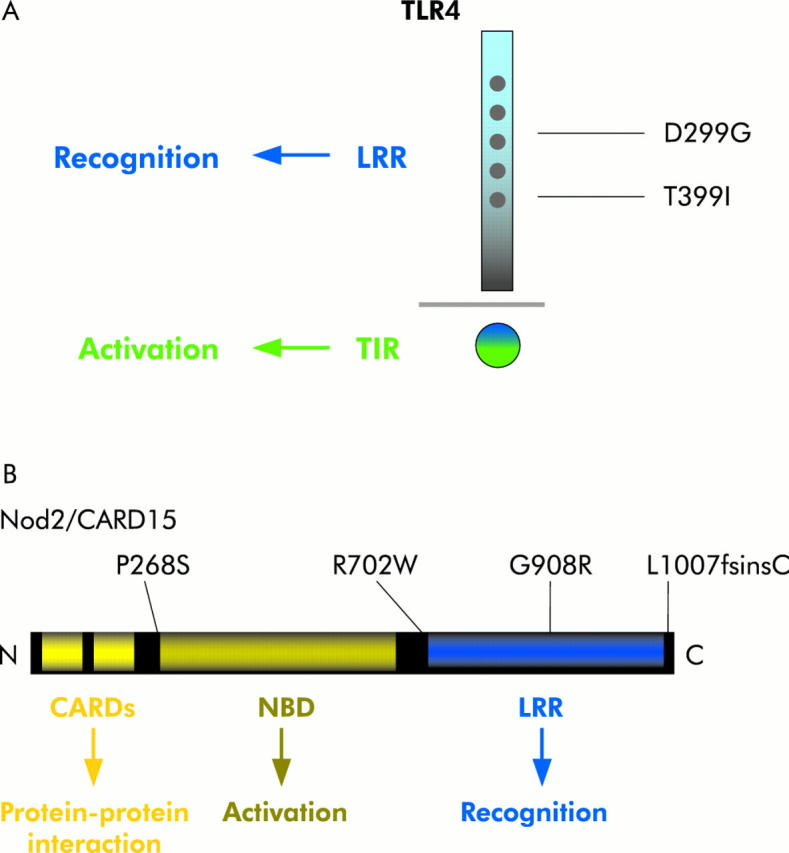
Toll-like receptor 4/nucleotide binding oligomerisation domain 2 (TLR4/NOD2) structure and inflammatory bowel disease associated common variants. (A) Mammalian TLRs are a family of type I transmembrane receptors which are all characterised by three common structural features, as exemplified for TLR4 here: a divergent ligand binding extracellular domain with multiple leucine rich repeats (LRR), a short transmembrane region and a highly homologous cytoplasmic Toll-interleukin 1 receptor (TIR) domain. Two common cosegregating missense mutations (D299G and T399I) that affect the extracellular ligand recognition domain of the TLR4 receptor are associated with deregulated immune responses to lipopolysaccharide in humans. (B) NOD1 and NOD2 are members of a family of intracellular proteins that contain an N terminal caspase recruitment domain (CARD), a centrally located nucleotide binding domain (NBD), and a C terminal regulatory domain. As shown here, NOD2 protein contains two N terminal CARDs fused to a central NBD domain followed by 10 tandem LRRs at the C terminus. Three main NOD2 variants (R702W, G908R, and L1007fsinsC) were confirmed to be associated with susceptibility to some types of Crohn’s disease.
recognition of molecular patterns present on pathogens;
expression at the interface with the “environment” of the gastrointestinal lumen;
induction of secretion of pro/anti-inflammatory cyto- and chemokines that link to the adaptive immune system; and
induction of antimicrobial effector pathways.
Recent studies have greatly advanced our understanding of these mechanisms through which the gastrointestinal innate immune system can mediate differential host-microbial interactions in recognition and sorting of the broad luminal spectrum of diverse microbial products. Furthermore, related findings propose that mammalian TLRx and NOD2 dysfunctions play a key role in the pathophysiology of IBD.
MOLECULAR BASIS OF BACTERIAL-MUCOSAL INTERACTIONS
Toll-like receptors (TLRs)
Structure
Mammalian TLRs comprise a family of (so far) 11 individual type I transmembrane receptors which are characterised by three common structural features (fig 1A ▶): a divergent ligand binding extracellular domain with leucine rich repeats (LRR), a short transmembrane region, and a highly homologous cytoplasmic Toll/interleukin 1 receptor (TIR) domain, similar to that of the interleukin 1 receptor family and essential for initiation of downstream signalling cascades.2
Expression pattern
TLRs are differentially (inducibly or constitutively) expressed by many distinct cell types throughout the whole gastrointestinal tract in vitro and in vivo, including (mature and immature) epithelial cells of the stomach, small intestine, and colon,3–14 as well as intestinal monocytes/macrophages15,16 and dendritic cells (Stagg AJ, personal communication, 2005) of the lamina propria and myofibroblasts,17 endothelial cells,18 and adipocytes (Siegmund B, personal communication, 2005) of the intestinal submucosa.
Some inter cell line and inter laboratory differences in detection levels of constitutive TLRx expression have been described for various intestinal epithelial tumour cell lines in vitro.3,8,10,11,19–21 It is a common, if not expected, problem that epigenetic drifts from the original progenitor lineage may occur in such cell lines with each cell culture passage. Different phenotypes of TLRx expression may also derive from distinct cell culture conditions which alter differentiation state in vitro.22 Regarding TLR expression in primary intestinal epithelial cells (IEC), there is field wide consensus that TLR2 and TLR4 are present only in small amounts on IEC in vivo, thus minimising recognition of lumenal bacteria in the healthy intestine.4,7,9,11,14,16 In contrast, TLR4 is significantly increased in primary IEC throughout the lower gastrointestinal tract in active disease of both Crohn’s disease (CD) and ulcerative colitis (UC)4 and murine colitis.9,14
Ligand specificity
Different pathogen associated molecular patterns selectively activate different TLRs (that is, each TLR binds specific “molecular signatures” of different classes of microorganisms or individual features present on diverse commensals or pathogens) (fig 2 ▶). TLR2, for example, recognises bacterial lipopeptides and lipoteichoic acid which are found abundantly in cell walls of Gram positive bacteria.23 TLR2 may cooperate with TLR6 and TLR1, suggesting an essential mechanism for diversifying the repertoire of TLR mediated responses.24 RNA from double stranded and “sense” single stranded viruses activates TLR3,25,26 whereas RNA from “antisense” single stranded viruses activates TLR7 and TLR8.27,28 TLR4 is the major receptor for lipopolysaccharide (LPS) activation29 which may require the presence of accessory proteins, such as MD-2, CD14 and LPS binding protein. Flagellin and flagellated bacteria have been identified as specific ligands for TLR5.30 Unmethylated CpgDNA found in prokaryotic genomes and DNA viruses modulates TLR931 and TLR11 is activated by uropathogenic bacteria,32 but the specific ligand has yet to be determined. Importantly, species specific differences in distinct TLRx-ligand recognition appear to exist.33
Figure 2.

TLRx ligand diversity. Different pathogen associated molecular patterns (PAMPs) selectively activate different Toll-like receptors (TLRs) (that is, each TLR binds specific “molecular signatures” of different classes of microorganisms or individual features present on diverse commensals or pathogens). A short list of the main ligands is presented.
Furthermore, endogenous mediators may regulate various TLRs, such as heat shock proteins34 or fibronectin35 but it is controversial as to whether such bona fide ligands may directly activate TLRs under physiological conditions, or rather potential contaminants in the preparations.36,37 Nevertheless, TLRs may indeed be activated by endogenous ligands under pathophysiological conditions, such as “self”-DNA complexes. CpG sequences in “self”-DNA are an important potential trigger for autoantibody secretion in systemic autoimmune disorders. In rheumatoid arthritis, autoantibodies may complex to chromatin, leading to exaggerated B cell activation via TLR9.38 In systemic lupus erythematosus, DNA containing immune complexes within lupus serum stimulate plasmacytoid dendritic cells to produce cytokines and chemokines via TLR9 and CD32.39 In primary biliary cirrhosis, CpG DNA induced IgM production by activated B cells via TLR940 may drive IgG autoantibody responses and complex formation in a similarly relentless autodysregulatory loop. Thus impaired “self” versus “non-self” discrimination by TLRs in autoimmune disease (including IBD) may lead to self directed immune responses contributing to exaggerated production of proinflammatory cytokines and subsequent tissue damage.
Some reports suggest that TLR4 ligand binding might trigger assembly of a multi-receptor complex in which several components apart from the central ligand binding receptor contribute to LPS signalling. It is widely accepted that TLR4 forms a functional LPS recognition complex together with MD-2 and CD14 (see Gangloff and Gay 41 for review) but whether several additional molecules (for example, an LPS binding complex of Hsp70-Hsp90-GDF5-CXCR442 or CD5543) which reside in lipid rafts next to TLR4 play a direct or, if at all, an auxiliary role in LPS binding and downstream signalling is not yet clear. Similarly, it has been proposed that CD36 may act as a facilitator or co-receptor to TLR2/6 for selective di-acylglyceride recognition.44 Further studies aimed at defining more complete models of functional interactions between these (and possibly other) molecules and the TLR4/MD-2/CD14 (or TLR1/2/6) cores, respectively, will hopefully soon elucidate potential fine tuning mechanisms of ligand recognition by such cooperative or competitive multi-receptor complexes.
Subcellular distribution
Subcellular compartmentalisation of TLRs appears to be a critical determinant of immune responsiveness. Thus based on the optimal site of bacterial ligand interaction, TLRs are strategically localised on the cell surface as well as in different subcellular compartments. Dynamic redistribution may be regulated by a state of differentiation as well as ligand exposure. TLR2 and TLR4 are mainly expressed at the apical pole of differentiated IEC in vitro, thus well positioned to monitor the lumenal frontline of bacterial components whereas both receptors are present mostly in cytoplasmic compartments in non-differentiated IEC.4,22 There are several reports suggesting that LPS-TLR4 redistributes between plasma membrane and endosomal structures which have been identified as part of the Golgi apparatus.22,45–47 Yet the functional consequence of recycling and Golgi recruitment of TLR4 on LPS recognition and signalling seems to be ambiguous,48,49 possibly reflecting cell type or species specific differences. TLR9 is retained in the endoplasmic reticulum50 but readily cycles to sites of CpG DNA after cellular uptake.51 In contrast, TLR5 is stably expressed at the basolateral pole of intestinal epithelia in vitro,5 the major scene of action of Salmonella translocated flagellin but in vivo, it seems that TLR5 is expressed on both poles of IEC.4,52
Signalling
Individual TLRs differentially activate distinct signalling events via diverse cofactors and adaptor proteins mediating specific immune responses. To date, at least five different adaptor proteins have been identified in humans: MyD88, Mal/TIRAP, TRIF/TICAM-1, TRAM/Tirp/TICAM-2, and SARM (for review see O’Neill and colleagues53). The first identified so-called “classical” pathway mediated by the conserved TIR cytoplasmic domain in TLRs involves recruitment of the adaptor molecule MyD88, activation of the serine/threonine kinases of the interleukin 1 receptor associated kinase (IRAK) family, subsequently leading to degradation of inhibitor κB (IκB) and translocation of nuclear factor κB (NFκB) to the nucleus (fig 3 ▶). Downstream, different signalling modules and partially interacting complexes result in activation of several transcription factors, including NFκB, AP-1, Elk-1, CREB, STATs, and the subsequent transcriptional activation of genes encoding pro- and anti-inflammatory cytokines and chemokines as well as induction of costimulatory molecules. All of these various downstream effects are critically involved in the control of pathogen elimination, commensal homeostasis, and linkage to the adaptive immunity. The mechanisms of agonist recognition and engagement with cell signalling may show considerable variation between TLRs and for each TLR perhaps also show variations between cell types and organ origin. Thus analysis of cytokine gene expression to distinct ligands in macrophages has identified important differences in biological responses induced by individual TLRs. Signalling through different TLRs can result in considerable qualitative differences in TH dependent immune responses by differential modulation of MAPKs and the transcription factor c-FOS.54 However, the precise molecular mechanisms that differentially influence cell type dependent outcome of TLRx specific signalling effects are not yet fully understood.
Figure 3.

TLRx/NOD2 signalling pathways. Toll-like receptor (TLR) signalling is mediated by a complexity of various selective pathways. Rip2 is a direct downstream signal transducer of both TLRx and nucleotide binding oligomerisation domain (NOD2), thus possibly allowing regulatory cross talk between these two distinct pathways. IFN-γ, interferon γ; IRAK, interleukin 1 receptor associated kinase; IL, interleukin; TNF-α, tumour necrosis factor α; NFκB, nuclear factor κB.
NODs
Structure
The NOD (nucleotide binding oligomerisation domain) family (also called the CATERPILLAR family) comprises at present more than 20 different mammalian NOD-LRR proteins which mostly contain three distinct functional domains (fig 1B ▶): a carboxy terminal ligand recognition (LRR) domain, a centrally located nucleotide binding domain (NBD), and a structurally variable amino terminal effector binding domain which consists of protein-protein interaction domains, such as caspase recruitment domains (CARDs) or pyrin domains (reviewed by Inohara and Nunez55 and Chamaillard and colleagues56). Recent research has mostly focused on two cytosolic receptors of this family, NOD1 (also designated CARD4) and NOD2 (CARD15), which both play a major role in intestinal regulation of proinflammatory signalling through NFκB in response to distinct bacterial ligands. NOD2 shares significant homology with NOD1, but contains two, instead of one, CARD domains at its N terminus.
Toll-like receptors (TLRs).
11 mammalian, type I transmembrane receptors with divergent LRR-extracellular domain, homologous TIR.
Each TLR binds distinct “molecular signatures” present on diverse commensals/pathogens.
NODs.
20 mammalian intracellular proteins with C terminal LRR, central NBD, and N terminal CARD(s).
PGN derived ligands: NOD2—muramyl dipeptide; NOD1—iE-DAP
Both.
Constitutively or inducibly expressed by many different cells throughout the gastrointestinal tract.
Downstream activation of pro/anti-inflammatory cytokine secretion and/or apoptotic cascades.
Expression pattern
NOD2 has been shown to be constitutively or inducibly expressed in monocytes, macrophages, T and B cells, dendritic cells,57 as well as IEC,58–61 including Paneth cells.62,63 NOD1 is ubiquitously expressed in many tissues and cells.
Ligand specificity
A specific motif of peptidoglycan (PGN), muramyl dipeptide (MDP), has been identified as (so far) the sole ligand of NOD2,64,65 whereas NOD1 senses a Gram negative PGN derivative, y-D-glutamyl-meso-diamino-pimelic acid (iE-DAP).66–68 Based on these studies, it has become evident that neither NOD1 nor NOD2 sense LPS directly but rather detected contaminations of these PGN motifs within non-purified LPS preparations in earlier studies.69,70 Initially it was also believed that PGN is co-recognised, independently of its MDP or iE-DAP components, by TLR2. But TLR2 mediated cell activation induced by commercially available Staphylococcus aureus PGN preparations results from significant impurities.71
It remains to be determined whether MDP or iE-DAP directly bind to NODs through LRRs or rather through—as yet unidentified—interim co-mediators. Another essential question is how muropeptides from non-invasive bacteria gain entry into the cytosol and thus access to intracellular NODs. A recent study provided the first answer by demonstrating that non-invasive Helicobacter pylori delivers PGN to intracellular NOD1 in epithelial cells only in the presence of a functional cag pathogenicity island encoding a bacterial type IV secretion system.72 In addition, enzymes present in the lumen or produced from phagocytic cells in the lamina propria may possibly digest bacterial cell wall PGN, resulting in release and subsequent cellular uptake of muropeptides.
Subcellular distribution
NOD2 protein has been detected throughout the cytoplasm of IEC in vivo59 but the exact subcellular organelle localisation of NOD1/NOD2 as well as the place of interaction with PGN fragments have not yet been identified.
Signalling
Despite enormous research developments and advances in this field during the last few years, current knowledge and understanding regarding detailed NOD1/2 signalling pathways is still limited (fig 3 ▶). On ligand stimulation, both NOD1 and NOD2 enter into CARD-CARD interactions with the serine-threonine kinase Rip2/RICK/CARDIAK which leads to NFκB activation73 and augmentation of caspase induced apoptotic cascades. An intermediate region that is located between CARD and the kinase domain of Rip2 interacts with IKKγ (also called NEMO), thereby linking NOD1 and NOD2 to the regulatory subunit of the IKK complex which initiates its ubiquitinylation.74,75 Functional consequences deriving from NOD2 (and mutant variants) signalling events are discussed below.
PHYSIOLOGICAL IMPACT OF BACTERIAL-MUCOSAL INTERACTIONS: BENEFICIAL EFFECTS FOR THE HOST—LINK TO MUCOSAL HOMEOSTASIS
Tolerance
In the intestine, tolerance is an essential mucosal defence mechanism maintaining hyporesponsiveness to harmless lumenal commensals and their products. Exaggerated inflammatory responses in the absence of pathogenic bacteria would be otherwise deleterious. Several molecular immune mechanisms that ensure tolerance via TLRs in IEC have recently been described (see detailed review by Cario and Podolsky76): decreased surface receptor expression which limits frontline recognition,4,19 high expression levels of the downstream signalling suppressor Tollip which inhibits IRAK activation,11 ligand induced activation of peroxisome proliferator activated receptor γ (PPARγ) which uncouples NFκB dependent target genes in a negative feedback loop,77,78 and external regulators which may suppress TLR mediated signalling pathways. Additional tolerising mediators negatively modulating immune responses by interference with the TLR signalling complex have recently been identified in other cell systems, such as SIGIRR (single immunoglobulin interleukin 1R related molecule; also known as TIR8),79 TRIAD3A,80 the zinc finger protein A20,81 and ST2.82 Interestingly, TIR8 deficient mice are more susceptible to developing intestinal inflammation, suggesting a crucial role for TIR8 in tuning mucosal tolerance towards commensals.83 The ubiquitin ligase TRIAD3A directly enhances proteolytic degradation of TLR4 and TLR9.80 A20 also downregulates the TLR4 pathway by deubiquitination of TRAF6 and blocking NFκB activation84 as well as the TLR3 pathway by inhibition of interferon (IFN)-β gene expression via TRIF.85
Commensal bacteria may assist the host in maintaining mucosal homeostasis by suppressing inflammatory responses and inhibiting specific intracellular signal transduction pathways,86 potentially directly via TLR4 through elevation of PPARγ expression, uncoupling NFκB dependent target genes in a negative feedback loop77 which may lead to attenuation of colonic inflammation.78 Administration of CpG-DNA ameliorates the severity of dextran sodium sulphate (DSS) induced colitis87,88 via TLR989 and limits cytokine derived intestinal epithelial proinflammatory immune responses.90
Commensals may also play a key role in preventing allergic sensitisation via TLR activation (reviewed by Prioult and Nagler-Anderson 91). TLR4 dependent signals by the intestinal microflora protect the host by inhibiting allergic responses to food antigens, while TLR4 mutant/knockout mice are highly susceptible to develop food allergy, which correlates with high levels of TH2 cytokines, interleukin (IL)-4 and IL-13.92 Thus LPS stimulation may prevent allergen induced TH2-type inflammation by upregulation of TH1 responses via TLR4 in regulatory T cells.93
Intestinal mucosal “intolerance” (that is, exaggerated immune responsiveness towards commensals) may occur as a consequence of endogenously or exogenously induced disturbance of any of the TLR dependent signalling mechanisms of tolerance described above. Further mechanistic understanding of such host beneficial TLR/NOD signalling mechanisms of commensal mediated suppression of intestinal inflammation and how imbalance in such signalling events may lead to intestinal disease could potentially provide an efficient immunoadjuvant therapeutic approach in IBD (and atopic diseases).
Mucosal barrier protection
Commensals are able to not only suppress but also actively induce expression of host genes that participate in important physiological functions, including cell differentiation and maturation.94 In order to maintain mucosal homeostasis against tissue damage, commensal induced host modulatory effects seem to require functional TLRx and NOD.95,96 “Healthy” gut conditions appear to demand constant exposure of the intestinal surface to commensal derived TLRx ligands and basal state of activation of downstream signalling pathways, thus ensuring rapid restitution and limited inflammatory responses.95 Deficient TLRx or NOD signalling may imbalance commensal dependent mucosal homeostasis, facilitating injury and leading to disease. Emerging evidence exists that commensals directly assist the host to strengthen intestinal epithelial barrier resistance.94,97,98 Our group has recently described an important role for commensal induced TLR2 signalling in enhancement of intestinal epithelial barrier function, which correlated with distinct tight junction associated morphological changes.99 However, our results also suggested that impaired function of TLR2 itself did not lead to increased intestinal epithelial permeability. In this context, further studies are needed to investigate whether lack of host protective TLR2 (or any other TLRx) ligands in the lumen that may be present in the resident microflora could lead to loss of barrier protection, facilitating invasion of pathogenic bacteria in disease.
Bacterial clearance and antimicrobial activity
NOD2 has been found to exert antibacterial activity in intestinal epithelial cells limiting survival of enteric bacteria after invasion. Bacterial clearance of Salmonella typhimurium is strongly accelerated in IEC expressing a functional NOD2 protein, whereas L1007fsinsC mutant expressing IEC are virtually unable to clear the pathogen in vitro.61 NOD2, as well as RIP2 (as downstream component of the NOD2 signalling pathway) knockout mice, exhibit a profoundly decreased ability to clear intracellular Listeria monocytogenes,73,96 inducing persistent immune activation by combined loss of antibacterial activity, dysregulation of cytokine production, and imbalance of T cell activation. Although the NOD2 induced gene targets that lead to elimination of invasive intracellular bacteria are not yet identified in detail, recent reports suggest that TLR100,101 as well as NOD96,102 signalling pathways may critically be involved in commensal induced antimicrobial peptide production, such as defensins. These natural antibiotic peptides represent an important mechanism of host defence in innate immunity by efficiently killing phagocytosed microbes, thus helping to prevent pathogenic bacteria from crossing the mucosal barrier (reviewed by Ganz103). Interestingly, β-defensin 2 may directly act as an endogenous ligand for TLR4,104 thus possibly exponentiating bactericidal activity in a reverse autocrine loop.
However, the full spectrum of host beneficial signalling pathways activated by commensal derived TLR/NOD ligands which may balance responsiveness and survival and confer integrity of the healthy intestinal mucosa (fig 4 ▶) remains to be identified in depth.
Figure 4.

TLRx/NOD2 physiology and pathophysiology. In the healthy gut, both Toll-like receptors (TLRx) and nucleotide binding oligomerisation domain 2 (NOD2) are majorly involved in host defence and tissue repair responses, thus maintaining mucosal as well as commensal homeostasis. TLRx signalling protects the intestinal epithelial barrier, confers tolerance, and promotes healing. NOD2 exerts antimicrobial activity through defensin production and prevents intracellular bacterial invasion. When commensal and/or mucosal homeostasis are impaired due to genetic and/or environmental triggers, disease may develop: bacterial dysrecognition and intolerance through aberrant TLR and/or NOD signalling stimulates exaggerated proinflammatory responses leading to chronic inflammation via cytokine and chemokine production. TLR dysfunction induces tissue damage and barrier destruction by loss of commensal mediated colonic epithelial progenitor responses. NOD2 dysfunction leads to defective sensing of microbial threats and loss of bactericidal responses, thus allowing bacterial invasion into the host.
Physiological effects of TLRs and NODs in the healthy gastrointestinal tract.
Tolerance (that is, maintaining hyporesponsiveness to harmless lumenal commensals).
Inhibition of allergic responses to food antigens.
Protection of intestinal epithelial barrier function.
Bacterial clearance by induction of antimicrobial peptide production.
▸Maintenance of commensal and mucosal homeostasis.
PATHOPHYSIOLOGICAL IMPACT OF BACTERIAL-MUCOSAL INTERACTIONS: GENETIC ABERRATIONS—LINK TO MUCOSAL DISEASE
Toll-like receptor polymorphisms associated with IBD
Chronic recurrent intestinal inflammation in IBD may result from undue stimulation of the mucosal immune system by the resident microflora. “Healthy” intestinal mucosa expresses low concentrations of TLR2 or TLR4 protein in vivo. However, TLR4 expression is significantly increased in IEC and lamina propria mononuclear cells throughout the lower gastrointestinal tract in association with IBD4 and murine colitis.9,14 TLR4 upregulation could also result from ligands other than lumenal LPS. T cell derived cytokines, such as IFN-γ and tumour necrosis factor α (TNF-α), which play significant pathophysiological roles in triggering IBD, have been found to upregulate intestinal epithelial TLR4 expression in vitro.7,10 It remains to be shown whether upregulated TLR4 confers functional hyperresponsiveness of the intestinal epithelium to LPS or rather reflects a loss of response.
The TLR4 gene is localised on chromosome 9 (q32–33),105 a genomic region in which a CD susceptibility gene has been implicated.106 In active IBD, variant alleles in the TLR4 gene could induce functional dysregulation of the LPS receptor. “Gain of function” mutations could functionally exhibit proinflammatory effects in response to physiological concentrations of LPS. Two common mutations in the human TLR4 gene, D299G and T399I, have been observed to occur at a general frequency of between 6% and 10% in Caucasian populations.107 D299G polymorphism has been associated with CD as well as UC in a Belgian population,108 but no association was found in Scottish109 or German110 populations. Increased susceptibility to IBD has been associated with coexistence of TLR4 and/or CD14 and NOD2 mutated alleles in a Greek population.111
Although the D299G mutation (but not the T399I mutation) has been shown to interrupt TLR4 mediated LPS signalling in vitro,112 the functional phenotypic consequence remains unresolved in IBD. Highly variable TLR4 gene mutations have been identified in various mice strains which exhibit a broad distribution of different phenotypic responses to LPS, ranging from hyper- to hyposensitivity,113 suggesting that additional gene interactions of the diverse strain backgrounds were involved. TLR4 knockout mice exhibit diminished TH1 driven immune responses and are therefore highly resistant to develop chronic nematode infections of the gut.114 Spontaneous colitis occurring in STAT3 knockout mice does not develop when these mice are crossed with TLR4 knockout mice, suggesting that aberrant TLR4 signalling in response to the indigenous intestinal flora contributes to the development of intestinal inflammation through the TH1 pathway.115 The role of TLR4 in TH1 dominant TNBS colitis has not yet been examined. These studies so far suggest that commensal mediated TLR4 signalling of mucosal T cells can be detrimental, leading to some forms of murine mucosal inflammation associated with excess TH1 responses (fig 5 ▶). Of note, TH1 type shifts appear to have a significant pathogenetic role in CD. However, other data from a different murine colitis model also support the fact that TLR4 signalling may exert cytoprotective characteristics, at least against chemically induced tissue injury in the intestinal mucosa. TLR4 mutant (C3H/HeJBir) and TLR4 deficient mice show increased susceptibility to DSS colitis in comparison with wild-type mice,95,116 suggesting that commensal mediated protection of the intestinal epithelial barrier (together with other mucosal cells) might be significantly impaired in TLR4 dysfunction after toxic DSS damage. Appropriate regeneration and restitution following epithelial wounding with DSS may require commensal mediated amplification of colonic epithelial progenitor responses which are specifically impaired in mice lacking MyD88,117 downstream of TLR4.
Figure 5.

Current concept of how TLRx/NOD2 dysfunctions may contribute to the pathophysiology of Crohn’s disease. Toll-like receptor (TLRs) and nucleotide binding oligomerisation domains (NODs) are present in the intestinal epithelium, the frontline of the mucosal immune system. “Gain of dysfunction” receptor variants may allow commensals to cross the intestinal epithelial barrier and gain access to the antigen presenting cells (APC) of the underlying mucosa. Subsequent commensal mediated stimulation of APC signalling may induce nuclear factor κB (NFκB) dysregulation leading to exaggerated TH1 responses and perpetuation of apoptosis.
“Loss of function” TLR4 mutations diminish sensitivity to LPS in C3H/HeJ mice29 but paradoxically predispose to Gram negative infections, such as live Salmonella typhimurium.118 Thus D299G mutation could confer hypersensitivity to Escherichia coli or any other Gram negative commensal or pathogenic ligand causing unwanted inflammation in IBD. Flagellin, the structural component of bacterial flagella, is secreted by several commensal and pathogenic bacteria, including Salmonella typhimurium, and induces intestinal epithelial chemokine secretion via TLR5 which will, in turn, initiate dendritic cell migration and recruitment to the mucosal site of inflammation.5 Interestingly, dominant antigens in sera from colitic C3H/HeJBir (TLR4 mutant) mice seem to be flagellins.119 Similar hyperreactivity to flagellins was also observed in sera from patients with CD.119,120 Although it is unclear whether there was any significant correlation with TLR4 (or NOD2) mutations in these CD patients, one may speculate that TLR4 mutations (similar to NOD2) could lead to impaired bacterial clearance and thus greater presence of bacterial antigens in the lumen, including flagellins. It is also possible that TLR4 mutation may induce genetic disequilibrium of TLR5 (or other TLRx) leading to unwanted inflammation through flagellin (or other TLRx specific ligand) dysrecognition.
Results from a preliminary report suggest that CD is also associated with TLR9 promoter polymorphisms in a single German cohort.121 Any association between IBD and TLR2 R753Q polymorphism which has recently been identified in septic patients has not yet been investigated. Although MyD88 deficient mice exhibit increased susceptibility to DSS induced colitis and tissue damage,95,122 mutations within TLR efferent signalling proteins have not yet been described for IBD (or other intestinal diseases).
NOD2—a major susceptibility gene for Crohn’s disease
In 2001, NOD2 was mapped via linkage disequilibrium to chromosome 16q12 as an excellent candidate gene for CD (IBD1). Consequently, certain genetic variations have been associated with increased susceptibility to some types of CD.123,124 Three major variants of the LRR region (within or nearby) include one frameshift mutation (L1007fsinsC) and two missense mutations (R702W and G908R), suggesting that a defect in bacterial recognition may be associated with CD. Initial studies demonstrated that up to 8% of Caucasian CD patients possessed at least one allele of the most common mutation, L1007fsinsC, causing a truncated NOD2 protein lacking the last 33 amino acids. In CD, NOD2 expression in Paneth cells is increased, possibly secondarily induced by proinflammatory cytokines.60,61,125
TLR4 alteration associated with IBD.
Upregulation of mucosal TLR4 expression in active human IBD.
Association of TLR4D299G-polymorphism with IBD in selective populations.
Murine TLR4 “gain of function”: spontaneous intestinal inflammation with TH1 excess.
Murine TLR4 “loss of function”: increased susceptibility to DSS colitis and Gram infections.
Previous studies have started to reveal the molecular mechanisms involved by which NOD2 may influence innate immune responses in the intestinal mucosa. It seems that different NOD2 mutations may span a spectrum of diverse phenotypes, ranging from complete “loss of function” to maximal “gain of function”. NOD2 mutations within NBD lead to constitutive ligand independent NFκB activation, causing a chronic systemic inflammatory disorder known as “Blau syndrome”.55 Conversely, it has been suggested that CD associated NOD2 mutants which are predominantly found in the microbial ligand dependent LRR domain rather reflect “loss of function” phenotypes. Several in vitro transfection studies showed that human CD associated NOD2 mutants significantly abolish NFκB activation in response to MDP.64,65,126 However, paradoxically, macrophages within the intestinal lamina propria of CD patients overproduce NFκB targets, including exaggerated production of pro-inflammatory cytokines, such as TNF-α and IL-1β (reviewed by Podolsky127). Accordingly, a recent in vivo study now demonstrates that MDP stimulated macrophages isolated from mice generated with a murine NOD22932iC variant, homologous to the human NOD23020insC ( = L1007fsinsC) variant, exhibit enhanced NFκB activation, increased apoptosis, and elevated IL-1β secretion,128 possibly implying an important mechanism of how dysfunctional NOD2 may trigger intestinal inflammation in some types of CD (fig 5 ▶). Thus this murine “gain of function” NOD2 frameshift mutation in the LRR region may imbalance functions of both terminal parts of the whole protein: bacterial dysrecognition through the impaired LRR domain, ligand independent NFκB activation, as well as uncontrolled apoptosis and subsequent induction of IL-1β processing and release through the hyperactive CARD domains.
In another in vivo study which was simultaneously published by a different group, it was shown that mice lacking full length NOD2 protein (NOD2 knockout) are more susceptible to oral infection with the bacterial pathogen Listeria monocytogenes.96 The NOD2 gene product is most abundant in ileal Paneth cells62,63 which express a diverse population of microbicidal defensins restricting colonisation or invasion of small intestinal epithelium by bacteria.129 Stimulation with the NOD2 ligand MDP elicits cryptidin secretion from murine NOD2 expressing Paneth cells.130 Production of a subgroup of mucosal antimicrobial peptides, known as cryptidins, is significantly diminished in NOD2 knockout animals,96 potentially supporting the recently proposed concept of a “defensin deficiency” defect in human IBD.131 Expression of defensin related cryptidin 4, which is mostly secreted from Paneth cells in the distal small intestine,132 is particularly decreased in NOD2 knockout mice96 whereas ileal expression of two human α-defensins (HD-5 and HD-6) is significantly diminished in NOD2 mutant CD patients.102 Interestingly, human mutations in the NOD2 gene are strongly associated with distinct phenotypic expressions of ileal disease (corresponding to the location of Paneth cells), mainly involving fibrostenosing complications,133–136 but not formation of epitheloid granulomas.137 However, in heterogeneous CD, it is likely that Paneth cell dysfunction is only one pathophysiological mechanism which is specifically associated with a certain phenotypical subtype of inflammatory disease with severe hyperproliferative changes in the lower ileum in response to, yet unidentified, pathogenic or commensal organisms.
NOD2 mutations associated with some forms of Crohn’s disease or colitis.
Upregulation of NOD2 expression in Paneth cells in active CD.
Worldwide genotypic and phenotypic heterogeneity (major mutations: L1007fsinsC, R702W, G908R).
CD associated mutant transfectants abolish NFκB activation in response to MDP in vitro.
Murine NOD22932iC: “gain of function” (induction of NFκB and IL-1β, uncontrolled apoptosis).
Murine NOD2−/−: “loss of function” (decreased bacterial clearance due to lack of cryptidin production).
It is important to note that neither NOD2 knockout nor NOD2 frameshift mutant mice develop intestinal inflammation spontaneously.57,96,128 However, in contrast with NOD22932iC mutant mice, NOD2 knockout mice do not demonstrate significantly enhanced susceptibility to colitis in the DSS induced model, implying possibly variable mechanistic phenotypes of innate host defence. Genotypic heterogeneity at the single NOD2 locus could not only lead to intraspecies but also interspecies variability of phenotypes. Thus direct proof is needed of whether the described signalling targets and effects of murine NOD2 dysfunctions indeed represent phenotypic consequences of NOD2L1007fsinsC (and other human NOD2 mutants (in association with or without TLRx mutations)) which directly contribute to the complex pathophysiology of IBD in humans.
TLRx/NOD2 synergy?
From a physiological point of view, it is conceivable that TLR and NOD pathways may cooperate in (positive and/or negative) regulatory feedback loops to modulate immunological responses. Synergistic effects between different sensing events could be advantageous for the host by exhibiting more controlled and immediate innate immune responses to potential threats. On the other hand, non-synergistic states of such interactions could lead to disease by uncontrolled actions of the innate immune system. Ligand dysrecognition and subsequent disequilibrium of signalling events could thus induce imbalance of pro- versus antiapoptosis and disturbance of bacterial clearance in the intestinal mucosa. TAK-1138,139 and Rip-273,140 have previously been identified as common regulatory checkpoints of NOD2 and TLRx signalling pathways (fig 3 ▶) which could combine multiple signalling pathways (including TNFR/IL1R/IL18R) of both the innate and adaptive immune systems. In addition, negative cross talk between TLR2 and NOD2 has recently been proposed. Stimulation with the NOD2 ligand MDP inhibited TLR2 driven TH1 cytokine production by tackling NFκB. Conversely, in NOD2 knockout mice, stimulation with synthetic lipopeptide was observed to result in NFκB dysregulation and subsequent imbalance of cytokine production (increase in IL-12, decrease in IL-10) via TLR2.141,142 Thus NOD2 may exhibit anti-inflammatory activities limiting NFκB activation after bacterial stimulation of the TLR2 pathway, suggesting that the hyperinflammatory mucosal response in CD may be due to lack of the inhibitory function of NOD2 on proinflammatory TLR2 signalling to bacterial ligands in the lumen. However, two other recent studies failed to observe a similar negative regulatory effect of NOD2 on TLR2.96,128 These contrasting results may imply ambiguous interrelations between these two pathways under different experimental conditions which remain to be clarified. In this context, one may also consider that PGN was used as a presumed TLR2 ligand in some of these studies but, when highly purified, PGN sensing does not seem to occur via TLR2.71 Taken together, discussion of potential TLRx-NOD2 interrelations and their functional impact remains unresolved at this point of the investigation and requires further study.
Genetic and phenotypic heterogeneity of TLR4/NOD2 carriers
Mutations of PRRs may modify detection of pathogens, leading to altered susceptibility to disease. As outlined above, certain mutations of TLR4 and NOD2 have been strongly associated with CD, implying that PRR dysfunction may be involved in the pathogenesis of some types of CD (fig 5 ▶). NOD2 mutants have been associated with fibrostenosing ileal disease in CD but at this point in the investigation it remains elusive whether NOD2 genotyping would be helpful in clinical practice to predict the severity of the disease course or treatment response.143 Compelling evidence emerges that genetic heterogeneity between ethnic populations exists for TLR4 and NOD2 variant alleles, which may reflect phenotypic heterogeneity in IBD worldwide.144,145 Interestingly, no common NOD2 variant was found in Japanese CD patients.146 Therefore, absence of a mutation in a healthy individual may not exclude development of intestinal disease later on, while on the other hand, a healthy individual may carry NOD2 variants, without ever developing intestinal disease. But protective gene-environment or gene-gene interactions at this locus remain to be identified. Therefore, recognition that the contribution of NOD2 mutations to disease susceptibility may significantly vary in different racial and ethnic populations in geographical areas has led to increased efforts towards identifying potential genetic, environmental, and immunological cofactors that may determine manifestation or resistance to disease in susceptible individuals. Phenotypic alterations of the innate immune response in different individuals may be caused by diverse mutational pressure and selectivity within imbalanced interplays of several gene combinations, but not all genes that may modulate innate immune responsiveness have yet been functionally identified. Thus future studies will need to extend “forward genetic” approaches in this field (that is, starting with the immunological difference of healthy versus diseased NOD2/TLR4-mutant carriers and then proceed from the top down to identify the causative gene complex(es)). It will be important to dissect the multi-factor causes of the detrimental shift from host-beneficial to host-threatening innate immune recognition of commensals via TLRx/NOD2 in human relevant studies in vivo.
Role of TLRx/NOD2 in gastrointestinal mucosal diseases other than IBD?
Coeliac disease is a HLA linked inflammatory disorder of the small intestine that is triggered by gluten peptides and strongly associated with circulating mucosal IgA autoantibodies to tissue transglutaminase. Twin studies led to the conclusion that coeliac disease is strongly linked to genetic factors but it does not necessarily strike both twins of an identical pair,147 suggesting that environmental factors may be involved in the onset of the disease. Commensals or pathogens could potentially provide such required cofactors for the development of this T cell mediated disease.148 A recent study provided first evidence of attachment of rod shaped (not further characterised) bacteria to the intestinal epithelium in the upper small bowel of some untreated and treated coeliac disease patients, but not in healthy controls, suggesting that bacteria may be involved in the pathogenesis of coeliac disease.149 It is possible that bacterial products may sensitise CD4+ T cells to gluten via TLRx.150 Interestingly, certain gluten fragments elicit a direct innate response, while others preferentially drive adaptive responses.151 Thus one may also speculate that dysfunctional TLRx (or other PRRs) could directly recognise dietary gluten peptides as “non-self” leading to release of IL-15 (see comment by Schuppan and colleagues152). But no study has yet clearly examined the potential role of TLRs and their specific ligands in the pathophysiology of coeliac disease.
Helicobacter pylori induces NFκB activation in gastric epithelial and monocytic cells but it is controversial as to which specific TLR(s) may be involved in this immune response—TLR2,153 TLR4,12,153–155 or TLR5,155,156 whereas others have suggested that NOD1 may act as a sensor.72
Further evaluation of potential involvement of TLRx/NODx in other pathogenic infections of the intestine will be of clinical importance.
CONCLUSIONS AND PERSPECTIVES
Recent studies have begun to define the mechanisms through which crucial PRRs may regulate intestinal innate immunity. Cooperative as well as competitive interactions may occur between different bacterial and non-bacterial ligands via TLRs and NODs or other components of the innate immune system leading to differential pro- as well as anti-inflammatory immune responses in different cell types. Both TLRx and NOD2 are significantly involved in host defence and tissue repair responses, thus crucially maintaining mucosal homeostasis. TLRx signalling protects intestinal epithelial barrier and maintains tolerance while NOD2 signalling exerts antimicrobial activity and prevents bacterial invasion. Thus both receptors collectively exhibit distinct features that ensure commensal as well as mucosal homeostasis. Imbalance of the complex interrelations between commensals and PRRs may result in tissue injury and subsequent inflammation of the intestinal mucosa (fig 4 ▶). Aberrant TLR and/or NOD signalling may stimulate diverse inflammatory responses leading to chronic intestinal inflammation with many different clinical phenotypes.
Perspectives of current and future studies.
Characterisation of the distinct phenotypic consequences of NOD2L1007fsinsC and TLR4D299G mutations, possibly resulting in specific subforms of IBD.
Identification of genetic, environmental, and immunological cofactors that determine manifestation or resistance to IBD in susceptible individuals.
Clarification of potential positive/negative TLRx-NOD2 synergy.
Investigation of PRR involvement in other gastrointestinal disorders, such as coeliac disease or enteric infections.
Evaluation of the potential value of PRRs (activation versus inactivation) as therapeutic targets.
Further studies of the physiological and pathophysiological mechanisms within this network of possible cell-cell, ligand-ligand, and PRR-PRR signalling interactions that may favour or prevent inflammatory disease could lead to promising novel approaches that may differentially exploit the TLR/NOD pathways as a means of inducing salutary immune responses for treatment of IBD. It is likely that differential therapeutic strategies will need to include agonists as well as antagonists of PRRs, taking into account differences in PRR pathophysiology at different stages of disease as well as phenotypic and genotypic heterogeneity between distinct subgroups of IBD patients. Prophylactic application of selective TLR/NOD2 ligands could enhance desired commensal mediated tissue protective processes in order to prevent disease. Once an acute inflammatory episode has broken out, some of the untoward effects of intestinal inflammation could be abrogated by blocking uncontrolled signal transduction by specific TLR/NOD2 inhibitors, thus dampening tissue destructive effects. One key element to this disease modifying approach might be to blunt, rather than entirely eliminate, the dysregulated innate responses in acute IBD. In this context, careful assessment of adverse effects will be critical when modulating such fundamental host defence pathways of innate immunity. Given the rapid and exciting advancements of research in this field over the last few years, it is reasonable to presume that more immunological evidence and concrete directions for the potential value of these PRRs as therapeutic targets in IBD (and possibly other intestinal diseases) will emerge in the near future.
Acknowledgments
Supported by grants from the Deutsche Forschungsgemeinschaft (Ca226/4-1; Ca226/5-1/SPP Innate Immunity) and the Medical Faculty, University Hospital of Essen (IFORES). I thank Drs Andrew Gewirtz, Cathy Nagler-Anderson, Dana Philpott, Dan Podolsky, and Eduard Stange for interesting comments and discussions.
Published online first 19 April 2005
Conflict of interest: None declared.
REFERENCES
- 1.Medzhitov R, Janeway CA Jr. Decoding the patterns of self and nonself by the innate immune system. Science 2002;296:298–300. [DOI] [PubMed] [Google Scholar]
- 2.Xu Y, Tao X, Shen B, et al. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature 2000;408:111–15. [DOI] [PubMed] [Google Scholar]
- 3.Cario E, Rosenberg IM, Brandwein SL, et al. Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing Toll-like receptors. J Immunol 2000;164:966–72. [DOI] [PubMed] [Google Scholar]
- 4.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun 2000;68:7010–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gewirtz AT, Navas TA, Lyons S, et al. Cutting edge: bacterial flagellin activates basolaterally expressed tlr5 to induce epithelial proinflammatory gene expression. J Immunol 2001;167:1882–5. [DOI] [PubMed] [Google Scholar]
- 6.Fusunyan RD, Nanthakumar NN, Baldeon ME, et al. Evidence for an innate immune response in the immature human intestine: toll-like receptors on fetal enterocytes. Pediatr Res 2001;49:589–93. [DOI] [PubMed] [Google Scholar]
- 7.Abreu MT, Arnold ET, Thomas LS, et al. TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J Biol Chem 2002;277:20431–7. [DOI] [PubMed] [Google Scholar]
- 8.Haller D, Russo MP, Sartor RB, et al. IKK beta and phosphatidylinositol 3-kinase/Akt participate in non-pathogenic Gram-negative enteric bacteria-induced RelA phosphorylation and NF-kappa B activation in both primary and intestinal epithelial cell lines. J Biol Chem 2002;277:38168–78. [DOI] [PubMed] [Google Scholar]
- 9.Ortega-Cava CF, Ishihara S, Rumi MA, et al. Strategic compartmentalization of Toll-like receptor 4 in the mouse gut. J Immunol 2003;170:3977–85. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki M, Hisamatsu T, Podolsky DK. Gamma interferon augments the intracellular pathway for lipopolysaccharide (LPS) recognition in human intestinal epithelial cells through coordinated up-regulation of LPS uptake and expression of the intracellular Toll-like receptor 4-MD-2 complex. Infect Immun 2003;71:3503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otte J - M, Cario E, Podolsky DK. Mechanisms of cross hyporesponsiveness to Toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology 2004;126:1054–70. [DOI] [PubMed] [Google Scholar]
- 12.Ishihara S, Rumi MA, Kadowaki Y, et al. Essential role of MD-2 in TLR4-dependent signaling during Helicobacter pylori-associated gastritis. J Immunol 2004;173:1406–16. [DOI] [PubMed] [Google Scholar]
- 13.Cetin S, Ford HR, Sysko LR, et al. Endotoxin inhibits intestinal epithelial restitution through activation of Rho-GTPase and increased focal adhesions. J Biol Chem 2004;279:24592–600. [DOI] [PubMed] [Google Scholar]
- 14.Singh JC, Cruickshank SM, Newton DJ, et al. Toll-like receptor-mediated responses of primary intestinal epithelial cells during the development of colitis. Am J Physiol Gastrointest Liver Physiol 2005;288:G514–24. [DOI] [PubMed] [Google Scholar]
- 15.Smith PD, Smythies LE, Mosteller-Barnum M, et al. Intestinal macrophages lack CD14 and CD89 and consequently are down-regulated for LPS- and IgA-mediated activities. J Immunol 2001;167:2651–6. [DOI] [PubMed] [Google Scholar]
- 16.Hausmann M, Kiessling S, Mestermann S, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology 2002;122:1987–2000. [DOI] [PubMed] [Google Scholar]
- 17.Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology 2003;124:1866–78. [DOI] [PubMed] [Google Scholar]
- 18.Maaser C, Heidemann J, von Eiff C, et al. Human intestinal microvascular endothelial cells express Toll-like receptor 5: a binding partner for bacterial flagellin. J Immunol 2004;172:5056–62. [DOI] [PubMed] [Google Scholar]
- 19.Abreu MT, Vora P, Faure E, et al. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol 2001;167:1609–16. [DOI] [PubMed] [Google Scholar]
- 20.Naik S, Kelly EJ, Meijer L, et al. Absence of Toll-like receptor 4 explains endotoxin hyporesponsiveness in human intestinal epithelium. J Pediatr Gastroenterol Nutr 2001;32:449–53. [DOI] [PubMed] [Google Scholar]
- 21.Bocker U, Yezerskyy O, Feick P, et al. Responsiveness of intestinal epithelial cell lines to lipopolysaccharide is correlated with Toll-like receptor 4 but not Toll-like receptor 2 or CD14 expression. Int J Colorectal Dis 2003;18:25–32. [DOI] [PubMed] [Google Scholar]
- 22.Cario E, Brown D, McKee M, et al. Commensal-associated molecular patterns induce selective toll-like receptor-trafficking from apical membrane to cytoplasmic compartments in polarized intestinal epithelium. Am J Pathol 2002;160:165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 1999;11:443–51. [DOI] [PubMed] [Google Scholar]
- 24.Ozinsky A, Underhill DM, Fontenot JD, et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A 2000;97:13766–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alexopoulou L, Holt AC, Medzhitov R, et al. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001;413:732–8. [DOI] [PubMed] [Google Scholar]
- 26.Wang T, Town T, Alexopoulou L, et al. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 2004;10:1366–73. [DOI] [PubMed] [Google Scholar]
- 27.Lund JM, Alexopoulou L, Sato A, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A 2004;101:5598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004;303:1526–9. [DOI] [PubMed] [Google Scholar]
- 29.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998;282:2085–8. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi F, Smith KD, Ozinsky A, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001;410:1099–103. [DOI] [PubMed] [Google Scholar]
- 31.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000;408:740–5. [DOI] [PubMed] [Google Scholar]
- 32.Zhang D, Zhang G, Hayden MS, et al. A toll-like receptor that prevents infection by uropathogenic bacteria. Science 2004;303:1522–6. [DOI] [PubMed] [Google Scholar]
- 33.Grabiec A, Meng G, Fichte S, et al. Human but not murine toll-like receptor 2 discriminates between tri-palmitoylated and tri-lauroylated peptides. J Biol Chem 2004;279:48004–12. [DOI] [PubMed] [Google Scholar]
- 34.Vabulas RM, Ahmad-Nejad P, da Costa C, et al. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem 2001;276:31332–9. [DOI] [PubMed] [Google Scholar]
- 35.Okamura Y, Watari M, Jerud ES, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 2001;276:10229–33. [DOI] [PubMed] [Google Scholar]
- 36.Bausinger H, Lipsker D, Ziylan U, et al. Endotoxin-free heat-shock protein 70 fails to induce APC activation. Eur J Immunol 2002;32:3708–13. [DOI] [PubMed] [Google Scholar]
- 37.Gao B, Tsan MF. Recombinant human heat shock protein 60 does not induce the release of tumor necrosis factor alpha from murine macrophages. J Biol Chem 2003;278:22523–9. [DOI] [PubMed] [Google Scholar]
- 38.Leadbetter EA, Rifkin IR, Hohlbaum AM, et al. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 2002;416:603–7. [DOI] [PubMed] [Google Scholar]
- 39.Means TK, Latz E, Hayashi F, et al. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 2005;115:407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kikuchi K, Lian ZX, Yang GX, et al. Bacterial CpG induces hyper-IgM production in CD27+ memory B cells in primary biliary cirrhosis. Gastroenterology 2005;128:304–12. [DOI] [PubMed] [Google Scholar]
- 41.Gangloff M, Gay NJ. MD-2: the Toll ‘gatekeeper’ in endotoxin signalling. Trends Biochem Sci 2004;29:294–300. [DOI] [PubMed] [Google Scholar]
- 42.Triantafilou M, Triantafilou K. Receptor cluster formation during activation by bacterial products. J Endotoxin Res 2003;9:331–5. [DOI] [PubMed] [Google Scholar]
- 43.Heine H, El-Samalouti VT, Notzel C, et al. CD55/decay accelerating factor is part of the lipopolysaccharide-induced receptor complex. Eur J Immunol 2003;33:1399–408. [DOI] [PubMed] [Google Scholar]
- 44.Hoebe K, Georgel P, Rutschmann S, et al. CD36 is a sensor of diacylglycerides. Nature 2005;433:523–7. [DOI] [PubMed] [Google Scholar]
- 45.Thieblemont N, Wright SD. Transport of bacterial lipopolysaccharide to the golgi apparatus. J Exp Med 1999;190:523–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Visintin A, Mazzoni A, Spitzer JA, et al. Secreted MD-2 is a large polymeric protein that efficiently confers lipopolysaccharide sensitivity to Toll-like receptor 4. Proc Natl Acad Sci U S A 2001;98:12156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hornef MW, Frisan T, Vandewalle A, et al. Toll-like receptor 4 resides in the Golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J Exp Med 2002;195:559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Latz E, Visintin A, Lien E, et al. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J Biol Chem 2002;277:47834–43. [DOI] [PubMed] [Google Scholar]
- 49.Hornef MW, Normark BH, Vandewalle A, et al. Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J Exp Med 2003;198:1225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leifer CA, Kennedy MN, Mazzoni A, et al. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol 2004;173:1179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Latz E, Schoenemeyer A, Visintin A, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol 2004;5:190–8. [DOI] [PubMed] [Google Scholar]
- 52.Bambou JC, Giraud A, Menard S, et al. In vitro and ex vivo activation of the TLR5 signaling pathway in intestinal epithelial cells by a commensal Escherichia coli strain. J Biol Chem 2004;279:42984–92. [DOI] [PubMed] [Google Scholar]
- 53.O’Neill LA, Fitzgerald KA, Bowie AG. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol 2003;24:286–90. [DOI] [PubMed] [Google Scholar]
- 54.Agrawal S, Agrawal A, Doughty B, et al. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J Immunol 2003;171:4984–9. [DOI] [PubMed] [Google Scholar]
- 55.Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol 2003;3:371–82. [DOI] [PubMed] [Google Scholar]
- 56.Chamaillard M, Girardin SE, Viala J, et al. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell Microbiol 2003;5:581–92. [DOI] [PubMed] [Google Scholar]
- 57.Pauleau AL, Murray PJ. Role of nod2 in the response of macrophages to toll-like receptor agonists. Mol Cell Biol 2003;23:7531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gutierrez O, Pipaon C, Inohara N, et al. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem 2002;277:41701–5. [DOI] [PubMed] [Google Scholar]
- 59.Berrebi D, Maudinas R, Hugot JP, et al. Card15 gene overexpression in mononuclear and epithelial cells of the inflamed Crohn’s disease colon. Gut 2003;52:840–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosenstiel P, Fantini M, Brautigam K, et al. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology 2003;124:1001–9. [DOI] [PubMed] [Google Scholar]
- 61.Hisamatsu T, Suzuki M, Reinecker HC, et al. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology 2003;124:993–1000. [DOI] [PubMed] [Google Scholar]
- 62.Lala S, Ogura Y, Osborne C, et al. Crohn’s disease and the NOD2 gene: a role for paneth cells. Gastroenterology 2003;125:47–57. [DOI] [PubMed] [Google Scholar]
- 63.Ogura Y, Lala S, Xin W, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut 2003;52:1591–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem 2003;278:5509–12. [DOI] [PubMed] [Google Scholar]
- 65.Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 2003;278:8869–72. [DOI] [PubMed] [Google Scholar]
- 66.Girardin SE, Boneca IG, Carneiro LA, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 2003;300:1584–7. [DOI] [PubMed] [Google Scholar]
- 67.Chamaillard M, Hashimoto M, Horie Y, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol 2003;4:702–7. [DOI] [PubMed] [Google Scholar]
- 68.Girardin SE, Travassos LH, Herve M, et al. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem 2003;278:41702–8. [DOI] [PubMed] [Google Scholar]
- 69.Inohara N, Ogura Y, Chen FF, et al. Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J Biol Chem 2001;276:2551–4. [DOI] [PubMed] [Google Scholar]
- 70.Girardin SE, Tournebize R, Mavris M, et al. CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep 2001;2:736–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Travassos LH, Girardin SE, Philpott DJ, et al. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep 2004;5:1000–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Viala J, Chaput C, Boneca IG, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 2004;5:1166–74. [DOI] [PubMed] [Google Scholar]
- 73.Chin AI, Dempsey PW, Bruhn K, et al. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature 2002;416:190–4. [DOI] [PubMed] [Google Scholar]
- 74.Inohara N, Koseki T, Lin J, et al. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem 2000;275:27823–31. [DOI] [PubMed] [Google Scholar]
- 75.Abbott DW, Wilkins A, Asara JM, et al. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol 2004;14:2217–27. [DOI] [PubMed] [Google Scholar]
- 76.Cario E, Podolsky DK. Intestinal epithelial TOLLerance versus InTOLLerance of commensals. Mol Immunol 2005;42:887–93. [DOI] [PubMed] [Google Scholar]
- 77.Dubuquoy L, Jansson EA, Deeb S, et al. Impaired expression of peroxisome proliferator-activated receptor gamma in ulcerative colitis. Gastroenterology 2003;124:1265–76. [DOI] [PubMed] [Google Scholar]
- 78.Kelly D, Campbell JI, King TP, et al. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol 2004;5:104–12. [DOI] [PubMed] [Google Scholar]
- 79.Wald D, Qin J, Zhao Z, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol 2003;4:920–7. [DOI] [PubMed] [Google Scholar]
- 80.Chuang TH, Ulevitch RJ. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol 2004;5:495–502. [DOI] [PubMed] [Google Scholar]
- 81.Gon Y, Asai Y, Hashimoto S, et al. A20 inhibits Toll-like receptor 2 and 4-mediated interleukin-8 synthesis in airway epithelial cells. Am J Respir Cell Mol Biol 2004;31:330–6. [DOI] [PubMed] [Google Scholar]
- 82.Brint EK, Xu D, Liu H, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol 2004;5:373–9. [DOI] [PubMed] [Google Scholar]
- 83.Garlanda C, Riva F, Polentarutti N, et al. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc Natl Acad Sci U S A 2004;101:3522–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 2004;5:1052–60. [DOI] [PubMed] [Google Scholar]
- 85.Wang YY, Li L, Han KJ, et al. A20 is a potent inhibitor of TLR3- and Sendai virus-induced activation of NF-kappaB and ISRE and IFN-beta promoter. FEBS Lett 2004;576:86–90. [DOI] [PubMed] [Google Scholar]
- 86.Neish AS, Gewirtz AT, Zeng H, et al. Prokaryotic regulation of epithelial responses by inhibition of IkappaB-alpha ubiquitination. Science 2000;289:1560–3. [DOI] [PubMed] [Google Scholar]
- 87.Rachmilewitz D, Karmeli F, Takabayashi K, et al. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology 2002;122:1428–41. [DOI] [PubMed] [Google Scholar]
- 88.Obermeier F, Dunger N, Strauch UG, et al. Contrasting activity of cytosin-guanosin dinucleotide oligonucleotides in mice with experimental colitis. Clin Exp Immunol 2003;134:217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rachmilewitz D, Katakura K, Karmeli F, et al. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology 2004;126:520–8. [DOI] [PubMed] [Google Scholar]
- 90.Jijon H, Backer J, Diaz H, et al. DNA from probiotic bacteria modulates murine and human epithelial and immune function. Gastroenterology 2004;126:1358–73. [DOI] [PubMed] [Google Scholar]
- 91.Prioult G, Nagler-Anderson C. Mucosal immunity and allergic responses: lack of regulation and/or lack of microbial stimulation? Immunol Rev 2005; (in press). [DOI] [PubMed]
- 92.Bashir ME, Louie S, Shi HN, et al. Toll-like receptor 4 signaling by intestinal microbes influences susceptibility to food allergy. J Immunol 2004;172:6978–87. [DOI] [PubMed] [Google Scholar]
- 93.Tulic MK, Fiset PO, Manoukian JJ, et al. Role of toll-like receptor 4 in protection by bacterial lipopolysaccharide in the nasal mucosa of atopic children but not adults. Lancet 2004;363:1689–97. [DOI] [PubMed] [Google Scholar]
- 94.Hooper LV, Wong MH, Thelin A, et al. Molecular analysis of commensal host-microbial relationships in the intestine. Science 2001;291:881–4. [DOI] [PubMed] [Google Scholar]
- 95.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, et al. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004;118:229–41. [DOI] [PubMed] [Google Scholar]
- 96.Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005;307:731–4. [DOI] [PubMed] [Google Scholar]
- 97.Madsen K, Cornish A, Soper P, et al. Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology 2001;121:580–91. [DOI] [PubMed] [Google Scholar]
- 98.Otte JM, Podolsky DK. Functional modulation of enterocytes by gram-positive and gram-negative microorganisms. Am J Physiol Gastrointest Liver Physiol 2004;286:G613–26. [DOI] [PubMed] [Google Scholar]
- 99.Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology 2004;127:224–38. [DOI] [PubMed] [Google Scholar]
- 100.Birchler T, Seibl R, Buchner K, et al. Human Toll-like receptor 2 mediates induction of the antimicrobial peptide human beta-defensin 2 in response to bacterial lipoprotein. Eur J Immunol 2001;31:3131–7. [DOI] [PubMed] [Google Scholar]
- 101.Vora P, Youdim A, Thomas LS, et al. Beta-defensin-2 expression is regulated by TLR signaling in intestinal epithelial cells. J Immunol 2004;173:5398–405. [DOI] [PubMed] [Google Scholar]
- 102.Wehkamp J, Harder J, Weichenthal M, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 2004;53:1658–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol 2003;3:710–20. [DOI] [PubMed] [Google Scholar]
- 104.Biragyn A, Ruffini PA, Leifer CA, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 2002;298:1025–9. [DOI] [PubMed] [Google Scholar]
- 105.Rock FL, Hardiman G, Timans JC, et al. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci U S A 1998;95:588–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cho JH, Nicolae DL, Gold LH, et al. Identification of novel susceptibility loci for inflammatory bowel disease on chromosomes 1p, 3q, and 4q: evidence for epistasis between 1p and IBD1. Proc Natl Acad Sci U S A 1998;95:7502–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lorenz E, Frees KL, Schwartz DA. Determination of the TLR4 genotype using allele-specific PCR. Biotechniques 2001;31:22–4. [DOI] [PubMed] [Google Scholar]
- 108.Franchimont D, Vermeire S, El Housni H, et al. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut 2004;53:987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arnott ID, Nimmo ER, Drummond HE, et al. NOD2/CARD15, TLR4 and CD14 mutations in Scottish and Irish Crohn’s disease patients: evidence for genetic heterogeneity within Europe? Genes Immun 2004;5:417–25. [DOI] [PubMed] [Google Scholar]
- 110.Torok HP, Glas J, Tonenchi L, et al. Polymorphisms of the lipopolysaccharide-signaling complex in inflammatory bowel disease: association of a mutation in the Toll-like receptor 4 gene with ulcerative colitis. Clin Immunol 2004;112:85–91. [DOI] [PubMed] [Google Scholar]
- 111.Gazouli M, Mantzaris G, Kotsinas A, et al. Association between polymorphisms in the Toll-like receptor 4, CD14, and CARD15/NOD2 and inflammatory bowel disease in the Greek population. World J Gastroenterol 2005;11:681–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Arbour NC, Lorenz E, Schutte BC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet 2000;25:187–91. [DOI] [PubMed] [Google Scholar]
- 113.Lorenz E, Jones M, Wohlford-Lenane C, et al. Genes other than TLR4 are involved in the response to inhaled LPS. Am J Physiol Lung Cell Mol Physiol 2001;281:L1106–14. [DOI] [PubMed] [Google Scholar]
- 114.Helmby H, Grencis RK. Essential role for TLR4 and MyD88 in the development of chronic intestinal nematode infection. Eur J Immunol 2003;33:2974–9. [DOI] [PubMed] [Google Scholar]
- 115.Kobayashi M, Kweon MN, Kuwata H, et al. Toll-like receptor-dependent production of IL-12p40 causes chronic enterocolitis in myeloid cell-specific Stat3-deficient mice. J Clin Invest 2003;111:1297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Elson CO, Sartor RB, Tennyson GS, et al. Experimental models of inflammatory bowel disease. Gastroenterology 1995;109:1344–67. [DOI] [PubMed] [Google Scholar]
- 117.Pull SL, Doherty JM, Mills JC, et al. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci U S A 2005;102:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.O’Brien AD, Rosenstreich DL, Scher I, et al. Genetic control of susceptibility to Salmonella typhimurium in mice: role of the LPS gene. J Immunol 1980;124:20–4. [PubMed] [Google Scholar]
- 119.Lodes MJ, Cong Y, Elson CO, et al. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest 2004;113:1296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sitaraman SV, Klapproth JM, Moore ID, et al. Elevated flagellin-specific immunoglobulins in Crohn’s Disease. Am J Physiol Gastrointest Liver Physiol 2005;288:G403–6. [DOI] [PubMed] [Google Scholar]
- 121.Torok HP, Glas J, Tonenchi L, et al. Crohn’s disease is associated with a toll-like receptor-9 polymorphism. Gastroenterology 2004;127:365–6. [DOI] [PubMed] [Google Scholar]
- 122.Araki A, Kanai T, Ishikura T, et al. MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J Gastroenterol 2005;40:16–23. [DOI] [PubMed] [Google Scholar]
- 123.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001;411:599–603. [DOI] [PubMed] [Google Scholar]
- 124.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001;411:603–6. [DOI] [PubMed] [Google Scholar]
- 125.Hisamatsu T, Suzuki M, Podolsky DK. Interferon-gamma augments CARD4/NOD1 gene and protein expression through interferon regulatory factor-1 in intestinal epithelial cells. J Biol Chem 2003;278:32962–8. [DOI] [PubMed] [Google Scholar]
- 126.Chamaillard M, Philpott D, Girardin SE, et al. Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc Natl Acad Sci U S A 2003;100:3455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Podolsky DK. Inflammatory bowel disease. N Engl J Med 2002;347:417–29. [DOI] [PubMed] [Google Scholar]
- 128.Maeda S, Hsu LC, Liu H, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science 2005;307:734–8. [DOI] [PubMed] [Google Scholar]
- 129.Ouellette AJ, Hsieh MM, Nosek MT, et al. Mouse Paneth cell defensins: primary structures and antibacterial activities of numerous cryptdin isoforms. Infect Immun 1994;62:5040–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ayabe T, Satchell DP, Wilson CL, et al. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol 2000;1:113–18. [DOI] [PubMed] [Google Scholar]
- 131.Fellermann K, Wehkamp J, Herrlinger KR, et al. Crohn’s disease: a defensin deficiency syndrome? Eur J Gastroenterol Hepatol 2003;15:627–34. [DOI] [PubMed] [Google Scholar]
- 132.Ouellette AJ, Darmoul D, Tran D, et al. Peptide localization and gene structure of cryptdin 4, a differentially expressed mouse paneth cell alpha-defensin. Infect Immun 1999;67:6643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Abreu MT, Taylor KD, Lin YC, et al. Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn’s disease. Gastroenterology 2002;123:679–88. [DOI] [PubMed] [Google Scholar]
- 134.Hampe J, Grebe J, Nikolaus S, et al. Association of NOD2 (CARD 15) genotype with clinical course of Crohn’s disease: a cohort study. Lancet 2002;359:1661–5. [DOI] [PubMed] [Google Scholar]
- 135.Cuthbert AP, Fisher SA, Mirza MM, et al. The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology 2002;122:867–74. [DOI] [PubMed] [Google Scholar]
- 136.Lesage S, Zouali H, Cezard JP, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet 2002;70:845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pierik M, De Hertogh G, Vermeire S, et al. Epithelioid granulomas, pattern recognition receptors, and phenotypes of Crohn’s disease. Gut 2005;54:223–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen CM, Gong Y, Zhang M, et al. Reciprocal cross-talk between Nod2 and TAK1 signaling pathways. J Biol Chem 2004;279:25876–82. [DOI] [PubMed] [Google Scholar]
- 139.McDermott EP, O’Neill LA. Ras participates in the activation of p38 MAPK by interleukin-1 by associating with IRAK, IRAK2, TRAF6, and TAK-1. J Biol Chem 2002;277:7808–15. [DOI] [PubMed] [Google Scholar]
- 140.Kobayashi K, Inohara N, Hernandez LD, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 2002;416:194–9. [DOI] [PubMed] [Google Scholar]
- 141.Watanabe T, Kitani A, Murray PJ, et al. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol 2004;5:800–8. [DOI] [PubMed] [Google Scholar]
- 142.Netea MG, Kullberg BJ, de Jong DJ, et al. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn’s disease. Eur J Immunol 2004;34:2052–9. [DOI] [PubMed] [Google Scholar]
- 143.Vermeire S. NOD2/CARD15: relevance in clinical practice. Best Pract Res Clin Gastroenterol 2004;18:569–75. [DOI] [PubMed] [Google Scholar]
- 144.Arnott ID, Ho GT, Nimmo ER, et al. Toll-like receptor 4 gene in IBD: further evidence for genetic heterogeneity in Europe. Gut 2005;54:308. [PMC free article] [PubMed] [Google Scholar]
- 145.Franchimont D, Vermeire S, Deviere J, et al. Toll-like receptor 4 gene in IBD: further evidence for genetic heterogeneity in Europe. Gut 2005;54:308–16. [PMC free article] [PubMed] [Google Scholar]
- 146.Inoue N, Tamura K, Kinouchi Y, et al. Lack of common NOD2 variants in Japanese patients with Crohn’s disease. Gastroenterology 2002;123:86–91. [DOI] [PubMed] [Google Scholar]
- 147.Bardella MT, Fredella C, Prampolini L, et al. Gluten sensitivity in monozygous twins: a long-term follow-up of five pairs. Am J Gastroenterol 2000;95:1503–5. [DOI] [PubMed] [Google Scholar]
- 148.Londei M, Maiuri L. Gliadin as stimulator adaptive and innate immune responses in celiac disease. J Pediatr Gastroenterol Nutr 2004;39 (suppl 3) :S729. [DOI] [PubMed] [Google Scholar]
- 149.Forsberg G, Fahlgren A, Horstedt P, et al. Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am J Gastroenterol 2004;99:894–904. [DOI] [PubMed] [Google Scholar]
- 150.Sollid LM, Gray GM. A role for bacteria in celiac disease? Am J Gastroenterol 2004;99:905–6. [DOI] [PubMed] [Google Scholar]
- 151.Maiuri L, Ciacci C, Ricciardelli I, et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 2003;362:30–7. [DOI] [PubMed] [Google Scholar]
- 152.Schuppan D, Esslinger B, Dieterich W. Innate immunity and coeliac disease. Lancet 2003;362:3–4. [DOI] [PubMed] [Google Scholar]
- 153.Mandell L, Moran AP, Cocchiarella A, et al. Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect Immun 2004;72:6446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Maeda S, Akanuma M, Mitsuno Y, et al. Distinct mechanism of Helicobacter pylori-mediated NF-kappa B activation between gastric cancer cells and monocytic cells. J Biol Chem 2001;276:44856–64. [DOI] [PubMed] [Google Scholar]
- 155.Smith MF Jr, Mitchell A, Li G, et al. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-kappa B activation and chemokine expression by epithelial cells. J Biol Chem 2003;278:32552–60. [DOI] [PubMed] [Google Scholar]
- 156.Gewirtz AT, Yu Y, Krishna US, et al. Helicobacter pylori flagellin evades toll-like receptor 5-mediated innate immunity. J Infect Dis 2004;189:1914–20. [DOI] [PubMed] [Google Scholar]