

Abstract
Background and aims: Leptin, the product of the ob gene, has been suggested to increase the risk of colon cancer. However, we have shown that although leptin stimulates epithelial cell proliferation it reduces the development of carcinogen induced preneoplastic lesions in the rat colon. Here, we explored the effect of leptin in vitro on proliferation of human colon cancer cells, and in vivo on the growth of HT-29 xenografts in nude mice and the development of intestinal tumours in ApcMin/+ mice.
Methods: Proliferation of HT-29, LoVo, Caco2, and SW 480 cells was assessed in the absence or presence of leptin (20–500 ng/ml) by 3H-thymidine incorporation and cell count. Leptin (800 µg/kg/day) or its vehicle was delivered for four weeks to nude mice, inoculated with HT-29 cells on day 0, and for six weeks to ApcMin/+ mice.
Results: Leptin dose dependently stimulated cell DNA synthesis and growth in all cell lines. In nude mice, leptin caused a 4.3-fold increase in plasma leptin levels compared with pair fed controls. This hyperleptinaemia, despite leptin receptor expression in tumours, did not induce significant variation in tumour volume or weight. Tumour Ki-67 index was even inhibited. In leptin treated ApcMin/+ mice, a 2.4-fold increase in plasma leptin levels did not modify the number, size, or distribution of intestinal adenomas compared with pair fed controls.
Conclusions: Leptin acts as a growth factor on colon cancer cells in vitro but does not promote tumour growth in vivo in the two models tested. These findings do not support a pivotal role for hyperleptinaemia in intestinal carcinogenesis.
Keywords: hormone; carcinogenesis; animal models; cell lines; TdT, terminal deoxynucleotidyl transferase; TUNEL, terminal deoxynucleotidyl transferase mediated dUTP nick end labelling
Leptin, the product of the mouse ob gene,1 is a 167 amino acid peptide hormone involved in energy balance and regulation of food intake. It was initially found to be produced in adipocytes. Later, other sources of leptin have been described, notably the stomach.2,3 In humans, serum leptin levels are high in obese subjects and decrease with weight loss.4 Epidemiological studies have revealed that overweight raises the risk of colon adenomas5 and colorectal cancers.6,7 The effects of leptin are mediated through binding of specific cell surface receptors (Ob-R) coupled to activation of PI3 kinase and Jak/Stat signalling. Both pathways exert a critical role in the control of many cellular functions, including survival, proliferation, and differentiation.
In this context, several lines of evidence suggest that leptin may be involved in carcinogenesis. Indeed, in vitro, leptin can: (i) stimulate the proliferation of different types of cancer cell lines, (ii) induce angiogenesis through interaction with Ob-R expressed on the surface of endothelial cells,8,9 angiogenesis being essential for tumour growth, invasion, and metastasis, and (iii) increase the secretion of metalloproteinases, key enzymes for tumorous invasion.10 With regards to the digestive tract, the long isoform of the leptin receptor (Ob-Rb), which is the functional form, is expressed all along this tractus.3,11–14 This Ob-Rb expression is preserved in human colonic adenomas and carcinomas as well as in human colon cancer cell lines.11,13 The hypothesis that leptin is related to the development of digestive cancers is supported by the fact that leptin promotes the proliferation of several cell lines derived from human adenocarcinomas, such as Barrett’s and squamous oesophageal cancer cell lines,15,16 the AGS gastric cancer cell line,17 and the HT-29 colon cancer cell line.13,18 Leptin can also promote the invasiveness of human colon cancer cells in collagen gel11 and counteract sodium butyrate induced apoptosis in HT-29 cells.19
Nevertheless, in vivo, data concerning the action of leptin on intestinal epithelial cell growth are contradictory. Thus in humans, although in some reports there was no evidence of elevated leptin levels in patients with colorectal cancers,20–22 a recent study showed that the risk of colonic cancer, but not rectal cancer, increases with high serum leptin concentration.23 In mice, leptin injection stimulated13 or had no effect or even inhibited the proliferation of colonic epithelial cells.24 Recently, in rats, we confirmed the promoting effect of leptin on cell proliferation of the right, but not the left, colonic mucosa. More interestingly, in the same work, we showed that leptin significantly reduced the development in the colonic epithelium of aberrant crypt foci induced by azoxymethane, a colon carcinogen, aberrant crypts being considered preneoplastic lesions.25 This was intriguing and indicated that leptin exerts a more complex action on the gut than first suspected.
In the present study, in an effort to analyse further the relationship between leptin and intestinal cancer, we investigated the potential effect of leptin (i) in vitro, on the proliferation of HT-29 cells and three other colon cancer cell lines known to express the leptin receptor Ob-Rb,11,13 and (ii), in vivo, on the growth of HT-29 cell xenografts in nude mice, and on the progression of spontaneous intestinal tumorigenesis in ApcMin/+ mice.
MATERIAL AND METHODS
Cell lines and culture
The human colon cancer cell lines SW 480, LoVo, Caco-2, and HT-29, the latter used as controls, were routinely grown in 25 cm2 plastic flasks (Costar, Cambridge, Massachusetts, USA). Cells were maintained at 37°C in a humidified atmosphere of 5% CO2/95% air in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, New York, USA) containing 10% decomplemented fetal calf serum (FCS) (Gibco) for HT-29 and SW 480 cells, and 20% decomplemented FCS and 1% non-essential amino-acids for Caco-2 cells. LoVo cells were grown under the same conditions in Ham’s F12 medium (Gibco) containing 10% decomplemented FCS.
In vitro tritiated thymidine incorporation assay
Colon cancer cells (105 cells/well) were seeded in 12 well clusters (Costar) in medium supplemented with FCS and cultured for 24 hours to allow cell adhesion. After washing, cells were cultured for another 24 hours in serum free medium. Human recombinant leptin (R&D Systems Europe Ltd, Abingdon, UK) was then added in the same medium at a concentration of 20–500 ng/ml and cells cultured for further 48 hours. At the end of that time, 0.1 µCi of 3H-thymidine (specific activity 5 Ci/mmol; Amersham Biosciences, Saclay, France) was added to each well for two hours. Incorporated radioactivity was determined in cell pellets using a β scintillation counter. Results are expressed in dpm per well. For each leptin concentration, the experiment was performed in 3–6 wells. Two to three experiments were carried out for each cell type.
Assessment of cell growth in vitro
Cells (1×104 cells/well) were seeded in 96 well clusters and cultured for 24 hours in medium supplemented with 10% FCS for HT-29, SW 480, and LoVo cells or 20% FCS for Caco-2. Then, attached cells were washed and grown in their respective serum free culture medium in the absence or presence of leptin (20–500 ng/ml for eight days, the medium being renewed every 48 hours). Then, cells were harvested and those which excluded Trypan blue were counted with a haemacytometer. For each leptin concentration, the experiment was performed in 3–6 wells. For each cell type, 2–3 experiments were carried out.
Leptin receptor immunoprecipitation and phosphorylation assay
Sixty per cent confluent HT-29 cells were starved for 24 hours in serum free Dulbecco’s modified Eagle’s medium. Then, the medium was renewed and cells were incubated in the absence (control) or presence of 100 ng/ml of leptin for 30 seconds, two minutes, or five minutes. Thereafter, cells were harvested at 4°C. The pellet was resuspended and incubated in ice cold RIPA buffer containing a cocktail of protease inhibitors (Sigma, St Louis, Missouri, USA) and 1 mM of sodium orthovanadate and sodium fluoride. Ob-Rs were immunoprecipitated from cell lysates (200 µg) overnight at 4°C using M18 polyclonal anti-Ob-R antibody—cross reacting with human and murine leptin receptors (Santa Cruz Biotechnology, California, USA)—and protein G sepharose beads (Amersham). Immunoprecipitates were resolved by 7.5% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose sheets. Phosphorylated Ob-Rs were detected by western blot with an antiphosphotyrosine monoclonal antibody (4G10) (Upstate Biotechnologies, Lake Placid, New York, USA). Signals were visualised with the peroxidase labelled secondary antibody using enhanced chemiluminescence (ECL; Amersham).
Animals
Six week old male BALB/c nu/nu athymic mice, referred to as nude mice, were obtained from Iffa Credo (L’Arbresle, France) and housed in our animal quarters in individual cages, kept in a temperature controlled room with a 12 hour light-dark period. They were given standard mice food pellets (SAFE A 03; Scientific Animal Food, Villemoisson sur Orge, France) and water ad libitum until the beginning of the experiment, one week later.
C57BL/6J-ApcMin/+ mouse progenitors were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). Our breeding colony was established by crossing heterozygous male C57BL/6J-ApcMin/+ mice with wild-type female C57BL/6J mice. Genomic DNA was prepared from tail biopsies, using Qiagen DNAeasy kit (Qiagen SA, Courtaboeuf, France) and offspring characterised for the Min genotype by multiplex polymerase chain reaction (PCR), according to the recommendations of the Jackson Laboratory. We used the following sense (5′- GCC ATC CCT TCA CGT TAG-3′ (wild-type) and 5′-TTC TGA GAA AGA CAG AAG TTA-3′—final adenosine residue corresponds to the T to A transversion at nucleotide 2549 in C57BL/6J-ApcMin/+ mice) and antisense (5′-TTC CAC TTT GGC ATA AGG C-3′) oligonucleotides. The expected size of the PCR products was 600 bp for wild-type animals. For the C57BL/6J-ApcMin/+ mice, two additional bands of 340 bp and 1000 bp were obtained, corresponding to the amplification of the mutant allele and the heteroduplex (wild-type/mutant allele), respectively.
Animals were treated in accordance with European Community guidelines concerning the care and use of laboratory animals.
Tumorigenicity assay in nude mice
On day 0, exponentially growing HT-29 cells were harvested and resuspended at a concentration of 1×108/ml. Nude mice were anaesthetised by intraperitoneal injection of a mixture of Rompum and ketamine in NaCl. Cells (1×107) were inoculated in the right flank of mice. A miniosmotic pump (Alzet pump, model 2002; Alza Mountain View, California, USA) was implanted subcutaneously in the left flank of mice. Pumps delivered recombinant human leptin diluted in saline + 0.3% bovine serum albumin (BSA) or leptin vehicle (saline+BSA) alone (control). The dose of leptin, based on the initial concentration and specified delivery rate of pumps, was 800 µg/kg/day for two weeks. Pumps were renewed after 14 days. Food intake of leptin treated mice was monitored daily. Vehicle treated mice were pair fed, receiving the same amount of food as had been eaten by leptin treated mice the day before. Body weight was monitored twice a week in the two groups. Tumour development was followed daily by calliper measurement along two orthogonal axes, length (L) and width (W). The volume (V) of tumour was calculated by the equation for ellipsoid (V = L × W2 × π/6). At the time of killing (day 28), blood samples were collected for radioimmunoassays, and tumours were dissected out from neighbouring connective tissues and weighed. Then, they were divided into three parts and frozen in liquid nitrogen and stored at −80°C or fixed partly in Bouin’s fluid or in 4% paraformaldehyde and paraffin embedded.
Estimation of Ki-67 proliferative index in nude mice tumours
Paraffin blocks were cut into 4 µm thick sections. Cell proliferative activity was examined after immunohistochemical detection of Ki-67, a nuclear protein present in all phases of the cell cycle, except G0. We used a mouse monoclonal antibody (clone MIB-1; Dakopatt, Glostrup, Denmark) diluted 1:100. In each tumour, the total number of epithelial cell nuclei and that of Ki-67 immunostained nuclei were counted in five different representative areas, using a calibrated ocular grid at ×400 magnification. The proliferative index was estimated from a total of at least 1000 nuclei counted per tumour.
In situ labelling of apoptotic cells in nude mice tumours
Apoptotic cells were identified by a terminal deoxynucleotidyl transferase (TdT) mediated dUTP nick end labelling (TUNEL) method. After dewaxing, tissue sections were treated with proteinase K (20 μg/ml) for 10 minutes at room temperature. The TUNEL reaction was performed according to procedures provided with the ApopTag Plus Peroxydase In Situ Apoptosis Detection Kit (Serologicals Corporation, Biotech, Norcross, Georgia, USA). Peroxidase activity was revealed with diaminobenzidine substrate. A negative control was run, omitting TdT from the reaction mixture. As a positive control, sections of normal human lymph node tissues were used. Apoptotic cells were identified by their brown positively labelled nuclei (including apoptotic bodies) and counted outside the necrosis areas. The apoptotic index was estimated, using the ocular grid, from a total of at least 1000 nuclei counted per tumour.
Experimental design in ApcMin/+ mice
The study was performed on five successive couples of animals, 8–9 weeks old. For each individual experiment, control and leptin treated C57BL/6J-ApcMin/+ mice originated from the same littermate and were matched for weight and sex. Mice were equipped on day 0 with a miniosmotic pump model 2002 implanted subcutaneously and delivering murine leptin (R&D Systems) (800 µg/kg) or its vehicle (control). These pumps were renewed twice and mice killed after six weeks of treatment. Animals in the two groups were pair fed and their body weight monitored once a week. At the time of killing (day 42), blood samples and the entire small and large intestines were taken.
Examination of intestinal mucosa and adenoma scoring in ApcMin/+ mice
The small intestine was measured and immediately cut into six equal segments, numbered from the proximal duodenum to the distal ileum. The large intestine was cut into three segments from the caecum to the anal ring. The lumen of segments was filled with 10% neutral buffered formol solution. After several minutes, these segments were opened, carefully pinned flat on a paraffin wax block to examine the entire mucosa with the minimum of artefact, then fixed in formalin for two days. After fixation, they were measured in length and examined under a light microscope at 40× magnification. The number of adenomas per segment was quantified and their largest diameter measured using a calibrated ocular grid by two independent observers (TA and TL). Adenomas were classified as a function of their size (in 500 µm steps) into six classes: class 1, diameter ⩽500 µm; class 2, >500 µm up to 1 mm, and so on until class 6, diameter >2.5 mm.
Immunohistochemistry and immunoblotting of the leptin receptor Ob-R
For Ob-R immunohistochemistry, we used goat polyclonal antibodies (Santa Cruz Biotechnology) diluted 1:30 to 1:50—C-20 for nude mice tumour xenografts or M-18 for ApcMin/+ mice small intestinal and colonic adenomas—and the corresponding Vectastin ABC kit (Vector Labs, Burlingame, California, USA). C-20, raised against human leptin receptor, recognises the functional Ob-Rb isoform and M18, Ob-Rb, and Ob-Ra isoforms. No reaction was seen when the primary antiserum was omitted.
For western immunoblotting, frozen tissues were disrupted in ice cold RIPA buffer. Equal amounts of protein tumour lysates (75 µg) were separated on SDS-7.5% PAGE, and transferred to nitrocellulose membranes. They were probed with M18 antibody diluted 1:50 and with horseradish peroxidase conjugated antigoat IgGs (Santa Cruz Biotechnology) using ECL. The specificity of the signal was evaluated by peptide neutralisation of M18 antibody. The relative loading of protein tumour samples was assessed after reprobing the membrane with a monoclonal antibody directed towards α-tubulin (Sigma-Aldrich, France).
Leptin and insulin radioimmunoassays
After blood centrifugation, plasma was collected and kept at −80°C for analyses. Leptin and insulin plasma concentrations were measured using radioimmunoassay kits for multispecies leptin and rat insulin, respectively (Linco Research Inc., St Charles, Missouri, USA), the insulin assay recognising mice insulin.
Statistical analysis
All results are expressed as means (SEM). Differences in proliferation of cultured cells between control values and the different doses of leptin tested were evaluated by one way ANOVA or the non-parametric Kruskall-Wallis test for multiple comparisons, followed by the Student’s t test or the Mann-Whitney U test, respectively, if significant results were obtained. For the other variables studied, statistical comparisons between control and leptin treated groups were made with the t test or U test (unequal variances) when relevant. Relationships between variables were examined using the non-parametric Spearman’s rank correlation. The level of significance was set at p = 0.05.
RESULTS
Leptin stimulates DNA synthesis and growth of human colon cancer cells
Firstly, we checked the functional activity of the leptin receptor Ob-Rb isoform known to be present in HT-29 cells by the ability of leptin to induce its phosphorylation. Ob-Rb phosphorylation was maximal after two minutes of incubation of HT-29 cells in the presence of 100 ng/ml leptin (fig 1A ▶). Secondly, leptin added in serum free culture medium significantly stimulated DNA synthesis and proliferation in these cells with a maximal effect at 100 ng/ml (+46% and +58% compared with controls, respectively) (fig 1B ▶). A stimulating effect was also observed in the other three human cancer cell lines tested. Figure 1B ▶ shows representative experiments for each of these cell types. The leptin effect was maximal at 50 ng/ml for DNA synthesis and growth in LoVo cells (+145% and +210%, respectively), and at 500 ng/ml in Caco-2 cells (+220% and +87%, respectively). Despite Ob-Rb expression in SW 480 cells, as evidenced by reverse transcription-PCR and western blot (data not shown), the leptin effect was less clearcut in these cells and not quite significant (100% increase in cell number compared with control values at 50 ng/ml leptin; p<0.07).
Figure 1.
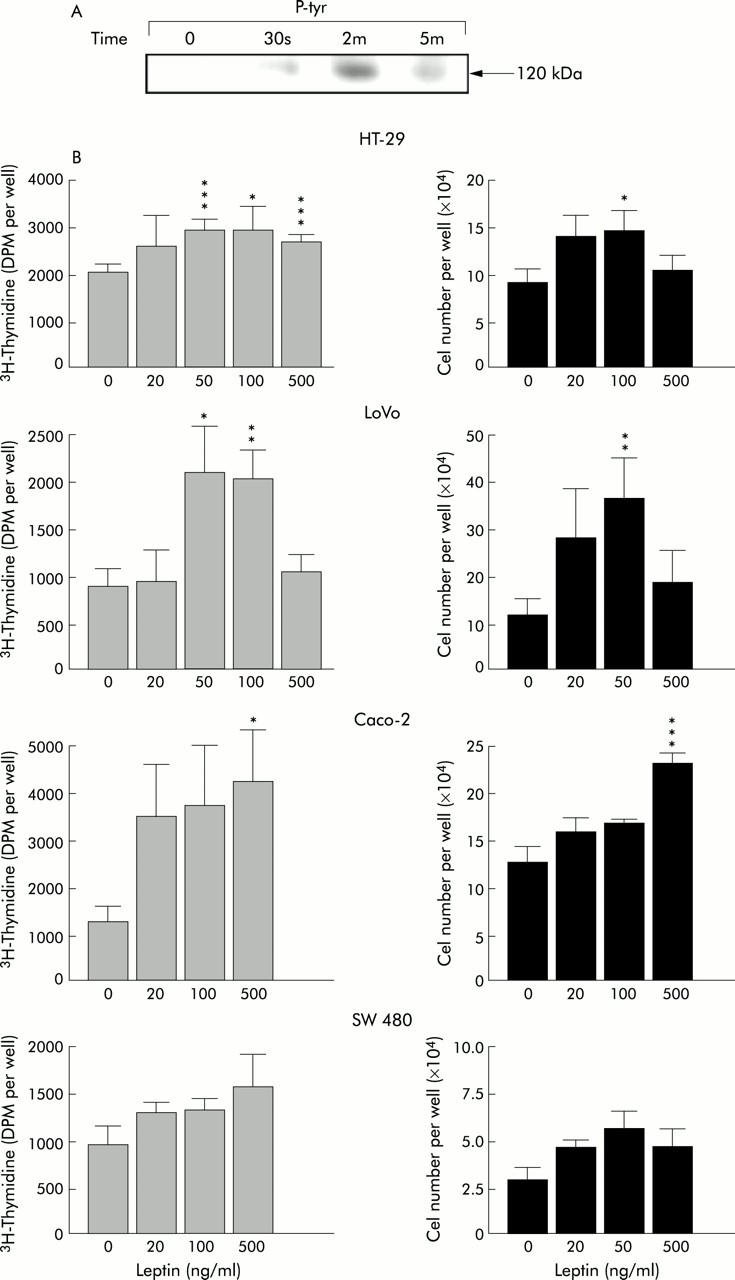
Biological effects of leptin in the human colonic HT-29, LoVo, Caco-2, and SW 480 cell lines. (A) Tyrosine phosphorylation assay of Ob-R leptin receptor in HT-29 cells in the absence or presence of 100 ng/ml of leptin. Ob-Rb was immunoprecipitated with the M18 anti-Ob-R antibody and its phosphorylation was detected by western blot with the 4G10 antiphosphotyrosine antibody (P-tyr). The functional isoform Ob-Rb was identified as a protein band of approximately 120 kDa. Its phosphorylation was maximal after two minutes of incubation with leptin. (B) DNA synthesis and cell growth of HT-29, LoVo, Caco-2, and SW 480 human colonic cancer cells cultured in serum free medium in the absence (control) or presence of 20–500 ng/ml leptin. Each histogram is representative of 2–3 experiments, except for HT-29 cells which served as controls and were studied only once. *p<0.05 ; **p<0.03 to p<0.02; ***p<0.005 to p<0.002 versus controls.
Leptin does not promote the growth of HT-29 cancer cell xenografts in nude mice
The promoting effect of leptin on in vitro growth of HT-29 cells led us to investigate whether this effect occurred in vivo in nude mice. Treatment with leptin or its vehicle (10 mice/group) began on the day of tumour cell inoculation (day 0). At the beginning of treatment, the mean weights of mice were identical in the two groups: 21.5 (0.6) g and 21.7 (0.7) g. During the experiment (28 days), leptin and vehicle treated mice were pair fed and had similar body weight curves, showing a decrease at the end of the experiment with a mean weight of 17.9 (0.8) g for leptin treated mice and 18.6 (0.7) g for vehicle treated mice. Two mice died in the course of the experiment, one control on day 15 and one leptin treated mouse on day 24.
Inoculation of HT-29 cells in nude mice resulted in the development of tumours, detectable at day 6. At that time, their volumes were significantly larger in leptin treated mice than in control mice (p = 0.02). Apart from this day, they were no significant differences from controls until the time of sacrifice, although tumour volume had a tendency to grow more quickly under leptin treatment (fig 2A ▶). Confirming the lack of effect of leptin on tumour volume, no variation was found in the weight of tumours at the end of the experiment (689 (99) mg for leptin treated mice and 663 (149) mg for vehicle treated mice) (fig 2B ▶). Autopsy of mice and histological examination of livers after fixation did not reveal any metastatic site of tumour development in either group. Histologically, HT-29 tumour xenografts were moderately differentiated adenocarcinomas which displayed very large areas of necrosis.
Figure 2.

Effect of four week continuous subcutaneous infusion of leptin on the growth of HT-29 cells xenografted in nude mice. (A) Tumour volume during the course of treatment. (B) Tumour weight on day 28, at the time of sacrifice. (C) Proliferative index. (D) Apoptotic index in epithelial tumour cells at day 28. *p = 0.02, **p<0.008 versus vehicle treated nude mice.
We verified by western immunoblots that leptin receptors remained expressed in HT-29 tumour cells (fig 3A ▶). Immunohistochemistry confirmed the presence of these receptors in tumours in the two groups of mice (fig 3B ▶). Then we examined the state of proliferation of epithelial tumour cells. The mean Ki-67 proliferative index was significantly lower in leptin treated mice (35.5 (5.5) than in controls 55.1 (3.4) (p<0.008) (fig 2C ▶, fig 3C ▶) whereas the mean apoptotic index was unchanged (1.90 (0.16) in leptin treated mice v 1.77 (0.15) in controls) (fig 2D ▶). Plasma leptin levels measured at the end of the experiment were however 4.3-fold higher in leptin treated than in vehicle treated mice (9.1 (2.4) v 2.1 (0.4) ng/ml, respectively; p<0.0003) (fig 4A ▶). We established that this hyperleptinaemia was biologically functional because plasma insulin levels were 4.1-fold decreased in hyperleptinaemic mice compared with controls (0.18 (0.04) v 0.74 (0.20) ng/ml, respectively; p = 0.008) (fig 4B ▶).
Figure 3.
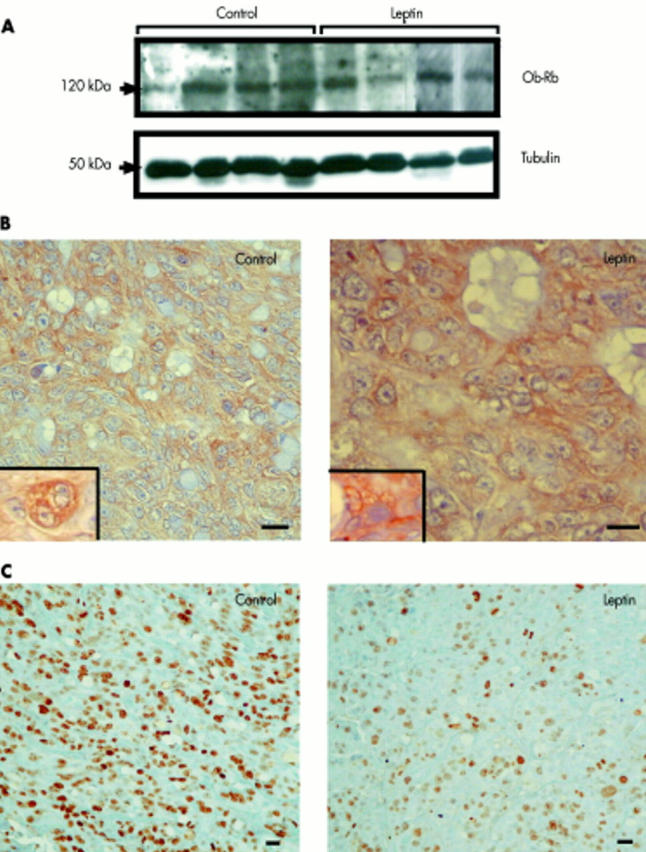
Effect of leptin treatment on expression of Ob-Rb and on the mitotic activity of HT-29 tumour xenografts. (A) Western immunoblots of Ob-Rb from homogenates of HT-29 tumour xenografts in leptin and vehicle treated nude mice using M18 Ob-R antibody. A band of approximately 120 kDa corresponding to the Ob-Rb functional isoform of the leptin receptor was identified in all tumours. This band disappeared after immunoneutralisation of Ob-R antibody with the corresponding blocking peptide (not shown). Membranes were reprobed with an antibody against α-tubulin (diluted 1:1000) to assess the relative equal loading of protein samples. (B) Ob-R immunohistochemistry. Insets illustrate detail of labelling on membranes and in the cytoplasm of tumour epithelial cells. (C) Ki-67 immunohistochemistry in tumours (MIB antibody) showing a higher proliferative index in the tumour of a vehicle treated mouse than in that of a leptin treated mouse. Bar = 20 µm.
Figure 4.

Effects of subcutaneous leptin treatment on leptinaemia and insulinaemia in nude mice and ApcMin/+ mice. (A) Plasma leptin and (B) plasma insulin after 28 days of subcutaneous continuous infusion of leptin or its vehicle in nude mice. *p = 0.008, **p<0.0003 versus vehicle treated mice. (C) Plasma leptin and (D) plasma insulin after 42 days of subcutaneous continuous infusion of leptin or its vehicle in ApcMin/+ mice. These variables were also measured in C57BL/6J wild mice studied as controls for Apc mutation. *p<0.03 versus vehicle treated mice (U test).
We concluded that continuous subcutaneous leptin delivery for four weeks in nude mice induced a strong and sustained hyperleptinaemia which exerted a clearcut effect on insulinaemia but had no noticeable effect on the growth of HT-29 colon cancer cell xenografts.
Leptin does not promote intestinal tumorigenesis in ApcMin/+ mice
We also investigated the possible influence of leptin in another tumour model, ApcMin/+ mice, which have a mutation in codon 850 of the Apc tumour suppressor gene, leading to the spontaneous development of intestinal adenomas and carcinomas.26 Three successive couples of male and two couples of female ApcMin/+ mice were divided into two groups with an identical weight at the beginning of the experiment (21.7 (0.35) g and 21.4 (0.34) g). After six weeks of continuous infusion of leptin or its vehicle, leptin treated mice weighed 22.1 (1.82) g and pair fed controls 22.1 (1.15) g. At that time, leptin levels were 2.4-fold higher in leptin treated than in vehicle treated ApcMin/+ mice (7.4 (2.3) ng/ml v 3.1 (0.4) ng/ml; p<0.03) (fig 4C ▶). To evaluate whether Apc mutation influenced leptinaemia, the latter was also determined in wild-type C57BL/6J counterparts restrained from feeding for a week and were found to be approximately of the same order as in vehicle treated ApcMin/+ mice (fig 4C ▶). Insulin levels were not significantly modified in leptin treated ApcMin/+ mice compared with vehicle treated ApcMin/+ mice. The latter had values similar to those of control wild-type C57BL/6J mice (fig 4D ▶). The small intestine exhibited numerous adenomas, most often solitary, but sometimes developing in narrow proximity and forming clusters (fig 5A ▶, B). The largest adenomas often displayed areas of high dysplasia (fig 5C ▶, D). Immunolabelling with Ob-R antibody (fig 5E ▶–H) indicated that epithelial cells of adenomas expressed leptin receptors but heterogeneously.
Figure 5.
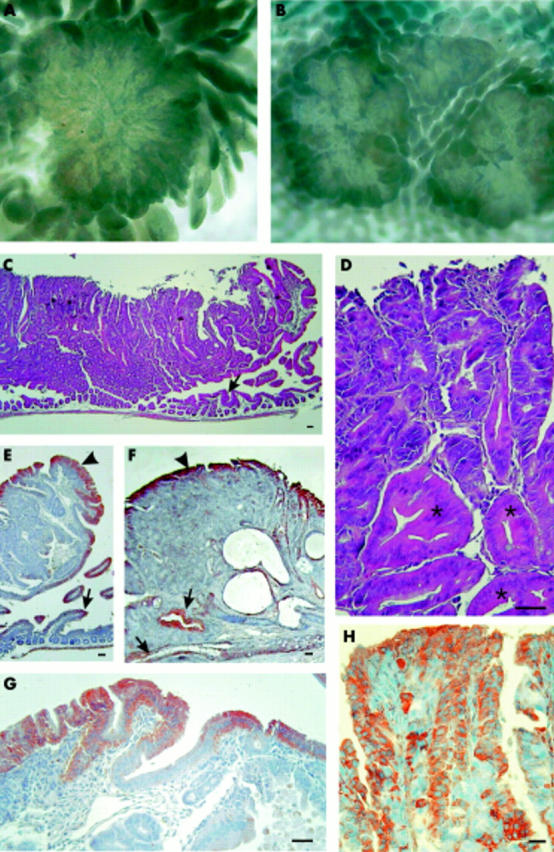
Characteristics and Ob-R expression in intestinal adenomas in ApcMin/+ mice. Macroscopic aspect of polyps, singly (A) or in clusters (B). (C) Histological pattern of adenoma (here a sessile duodenal polyp). The duodenal mucosa under the polyp appears atrophic (arrowhead). (D) Detail of the polyp showing areas of moderate to high dysplasia. Asterisks indicate glandular tubes only slightly modified. (E–H) Immunohistochemical Ob-R signal. (E) Part of the duodenal polyp. Labelling is seen in the epithelium surrounding the adenoma (arrowhead) and in the duodenal villi (arrow). (F) Colonic adenoma. Labelling is seen in the surrounding epithelium (arrowhead) and in some dysplastic glandular tubes (arrows). (G–H) Details of the reaction in the duodenal polyp showing heterogeneous Ob-R expression in the surrounding epithelium (G) and in epithelial cells of dysplastic glands in another area (H). Bar = 40 µm.
For all of the experiments, the mean numbers of tumours in the small intestine of leptin and vehicle treated ApcMin/+ mice were similar: 114.9 (14.3) and 123.0 (23.1), respectively (fig 6A ▶). No significant difference was found between control and leptin treated males or between control and leptin treated females. The density of adenomas per cm of intestine was also unchanged, whether small intestinal length was measured before fixation (3.19 (0.55) v 3.07 (0.59) per cm for controls and leptin treated mice, respectively) or after fixation (3.07 (0.42) v 3.25 (0.67) per cm) (fig 6A ▶). Moreover, the distribution profiles of tumour size were similar for the two groups of ApcMin/+ mice (fig 6B ▶). Tumours most frequently encountered measured between 500 µm and 1.5 mm in diameter. The distribution of tumours along the small intestine also did not differ between the two groups (table 1 ▶).
Figure 6.
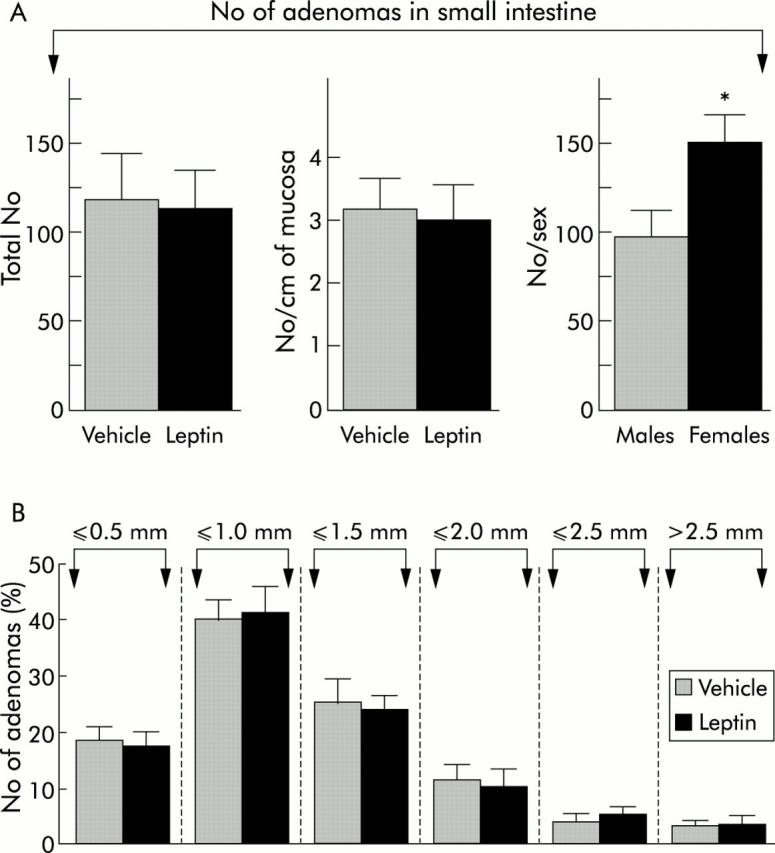
Numbers and size distribution of adenomas in the small intestine of leptin and vehicle treated ApcMin/+ mice. (A) No difference was observed in the total number or number per cm of intestine (here considering the length before fixation) between the two groups. By averaging values in all females and all males, the total number of adenomas was found to be significantly higher in the small intestine of females than in males (p<0.03). (B) Histogram showing the distribution profile of adenomas in the small intestine as a function of their size. Polyps were classified into six classes. There was no difference between leptin and vehicle treated ApcMin/+ mice for any class. In this study, the size of most polyps ranged between 500 µm and 1.5 mm.
Table 1.
Distribution of adenomas as a function of small intestinal segment localisation
| Segment 1 | Segment 2 | Segment 3 | Segment 4 | Segment 5 | Segment 6 | |
| Leptin | ||||||
| Total number | 8.4 (1.7) | 6.4 (0.5) | 22 (6.6) | 37.4 (9.3) | 31.3 (4.2) | 9.5 (5.1) |
| % | 7.9 (2.7) | 5.9 (0.8) | 18.2 (4.6) | 31.0 (4.6) | 28.2 (3.2) | 8.8 (4.8) |
| Controls | ||||||
| Total number | 9.5 (3.0) | 7.2 (1.8) | 25.1 (7.5) | 39.3 (11.3) | 33.1 (5.6) | 8.8 (2.5) |
| % | 7.7 (1.5) | 5.9 (1) | 19.0 (3.8) | 29.5 (5.7) | 28.6 (3.9) | 8.8 (3) |
The first half of segment 1 corresponds to the duodenum, the second half of segment 1, segments 2 and 3 to the jejunum, and segments 4–6 to the ileum. Approximately 77% of adenomas were found in segments 3–5.
Adenomas developed preferentially in the distal jejunum and proximal ileum, their number decreasing abruptly in the distal ileum. Adenomatous clusters had a tendency to be larger in vehicle treated than in leptin treated mice. As no difference was seen between leptin and vehicle treated mice, we analysed the number of tumours in the small intestine as a function of sex. Females had noticeably more tumours than males (150.9 (14.7) v 97.7 (12.8); p<0.03) (fig 6A ▶). Finally, when examining the relationship between total numbers of small intestinal adenomas and plasma leptin levels in mice, we observed that the two variables tended to be inversely correlated (r′ = −0.34, Spearman’s rank correlation) but this was not significant (p = 0.3). Adenomas were rare in the caecum, colon, and rectum, and their numbers did not differ: 1.3 (0.4) in control ApcMin/+ mice versus 0.9 (0.5) in leptin treated ApcMin/+ mice.
We concluded that hyperleptinaemia in ApcMin/+ mice did not enhance the development of adenomas in the small as in the large intestine, or the size and distribution of adenomas in the small intestine. Also, in these mice, hyperleptinaemia was not associated with a change in plasma insulin levels.
DISCUSSION
The present study has provided original data on the in vitro growth action of leptin in three human colon cancer cell lines characterised for their expression of functional leptin receptors and explored for the first time the potential role of hyperleptinaemia on intestinal tumorigenesis in two animal models, HT-29 xenografts in nude mice and ApcMin/+ mice. The results highlighted the lack of obvious effect of leptin on tumour growth in the two models.
We confirmed that leptin in vitro stimulates DNA synthesis and proliferation of HT-29 cells and thus is a growth factor for these cells, as previously reported.13,18,19 We also extended the effects of leptin to three other colon cancer cell lines, namely LoVo, Caco-2, and SW 480. Variability in the leptin response was noted in these cell lines which was apparently not related to p53 mutation or microsatellite instability. A differential effect of leptin on the in vitro proliferation of other cancer cell lines has also been reported.16 The doses tested here were in the range of those used by others.13,18 However, if we refer to mean serum leptin concentrations reported in obese human subjects (approximately 30 ng/ml, 40–100 ng/ml in some individuals),27 maximal effects on DNA synthesis and cell growth were obtained with doses corresponding to the highest levels measured in humans or with supraphysiological doses.
In nude mice, we showed that sustained delivery of exogenous leptin caused a 4.3-fold increase in plasma leptin levels compared with controls. This value is in the range of the 4.2-fold increase in plasma leptin levels reported between obese and normal weight humans.27 In these mice, hyperleptinaemia induced a decrease of the same order in plasma insulin levels. We and others have already observed such actions of leptin on insulin levels in Fisher 344 rats.25,28 This is in line with the majority of studies in rodents indicating that leptin inhibits insulin secretion.29 Although tumours seemed to develop somewhat more quickly in leptin treated than in vehicle treated mice, the difference did not reach significance, except on day 6, the day on which tumours began to be measurable. It should be noted that measurement of tumour volume through the skin entails some degree of imprecision inherent in the technique. At the end of the experiment, mean weight of tumours–which is a more exact variable—was in the same range in the two groups as there was only a 4% increase in leptin treated mice compared with controls. Thus leptin did not exert a noticeable action on the growth of xenografted HT-29 tumours. These results could not be attributed to the absence of leptin receptors because they remained expressed in tumour cells.
Estimation of the Ki-67 proliferative index in epithelial tumour cells is another way of assessing the influence of leptin on HT-29 xenografts. Surprisingly, this index was significantly lower whereas the apoptotic index was slightly higher in leptin treated mice than in controls. At first insight, the proliferative effect was opposite to the in vitro effect that we observed on HT-29 cells and in previous works.13,18,19 In contrast with the present in vivo data, we have previously shown an antiapoptotic effect of leptin on HT-29 cells in vitro.19 These data raise two hypotheses: (i) this decrease in tumour cell proliferation is directly due to leptin. However, although some authors have shown that leptin inhibits the mitotic activity of epithelial cells in the mouse large intestine in vivo,24 it is unlikely that leptin exerted such a direct effect on HT-29 cell proliferation as tumours in the leptin treated group grew slightly faster than in controls; (ii) more plausibly is the fact that consequent to their accelerated growth, tumours of leptin treated mice had begun an involution process at the time of sacrifice which was perhaps accompanied by reduced cell replication. Consistent with this hypothesis, histological examination of tumours revealed that necrotic areas were somewhat larger in tumours from leptin treated mice than from controls. This may result from an altered balance in tumour growth and angiogenesis. Nevertheless, freezing large samples from each tumour prevented us from doing a global evaluation of necrosis.
The lack of effect of leptin on the growth of HT-29 cells in nude mice may be further explained by the decrease in insulinaemia. Accordingly, insulin has been reported to stimulate in vitro the proliferation of colon cancer cell lines, among them HT-29 cells,30–33 to promote colon carcinogenesis in the rat34,35 and to be related to a higher risk of colonic neoplasia.31,36,37 Consequently, a decrease in insulin may counteract a potential growth effect of leptin. Alternatively, leptin might be not involved in cancer growth.
We found no evidence of any effect of leptin on the promotion of intestinal tumours in ApcMin/+ mice, as evaluated by the size, number, and distribution of adenomas. Hyperleptinaemia in leptin treated ApcMin/+ mice was significant but only moderate (2.4-fold increase) although a similar dose of leptin as for nude mice was used. Moreover, the lack of effect of leptin on tumorigenesis in ApcMin/+ mice could not be explained by a decrease in plasma insulin levels as the latter did not significantly change in leptin treated mice and were in the range of those in wild-type control mice. No significant effect of leptin on insulin release has previously been observed in a few rodent studies.29 Our data are closer to those recently reported in ApcMin/+ mice submitted to physical exercise.38 Colbert et al observed a significant decrease in plasma leptin levels in those animals compared with controls but no change in the total number and size of intestinal polyps. Also, they noted that leptin levels and total polyp numbers were significantly and inversely correlated.38 In our ApcMin/+ mice, we also found a trend towards an inverse correlation between plasma leptin levels and polyp numbers in the small intestine.
Hence the present data as well as our previous findings in the rat showing that leptin significantly reduced the development of carcinogen induced aberrant crypt foci in the colonic mucosa25 do not support a promoting role for leptin in intestinal tumorigenesis. Several reports seem opposed to this notion. Thus in bearing tumour animals (that is, rats inoculated with hepatoma cells and mice inoculated with Lewis lung carcinoma cells) the circulating leptin levels, measured in the absence of anorexia, were lower than in pair fed control animals.39 In some studies in human there was no evidence of increased leptin levels in patients with colorectal cancers.20–22 However, from a recent report, the risk of colonic cancer, but not rectal cancer, increased with increasing serum leptin concentrations.23 Nevertheless, in that study, it must be noted that the risk was significant only in individuals with the highest quartile leptin levels and, when colonic segments were studied separately, the risk was apparent only for the left colon. All of these facts suggest that leptin is not involved in intestinal tumorigenesis. Nevertheless, if leptin is implicated in this process, it is likely that very high hyperleptinaemia may play a role.
In conclusion, our results show that leptin exerts differential effects. In vitro, it acts as a growth factor for colon cancer cell lines. However, in vivo, in the rodent models tested and under our conditions, leptin exerted no influence on HT-29 colon cancer growth in nude mice or on the promotion of intestinal tumorigenesis in ApcMin/+ mice. These latter findings do not support a pivotal role for hyperleptinaemia on intestinal carcinogenesis.
Acknowledgments
This work was supported by the Institut de la Santé et de la Recherche médicale (INSERM) and grants from La Ligue contre le Cancer, Comité de Paris (No 75-02/RS-7 to T Lehy) and from the Association pour la Recherche contre le Cancer (ARC, France, to E Chastre). Thomas Aparicio was supported by the Assistance Publique des Hôpitaux de Paris, and Larissa Kotelevets by the ARC.
Abbreviations
FCS, fetal calf serum
PCR, polymerase chain reaction
BSA, bovine serum albumin
SDS-PAGE, sodium dodecyl sulphate-polyacrylamide gel electrophoresis
Published online first 27 April 2005
Conflict of interest: None declared.
REFERENCES
- 1.Zhang Y, Proenca R, Maffei M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature 1994;372:425–32. [DOI] [PubMed] [Google Scholar]
- 2.Bado A, Levasseur S, Attoub S, et al. The stomach is a source of leptin. Nature 1998;394:790–3. [DOI] [PubMed] [Google Scholar]
- 3.Sobhani I, Bado A, Vissuzaine C, et al. Leptin secretion and leptin receptor in the human stomach. Gut 2000;47:178–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maffei M, Halaas J, Ravussin E, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1995;1:1155–61. [DOI] [PubMed] [Google Scholar]
- 5.Kono S, Handa K, Hayabuchi H, et al. Obesity, weight gain and risk of colon adenomas in Japanese men. Jpn J Cancer Res 1999;90:805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giacosa A, Franceschi S, La Vecchia C, et al. Energy intake, overweight, physical exercise and colorectal cancer risk. Eur J Cancer Prev 1999;8 (suppl 1) :S53–60. [PubMed] [Google Scholar]
- 7.Murphy TK, Calle EE, Rodriguez C, et al. Body mass index and colon cancer mortality in a large prospective study. Am J Epidemiol 2000;152:847–54. [DOI] [PubMed] [Google Scholar]
- 8.Bouloumie A, Drexler HC, Lafontan M, et al. Leptin, the product of Ob gene, promotes angiogenesis. Circ Res 1998;83:1059–66. [DOI] [PubMed] [Google Scholar]
- 9.Sierra-Honigmann MR, Nath AK, Murakami C, et al. Biological action of leptin as an angiogenic factor. Science 1998;281:1683–6. [DOI] [PubMed] [Google Scholar]
- 10.Castellucci M, De Matteis R, Meisser A, et al. Leptin modulates extracellular matrix molecules and metalloproteinases: possible implications for trophoblast invasion. Mol Hum Reprod 2000;6:951–8. [DOI] [PubMed] [Google Scholar]
- 11.Attoub S, Noe V, Pirola L, et al. Leptin promotes invasiveness of kidney and colonic epithelial cells via phosphoinositide 3-kinase-, rho-, and rac-dependent signaling pathways. FASEB J 2000;14:2329–38. [DOI] [PubMed] [Google Scholar]
- 12.Barrenetxe J, Villaro AC, Guembe L, et al. Distribution of the long leptin receptor isoform in brush border, basolateral membrane, and cytoplasm of enterocytes. Gut 2002;50:797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardwick JC, Van Den Brink GR, Offerhaus GJ, et al. Leptin is a growth factor for colonic epithelial cells. Gastroenterology 2001;121:79–90. [DOI] [PubMed] [Google Scholar]
- 14.Mix H, Widjaja A, Jandl O, et al. Expression of leptin and leptin receptor isoforms in the human stomach. Gut 2000;47:481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Somasundar P, Riggs D, Jackson B, et al. Leptin stimulates esophageal adenocarcinoma growth by nonapoptotic mechanisms. Am J Surg 2003;186:575–8. [DOI] [PubMed] [Google Scholar]
- 16.Somasundar P, Yu AK, Vona-Davis L, et al. Differential effects of leptin on cancer in vitro. J Surg Res 2003;113:50–5. [DOI] [PubMed] [Google Scholar]
- 17.Schneider R, Bornstein SR, Chrousos GP, et al. Leptin mediates a proliferative response in human gastric mucosa cells with functional receptor. Horm Metab Res 2001;33:1–6. [DOI] [PubMed] [Google Scholar]
- 18.Liu Z, Uesaka T, Watanabe H, et al. High fat diet enhances colonic cell proliferation and carcinogenesis in rats by elevating serum leptin. Int J Oncol 2001;19:1009–14. [DOI] [PubMed] [Google Scholar]
- 19.Rouet-Benzineb P, Aparicio T, Guilmeau S, et al. Leptin counteracts sodium butyrate-induced apoptosis in human colon cancer HT-29 cells via NF-(kappa) B signaling. J Biol Chem 2004;279:16495–502. [DOI] [PubMed] [Google Scholar]
- 20.Arpaci F, Yilmaz MI, Ozet A, et al. Low serum leptin level in colon cancer patients without significant weight loss. Tumori 2002;88:147–9. [DOI] [PubMed] [Google Scholar]
- 21.Tessitore L, Vizio B, Jenkins O, et al. Leptin expression in colorectal and breast cancer patients. Int J Mol Med 2000;5:421–6. [DOI] [PubMed] [Google Scholar]
- 22.Wallace AM, Sattar N, McMillan DC. Effect of weight loss and the inflammatory response on leptin concentrations in gastrointestinal cancer patients. Clin Cancer Res 1998;4:2977–9. [PubMed] [Google Scholar]
- 23.Stattin P, Lukanova A, Biessy C, et al. Obesity and colon cancer: does leptin provide a link? Int J Cancer 2004;109:149–52. [DOI] [PubMed] [Google Scholar]
- 24.Chaudhary M, Mandir N, FitzGerald AJ, et al. Starvation, leptin and epithelial cell proliferation in the gastrointestinal tract of the mouse. Digestion 2000;61:223–9. [DOI] [PubMed] [Google Scholar]
- 25.Aparicio T, Guilmeau S, Goiot H, et al. Leptin reduces the development of the initial precancerous lesions induced by azoxymethane in the rat colonic mucosa. Gastroenterology 2004;126:499–510. [DOI] [PubMed] [Google Scholar]
- 26.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990;247:322–4. [DOI] [PubMed] [Google Scholar]
- 27.Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 1996;334:292–5. [DOI] [PubMed] [Google Scholar]
- 28.Barazzoni R, Zanetti M, Stebel M, et al. Hyperleptinemia prevents increased plasma ghrelin concentration during short-term moderate caloric restriction in rats. Gastroenterology 2003;124:1188–92. [DOI] [PubMed] [Google Scholar]
- 29.Kieffer TJ, Habener JF. The adipoinsular axis: effects of leptin on pancreatic beta-cells. Am J Physiol Endocrinol Metab 2000;278:E1–14. [DOI] [PubMed] [Google Scholar]
- 30.Bjork J, Nilsson J, Hultcrantz R, et al. Growth-regulatory effects of sensory neuropeptides, epidermal growth factor, insulin, and somatostatin on the non-transformed intestinal epithelial cell line IEC-6 and the colon cancer cell line HT 29. Scand J Gastroenterol 1993;28:879–84. [DOI] [PubMed] [Google Scholar]
- 31.Giovannucci E. Insulin, insulin-like growth factors and colon cancer: a review of the evidence. J Nutr 2001;131 (11 suppl) :3109–20S. [DOI] [PubMed] [Google Scholar]
- 32.Koenuma M, Yamori T, Tsuruo T. Insulin and insulin-like growth factor 1 stimulate proliferation of metastatic variants of colon carcinoma 26. Jpn J Cancer Res 1989;80:51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeoman LC, Wan CW, Zorbas MA. Transferrin and insulin enhance human colon tumor cell growth by differentiation class specific mechanisms. Oncol Res 1996;8:273–9. [PubMed] [Google Scholar]
- 34.Corpet DE, Jacquinet C, Peiffer G, et al. Insulin injections promote the growth of aberrant crypt foci in the colon of rats. Nutr Cancer 1997;27:316–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tran TT, Medline A, Bruce WR. Insulin promotion of colon tumors in rats. Cancer Epidemiol Biomarkers Prev 1996;5:1013–15. [PubMed] [Google Scholar]
- 36.Sandhu MS, Dunger DB, Giovannucci EL. Insulin, insulin-like growth factor-I (IGF-I), IGF binding proteins, their biologic interactions, and colorectal cancer. J Natl Cancer Inst 2002;94:972–80. [DOI] [PubMed] [Google Scholar]
- 37.Yang YX, Hennessy S, Lewis JD. Insulin therapy and colorectal cancer risk among type 2 diabetes mellitus patients. Gastroenterology 2004;127:1044–50. [DOI] [PubMed] [Google Scholar]
- 38.Colbert LH, Mai V, Perkins SN, et al. Exercise and intestinal polyp development in APCMin mice. Med Sci Sports Exerc 2003;35:1662–9. [DOI] [PubMed] [Google Scholar]
- 39.Lopez-Soriano J, Carbo N, Tessitore L, et al. Leptin and tumor growth in rats. Int J Cancer 1999;81:726–9. [DOI] [PubMed] [Google Scholar]