

Abstract
Background: Overexpression of cyclooxygenase 2 (COX-2) is frequently detected in gastric cancer and is believed to play a crucial role in gastric carcinogenesis.
Aim: We examined the chemopreventive effect of a COX-2 inhibitor in an animal model of stomach carcinogenesis.
Methods: Eighty six male Wistar rats were divided into six different treatment groups: group A, water alone (n = 5); group B, N-methyl-N′-nitro-N-nitrosoguanidine (MNNG 100 μg/ml) (n = 16); group C, indomethacin (3 mg/kg/day) (n = 16); group D, celecoxib (5 mg/kg/day) (n = 17); group E, celecoxib (10 mg/kg/day) (n = 16); and group F, celecoxib (20 mg/kg/day) (n = 16). Group B–F animals were treated with 10% sodium chloride (in the initial six weeks) and MNNG in drinking water to induce adenocarinoma in the stomach. All animals received treatment for 40 weeks, and were sacrificed after death or at 48 weeks. Gastric neoplasm was evaluated by histology.
Results: The incidences of gastric cancer were 0% in group A, 75% in group B, 68.8% in group C, 70.6% in group D, 18.8% in group E, and 31.3% in group F (p = 0.002, ANOVA). Compared with MNNG controls, treatment with celecoxib 10 mg/kg/day also showed lower tumour multiplicity (0.19 (0.40) v 1.00 (0.73); p = 0.004) and lower mean tumour volume (2.4 v 2805 mm3; p = 0.02). Although tumours had significantly higher COX-2 expression than their adjacent normal tissues (p<0.02), there was no significant difference in COX-2 levels among tumours in the different treatment groups. The lowest tumour prostaglandin E2 level was found in the indomethacin treated group, suggesting that the chemopreventive effect of celecoxib may be mediated by a COX independent pathway.
Conclusion: While treatment with indomethacin had no significant effect on tumour development, treatment with celecoxib reduced gastric cancer incidence and growth in rats.
Keywords: rats, celecoxib, cyclooxygenase-2, gastric cancer
Gastric cancer is the second leading cause of cancer related death worldwide.1 Notwithstanding the global declining incidence of gastric cancer, mortality is still rising in Asian countries. To date, there is no effective measure to prevent development of gastric cancer. Although Helicobacter pylori infection has been identified as the most important causative factor,2 there is little evidence to substantiate the fact that eradication of the bacterium alone can stop the process of gastric carcinogenesis.3,4
Since the observation from the Physician’s Health Study that usage of aspirin may reduce the risk of colorectal cancer,5 intense interest has been directed towards investigation of the anticancer properties of aspirin and non-steroidal anti-inflammatory drugs (NSAIDs). There are at least 12 published observational studies showing the protective effects of NSAIDs against colorectal cancer. More recently, studies in colonic cancer show that induction of cyclooxygenase 2 (COX-2) is associated with inhibition of apoptosis, increase in angiogenesis, and metastatic potential.6 Celecoxib, a COX-2 inhibitor, has been shown to reduce polyp formation in a cohort of patients with familial adenomatous polyposis syndrome.7
COX-2 expression is upregulated in H pylori induced mucosal inflammation.8 It is frequently expressed in gastric cancer9–12 as well as in premalignant gastric lesions.13 Inhibition of COX-2 in vitro results in growth inhibition of gastric cancer cells.14 Furthermore, the use of COX-2 inhibitors has been shown to suppress the growth of gastric cancer xenografts in nude mice.15 Unlike colorectal cancers, however, there are a lack of animal and human data demonstrating the effectiveness of COX-2 inhibition and NSAIDs in the prevention of gastric cancer.
In this study, we evaluated the use of celecoxib and indomethacin in the prevention of N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) induced gastric cancer in rats.
MATERIAL AND METHODS
Animals
Administration of MNNG in drinking water is a well established animal model for the study of the differentiated type of human stomach cancer.16 Eighty six four week old grade 2 male Wistar rats (approximately 50 g in weight) were obtained from the Laboratory Animal Centre of the Sun Yat-Sen University. Rats were kept in metal cages at 21°C, humidity 50%, with a 12 hour light-dark cycle. Rats had free access to regular chow pellets and drinking water. There was one week of acclimatisation prior to the initiation of this experiment. The study protocol was approved by the animal ethics committee of the Sun Yat-Sen University.
Chemicals
MNNG (Fluka, Germany) solution was prepared three times per week with distilled water at a concentration of 100 μg/ml. It was protected from light and given ad libitum to animals in their drinking water. In addition to MNNG, all animals were given 1 ml of 10% sodium chloride weekly by oral gavage in the initial six weeks to enhance gastric cancer development.17
Study design
Rats were allocated to one of six different groups (groups A–F). Group A was a control group whereas groups B–F were treated with MNNG. In addition, they were given water (control, group B), indomethacin (3 mg/kg/day, group C), or celecoxib (Pfizer Pharmaceuticals, New York) at 5 mg/kg/day (group D), 10 mg/kg/day (group E), or 20 mg/kg/day (group F). Drugs were administered by oral gavage daily from the age of six weeks for 40 weeks. All animals were monitored closely for general health during the study period and their body weights were recorded weekly. At week 48, all rats were sacrificed. Animals that died before the end of experiment were autopsied to determine the cause of death and presence of gastric tumours. At laparotomy, the stomach was opened along the greater curvature and was carefully examined, as were other organs. All dissections were performed by investigators blinded to the different treatment groups. The length (L), width (W), and depth (D) of gastric tumours were measured by callipers. Tumour volume was calculated using the formula, V = L×W×D×π/6.18
Histopathology
For histological examination, the stomach was fixed in 10% neutral buffered formalin. Paraffin embedded sections (5 μm) were cut and stained with haematoxylin and eosin for histological examination by a pathologist who was unaware of the treatment assignments. Adenocarcinoma, as defined by the presence of atypical glands that locally invaded the submucosa, muscularis propria, or serosa, was recorded.19
RNA extraction and quantitative PCR
Gastric tissue specimens were homogenised with an ultrasound homogeniser. Total RNA was extracted by RNA Tri Reagents (CINNA/MRC; Cincinnati, Ohio, USA). Total RNA (1 µg) was reverse transcribed into cDNA using dNTPs (1 mM), 5× reverse transcription buffer (500 mM Tris HCl, pH 8.3, 250 mM KCl, 50 mM MgCl2, and 50 mM DTT), 16 units RNasin, and 2.5 units of AMV reverse transcriptase (GibcoBRL, Life Technologies Gaithersburg, Maryland, USA).
Real time quantitative polymerase chain reaction (PCR) was performed on a ABI PRISM 7000 sequence detection system using Sybrgreen, PCR mastermix (Perkin Elmer, Branchburgh, New Jersey), and primers. Primer sequences were designed from the Genbank as follows: COX-2 (L25925) (forward, 1408–1435) 5′-ACAGGAGAGAAAGAAATGGCTGCAGAGT-3′, (reverse, 1598–1573) 5′-CAGTATTGAGGAGAACAGATGGGATT-3′; and β-actin (NM-031144) (forward, 476–500), 5′-TCACCCACACTGTGCCCATCTATGA-3′, (reverse, 633–610) 5′-GTCACGCACGATTTCCCTCTCAGC-3′. A 24 µl reaction mix was aliquoted with 1 µl/replicate of cDNA. A DNA free template control (containing water) was included and each sample was added in duplicate. Reaction tubes were sealed with optical caps, and the PCR reaction was run at 50°C for two minutes, 95°C for 10 minutes, followed by 40 cycles at 96°C for 45 seconds, 60°C for 45 seconds, and 72°C for one minute. The specificity of the PCR products was characterised by melting curve analysis and followed by gel electrophoresis. Quantification was determined by the threshold cycle. Actin was used as a housekeeping gene to normalise mRNA levels and compared against mRNA expression levels in normal control stomach.
PGE2 assay
Prostaglandin E2 (PGE2) levels were measured in snap frozen tissue specimens using an ELISA based assay (Amersham Pharmacia Biotech, Piscataway, New Jersey, USA). Briefly, approximately 10 mg of snap frozen tissues (mean weight 10.3 (SD 2.8)) were homogenised in 20 volumes of ethanol using a ground glass homogeniser cooled on ice. Ice cold water was added to give a final ethanol concentration of 15% and the mixture was centrifuged for 10 minutes at 400 g. A 10 µl volume of glacial acetic acid was added to each sample to pH 3.0 and followed by a five minute incubation period at room temperature. The supernatant was then applied to a pre-primed Amprep C18 mini column (Amersham Pharmacia Biotech), and the column was washed with distilled water and hexane. PGE2 was eluted with two 0.75 ml volumes of ethyl acetate. This fraction was evaporated to dryness under nitrogen and stored at −80°C. Samples were resuspended in 1 ml of buffer and assayed in 96 well plates. The quantity of PGE2 in supernatants was determined using ELISA.
Statistics
Body weight, tumour incidence (percentage of animals with tumour development), tumour multiplicity (mean number of tumours per animal), mean tumour volume (mean volume of tumour in tumour bearing rats), COX-2, and PGE2 levels were compared among animals fed MNNG control alone, indomethacin, and celecoxib. Parametric data were analysed by ANOVA with Bonferroni’s multiple comparison; non-parametric data were computed by χ2 test or Fisher’s exact test with Bonferroni’s correction. A p value of <0.05 was considered to be statistically significant.
RESULTS
General observation
Body weights of group A control animals were higher than those of the other groups in the early phase of the study (fig 1 ▶). However, there was no significant difference in body weight among other treatment groups during the whole study period. There were in total 26 deaths during the study period: none in group A, six in group B, six in group C, five in group D, six in group E, and three in group F. The causes of death are listed in table 1 ▶. Most animals died from gastric (n = 14) and small bowel (n = 8) cancer. Two animals died from intestinal haemorrhage after receiving the high dose (20 mg/kg/day) of celecoxib. Two animals died from non-digestive tract diseases.
Figure 1.

Body weight of animals in the different treatment groups. The body weight of group A control rats was higher than in the N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) treated groups in the initial phase of the experiments. However, there was no significant difference in body weight among all MNNG treated groups (groups B–F).
Table 1.
Tumour incidences and causes of death in the different groups of study animals
| Group | No of rats | Treatment | Causes of death (No of animals) |
| A | 5 | Control | |
| B | 16 | MNNG alone | Gastric cancer (6) |
| C | 16 | MNNG+indomethacin (3 mg/kg/day) | Gastric cancer (2), small bowel cancer (3), unknown (1) |
| D | 17 | MNNG+celecoxib (5 mg/kg/day) | Gastric cancer (5) |
| E | 16 | MNNG+celecoxib (10 mg/kg/day) | Small bowel cancer (5), lung cancer (1) |
| F | 16 | MNNG+celecoxib (20 mg/kg/day) | Gastric cancer (1), intestinal haemorrhage (2) |
MNNG, N-methyl-N′-nitro-N-nitrosoguanidine.
Tumour incidence
Table 2 ▶ summarises the incidences of MNNG induced gastric tumours in the six treatment groups. Seventy five per cent of rats treated with MNNG developed gastric cancer at the end of this study whereas none of the control rats in group A had a gastric tumour. There was a significant difference in tumour incidences among different treatment groups (p = 0.002). Rats treated with celecoxib 10 mg/kg/day (group E) had the lowest tumour incidence (18.8%) which was significantly lower than the MNNG group (75.0%; p = 0.004). The tumour incidence of group F rats (celecoxib 20 mg/kg/day) also tended to be lower than the MNNG control (group B) (31.3% v 75%; p = 0.052). The absolute risk reductions of gastric cancer in animals treated with celecoxib 10 mg/kg/day and 20 mg/kg/day were 56.3% (95% confidence interval (CI) 16.7–80.4%) and 43.8% (95% CI 3.9–71.8%), respectively. On the other hand, the numbers of gastric tumours were comparable between the other treatment groups (indomethacin or celecoxib at 5 mg/kg/day) and the MNNG control group. All gastric tumours were confirmed to be adenocarcinomas (fig 2 ▶) and the majority (90.7%) were high grade tumours.
Table 2.
Tumour incidences and multiplicity in the different treatment groups
| Group | Treatment | No of deaths (%) | No of rats with gastric tumours (%) (incidence)* | No of gastric cancers per rat (SD) (multiplicity)† |
| A | Control | 0 (0) | 0 (0) | 0 (0) |
| B | MNNG alone | 6 (37.5) | 12 (75.0) | 1.0 (0.7) |
| C | MNNG+indomethacin | 6 (37.5) | 11 (68.8) | 0.8 (0.80 |
| D | MNNG+celecoxib 5 mg/kg/day | 5 (29.4) | 12 (70.6) | 0.8 (0.6) |
| E | MNNG+celecoxib 10 mg/kg/day | 6 (37.5) | 3 (18.8) | 0.2 (0.4) |
| F | MNNG+celecoxib 20 mg/kg/day | 3 (18.8) | 5 (31.3) | 0.3 (0.5) |
*p = 0.002 (χ2): C versus B, p = 1.00; D versus B, p = 1.00; E versus B, p = 0.004; F versus B, p = 0.052.
†p = 0.001 (ANOVA): C versus B, p = 1.00; D versus B, p = 1.00; E versus B, p = 0.004; F versus B, p = 0.025.
Figure 2.
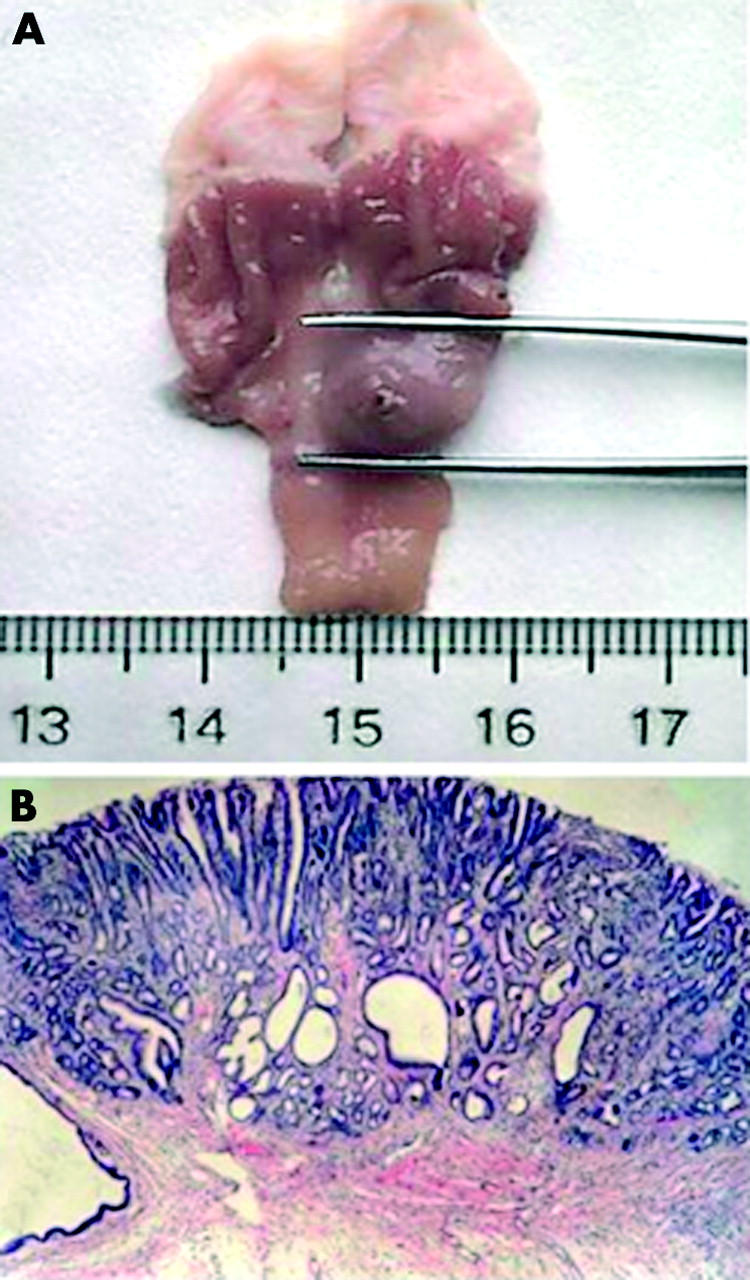
Macroscopic and microscopic appearance of N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) induced gastric tumour in a rat. (A) Macroscopic view of MNNG induced tumour formation in the distal stomach of a Wistar rat. (B) Haematoxylin-eosin staining of well differentiated gastric adenocarcinoma in the stomach (×20).
Moreover, premalignant gastric lesions such as dysplasia were frequently detected in MNNG treated rats. Of the 10 remaining viable rats in group B, nine had dysplasia on histological examination of the gastric mucosa. In contrast, only four of the 10 remaining rats in group E (celecoxib 10 mg/kg/day) had gastric dysplasia (p = 0.23). The frequencies of gastric dysplasia in rats treated with indomethacin and other doses of celecoxib were comparable with group B.
Tumour multiplicity
In addition to tumour incidence, there was a significant difference in tumour multiplicity, or number of cancers per rat, among the different treatment groups (p = 0.001, table 2 ▶). Compared with the MNNG group, rats fed celecoxib 10 mg/kg/day or 20 mg/kg/day had significantly lower tumour multiplicities compared with animals treated with MNNG alone (0.2 (0.4) v 1.0 (0.7), p = 0.004 and 0.3 (0.5) v 1.0 (0.7), p = 0.025). However, treatment with indomethacin (group C) and low dose celecoxib (5 mg/kg/day, group D) did not have any apparent suppressive effect on tumour multiplicity.
Tumour volume
Mean tumour volume was significantly different among the treatment groups (p = 0.009). Specifically, rats treated with celecoxib had a markedly reduced tumour volume compared with the MNNG control group (group B). Mean tumour volumes were significantly lower in animals treated with celecoxib 5 mg/kg/day (group D) (188.5 (377.8) mm3; p = 0.036), 10 mg/kg/day (group E) (2.4 (7.0) mm3; p = 0.022) and 20 mg/kg/day (group F) (38.9 (110.5) mm3; p = 0.025) compared with those treated with MNNG alone (group B) (2805 (5540.1) mm3). In contrast, mean tumour volume in indomethacin treated animals (group C) was only marginally lower than the MNNG control group (359 (859.8) mm3v 2805 (5540.1) mm3; p = 0.07).
Non-gastric tumours
There were eight animals that developed small bowel adenocarcinoma, three in the indomethacin treated group (group C) and five in the celecoxib treated group (group E). One animal in group E also developed lung cancer. Overall, there was no significant difference in the number of small bowel and lung tumours with different treatment allocations.
COX-2 and PGE2 levels
COX-2 was expressed at low levels in the stomach of control rats (0.53 (0.1)) (fig 3 ▶). In contrast, COX-2 was upregulated in tumours. Gastric tumours had higher COX-2 expression than their adjacent normal tissues in all treatment groups (p<0.02). Treatment with celecoxib or indomethacin did not reduce tumour COX-2 levels but COX-2 was significantly lower in adjacent normal tissues of celecoxib or indomethacin treated groups (p<0.01).
Figure 3.

Cyclooxygenase 2 (COX-2) mRNA expression levels of tumours and adjacent normal tissues in the different treatment groups. COX-2 mRNA expression levels were determined by quantitative reverse transcription-polymerase chain reaction. Mean (SEM) values are shown. There was upregulation of COX-2 in all tumours compared with adjacent normal tissues (p<0.02). However, there was no significant difference in COX-2 mRNA levels among tumours in different treatment groups. Adjacent normal tissues from the N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) treated group had the highest COX-2 levels (p<0.01). Indo, indomethacin; Cele5, 10, 20, celecoxib 5, 10, and 20 mg/kg/day. Significant differences: *p<0.02, **p<0.002, ***p<0.0001; between different tumours, p>0.05; between different normal tissues, p<0.0001; †p<0.0001, MNNG normal versus all other normal tissues.
In addition to induction of COX-2, PGE2 levels were increased in tumours (fig 4 ▶). Gastric tumours in all treatment groups tended to have higher PGE2 levels than their adjacent normal tissues but a significant difference was only observed in the low dose celecoxib (5 mg/kg/day) group (p = 0.015). Treatment with indomethacin (3 mg/kg/day) or high doses of celecoxib (>10 mg/kg/day) were associated with mildly reduced tumour PGE2 levels, but the difference did not reach statistical significance. Moreover, there was no significant difference in PGE2 levels of normal tissues among the different treatment groups.
Figure 4.

Prostaglandin E2 (PGE2) levels in tumours and adjacent normal tissues in the different treatment groups. PGE2 was measured by ELISA and mean (SEM) values are shown. PGE2 levels tended to be higher in tumours than in adjacent normal tissues. Treatment with indomethacin (Indo) or celecoxib (Cele) (> 10mg/kg/day) tended to reduce PGE2 in tumours but the difference did not reach statistical significance. There was no difference in PGE2 levels among normal tissues of the different treatment groups (p>0.05). Significant differences: *p<0.015; between different tumours, p>0.05; between different normal tissues, p>0.05.
DISCUSSION
In this study, we determined the role of COX-2 inhibition in the prevention of sodium chloride enhanced gastric carcinogenesis induced by MNNG in Wistar rats. MNNG induced gastric cancer is a well established animal model of stomach carcinogenesis.16 The mutagen, when given in drinking water, induces intestinal metaplasia and adenocarcinoma in the pyloric mucosa of Wistar rats.16,20 The histology of this induced gastric malignancy resembles the differentiated type of stomach cancer in humans. To enhance the carcinogenic effects of MNNG, highly concentrated sodium chloride solution was given to these animals in the initial six weeks.17 In the present study, 75% of MNNG treated animals developed gastric cancer at the end of 48 weeks, confirming that this is a highly successful model of gastric tumorigenesis.
Although the exact mechanism underlying MNNG induced gastric cancer remains poorly understood, previous studies showed that the genetic makeup of the animals may play a role.21 For example, ACI/N rats are highly susceptible to MNNG induced stomach carcinogenesis but BUF/Nac rats are relatively resistant.22 Recently, COX-2 and Bcl-2 were found to be coexpressed in the glandular corpus epithelium of rats treated with MNNG.23 This upregulated expression is associated with cell proliferation, atrophy, and intestinal metaplasia of the stomach. It is therefore logical to anticipate that treatment with a COX-2 inhibitor may have an antiproliferative and hence chemopreventive effect on MNNG induced gastric cancer.
The results of this study showed, for the first time, that both the incidence and multiplicity of MNNG induced gastric cancer can be significantly reduced in rats treated with celecoxib. The chemopreventive effect of celecoxib was demonstrated when a moderate dose (10 mg/kg/day) was given to these animals. With the use of celecoxib 10 mg/kg/day, there was an approximate 56% reduction in tumour incidence, 80% reduction in tumour multiplicity, and 1169-fold reduction in tumour volume. This remarkable degree of tumour suppression by celecoxib is comparable with that reported in the azoxymethane induced colon cancer model in rats.24 Moreover, it exceeds that previously reported in MNNG induced gastric cancer by other agents, such as genistein (a tyrosine kinase inhibitor),25 C-erbB-2/neu antisense oligonucleotide,26 and curcumin.27 However, this effect was not seen in animals treated with a lower dose of celecoxib (5 mg/kg/day), presumably due to suboptimal suppression of COX-2 expression in the gastric mucosa. It is interesting to note that the high dose of celecoxib (20 mg/kg/day) did not produce a further increase in the chemopreventive effect. In keeping with this observation, there was no further reduction in tumour PGE2 or COX-2 levels with the high dose of celecoxib (20 mg/kg/day) compared with 10 mg/kg/day, suggesting the effect had plateaued. Previous experiments in rat models of inflammation also suggest that the best anti-inflammatory effect of celecoxib is achieved at a dose of 10 mg/kg.28,29 This dosage is also comparable with the usual dose in humans (200–400 mg/day) for acute pain and inflammation. Conversely, we found that the high dose of celecoxib used in this study may be associated with more toxicity, such as intestinal haemorrhage. Moreover, the COX-2 selectivity of celecoxib may be lost at high doses, resulting in more COX-1 inhibition. Based on our data with high dose celecoxib and indomethacin, concurrent COX-1 inhibition may have a paradoxical effect on chemoprevention. It remains undetermined whether concurrent COX-1 inhibition has a promotional effect on tumour development.
In this study, indomethacin, a non-selective COX inhibitor, showed no apparent chemopreventive effect on MNNG induced gastric tumours in rats. There was only a tendency favouring a lower tumour volume in indomethacin treated rats compared with MNNG controls. The reason for these discrepancies between indomethacin and celecoxib is unclear. One plausible explanation may be related to the dose of indomethacin used in this study. Our selection of this dose was based on two facts. Firstly, the recommended dose of indomethacin in humans is 1–3 mg/kg/day. Secondly, previous animal studies demonstrated inhibitory effects on the formation of aberrant crypt foci in the colons of dimethyl hydrazine treated rats using a dose of 2 mg/kg/day.30 As shown in figure 4 ▶, tumour PGE2 levels in the indomethacin treated group were in fact among the lowest of all groups, suggesting that this dose of indomethacin was adequate for inhibition of COX activity. None the less, it was not associated with a parallel reduction in gastric tumour development. Thus it appears that the chemopreventive effects of celecoxib in this rat model of gastric carcinogenesis may not be solely related to COX-2 inhibition and PGE2 suppression. In this regard, there are emerging data to suggest that chemoprevention of cancer by NSAIDs or COX-2 inhibitors may not be mediated via COX dependent pathways.31 Firstly, compounds that do not inhibit COX-2, such as sulindac sulphone, also induce apoptosis and inhibit colorectal carcinogenesis in animal models.32 In contrast, a recent study showed that the use of piroxicam was ineffective in the prevention of carcinogen induced tumorigenesis in the rat oesophagus, despite upregulation of COX-2 in these tumours and the ability of piroxicam to suppress PGE2 levels.33 Moreover, the use of low dose aspirin (81 mg/day), which has virtually no COX-2 inhibitory effects, still possesses chemopreventive effects against colorectal adenoma in high risk individuals.34 Hence COX independent mechanisms are likely to be involved. One of the non-COX mediated pathways that may be involved in carcinogenesis is the nuclear factor κB (NFκB) signalling pathway.35 Activated NFκB translocates into the nucleus where it modulates expression of a variety of genes, mostly through IκB kinase dependent phosphorylation and subsequent degradation of its inhibitor. It is recognised that aspirin and sulindac, but not indomethacin, inhibit the activity of IκB kinase β in vitro.36 Therefore, indomethacin may not be able to inhibit IκB kinase β, resulting in less COX independent tumour suppression. Whether the difference in IκB kinase β inhibitory effect accounts for the differences in outcome between indomethacin and celecoxib warrants further investigation. Furthermore, it has recently been recognised that celecoxib enhances suppression of Akt activation and increases activation of caspase-9 and -3 in cholangiocarcinoma cells.37 This finding suggests that the Akt pathway may be another COX-2 independent pathway in suppressing growth and enhancing apoptosis of tumour cells.
The Mongolian gerbil was recently found to be a good animal model to study H pylori associated gastric carcinogenesis.38 Moreover, emerging data show that COX-2 is upregulated in the gerbil stomach after H pylori infection.39 It will be interesting to characterise the role of COX-2 inhibition in the chemoprevention of gastric cancer in this gerbil model. Another issue that is worth further study is the role of celecoxib in the therapy of established gastric cancer, as this drug was introduced at the same time as the carcinogen in this study. The exact therapeutic role of celecoxib against established cancer remains unknown and a study that introduces celecoxib at different time points may be useful in clarifying this point. Moreover, this type of study may help address the important question of the optimal time of intervention if it is found that celecoxib only prevents gastric cancer development but fails in the treatment of established cancer.
In summary, our study showed that treatment with celecoxib, a specific COX-2 inhibitor, suppressed MNNG induced gastric cancer in rats. This finding lends further support to the use of COX-2 inhibitors in the chemoprevention of gastric cancer. Whether this result can be translated into clinical benefit requires further confirmation in human clinical studies.
Acknowledgments
This study was supported by an unrestricted research grant from the Hong Kong Society of Digestive Endoscopy and the Natural Science Foundation of Guangdong Province of China (No 010713).
Abbreviations
COX-2, cyclooxygenase 2
MNNG, N-methyl-N′-nitro-N-nitrosoguanidine
NSAIDs, non-steroidal anti-inflammatory drugs
PCR, polymerase chain reaction
PGE2, prostaglandin E2
NFκB, nuclear factor κB,
REFERENCES
- 1.Pisani P, Parkin DM, Bray F, et al. Estimates of the worldwide mortality from 25 cancers in 1990. Int J Cancer 1999;83:18–29. [DOI] [PubMed] [Google Scholar]
- 2.Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001;345:784–9. [DOI] [PubMed] [Google Scholar]
- 3.Sung JJY, Lin SR, Ching JYL, et al. Atrophy and intestinal metaplasia one year after cure of H. pylori infection: a prospective, randomized study Gastroenterology 2000;119:7–14. [DOI] [PubMed] [Google Scholar]
- 4.Correa P, Fontham ETH, Bravo JC, et al. Chemoprevention of gastric dysplasia: randomized trial of antioxidant supplements and anti-Helicobacter therapy. J Natl Cancer Inst 2000;92:1881–8. [DOI] [PubMed] [Google Scholar]
- 5.Sturmer T, Glynn RJ, Lee IM, et al. Aspirin use and colorectal cancer: post-trial follow-up data from teh Physician’s Health Study. Ann Intern Med 1998;128:713–20. [DOI] [PubMed] [Google Scholar]
- 6.Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer 2001;1:11–21. [DOI] [PubMed] [Google Scholar]
- 7.Steinbach G, Lynch PM, Phlips RKS, et al. The effects of celecoxib, a cyclooxygenase 2 inhibitor in familial adenomatous polyposis. N Engl J Med 2000;342:1946–52. [DOI] [PubMed] [Google Scholar]
- 8.Chan FK, To KF, Ng YP, et al. Expression and cellular localization of COX-1 and -2 in Helicobacter pylori gastritis. Aliment Pharmacol Ther 2001;15:187–93. [DOI] [PubMed] [Google Scholar]
- 9.Ristimaki A, Honkanen N, Jankala H, et al. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res 1997;57:1276–80. [PubMed] [Google Scholar]
- 10.Yamamoto H, Itoh F, Fukushima H, et al. Overexpression of cyclooxygenase-2 protein is less frequent in gastric cancers with microsatellite instability. Int J Cancer 1999;84:400–3. [DOI] [PubMed] [Google Scholar]
- 11.Uefuji K, Ichikura T, Mochizuki H. Cyclooxygenase-2 expression is related to prostaglandin biosynthesis and angiogenesis in human gastric cancer. Clin Cancer Res 2000;6:135–8. [PubMed] [Google Scholar]
- 12.Leung WK, To KF, Ng YP, et al. Association between cyclo-oxygenase-2 overexpression and missense p53 mutations in gastric cancer. Br J Cancer 2001;84:335–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sung JJY, Leung WK, Go MYY, et al. Cyclooxygenase-2 expression in Helicobacter pylori-associated premalignant and malignant gastric lesions. Am J Pathol 2000;157:729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsuji S, Kawano S, Sawaoka H, et al. Evidences for involvement of cyclooxygenase-2 in proliferation of two gastrointestinal cancer cell lines. Prostaglandins Leukot Essent Fatty Acids 1996;55:179–83. [DOI] [PubMed] [Google Scholar]
- 15.Sawaoka H, Kawano S, Tsuji S, et al. Cyclooxygenase-2 inhibitors suppress the growth of gastric cancer xenografts via induction of apoptosis in nude mice. Am J Physiol 1998;274:G1061–7. [DOI] [PubMed] [Google Scholar]
- 16.Sugimura T, Fujimura S, Baba T. Tumor production in the glandular stomach and alimentary tract of the rat by N-methyl-N′-nitro-nitrosoguanidine. Cancer Res 1970;30:455–65. [PubMed] [Google Scholar]
- 17.Tatematsu M, Takahashi M, Fukushima S, et al. Effects in rats of sodium chloride on experimental gastric cancers induced by N-methyl-N-nitro-N-nitrosoguanidine or 4-nitroquinoline-1-oxide. J Natl Cancer Inst 1975;55:101–6. [DOI] [PubMed] [Google Scholar]
- 18.Rao CV, Rivernson A, Simi B, et al. Chemoprevention of colon carcinogenesis by sulindac, a nosteroidal anti-inflammatory agent. Cancer Res 1995;55:1464–72. [PubMed] [Google Scholar]
- 19.Saito T, Inokuchi K, Takayama S, et al. Sequential morphological changes in N-methyl-N′-nitro-N-nitrosoguanidine carcinogenesis in the glandular stomach of rats. J Natl Cancer Inst 1970;44:769–83. [PubMed] [Google Scholar]
- 20.Matsukura N, Kawachi T, Sasajima K, et al. Induction of intestinal metaplasia in the stomachs of rats by N-methyl-N′-nitro-N-nitrosoguanidine. J Natl Cancer Inst 1978;61:141–4. [DOI] [PubMed] [Google Scholar]
- 21.Ushijima T, Yamamoto M, Suzui M, et al. Chromosomal mapping of genes controlling development, histological grade, depth of invasion, and size of rat stomach carcinomas. Cancer Res 2000;60:1092–6. [PubMed] [Google Scholar]
- 22.Ohgaki H, Kawachi T, Matsukura N, et al. Genetic control of susceptibility of rats to gastric carcinoma. Cancer Res 1983;43:3663–7. [PubMed] [Google Scholar]
- 23.Loogna P, Franzen L, Sipponen P, et al. Cyclooxygenase-2 and Bcl-2 expression in the stomach mucosa of Wistar rats exposed to Helicobacter pylori, N-methyl-N′-nitro-N-nitrosoguanidine and bile. Virchows Arch 2002;441:77–84. [DOI] [PubMed] [Google Scholar]
- 24.Kawamori T, Rao CV, Seibert K, et al. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res 1998;58:409–12. [PubMed] [Google Scholar]
- 25.Tatsuta M, Iishi H, Baba M, et al. Attenuation by genistein of sodium-chloride-enhanced gastric carcinogenesis induced by N-methyl-N′-nitro-N-nitrosoguanidine in Wistar rats. Int J Cancer 1999;80:396–9. [DOI] [PubMed] [Google Scholar]
- 26.Uedo N, Tatsuta M, Baba M, et al. Inhibition by rat C-erbB-2/neu antisense oligonucleotide of gastric carcinogenesis induced by N-methyl-N′-nitro-N-nitrosoguanidine in Wistar rats. Int J Cancer 1999;83:670–3. [DOI] [PubMed] [Google Scholar]
- 27.Ikezaki S, Nishikawa A, Furukawa F, et al. Chemopreventive effects of curcumin on glandular stomach carcinogenesis induced by N-methyl-N′-nitro-N-nitrosoguanidine and sodium chloride in rats. Anticancer Res 2001;21:3407–11. [PubMed] [Google Scholar]
- 28.Pinheiro RM, Calixto JB. Effect of the selective COX-2 inhibitors, celecoxib and rofecoxib in rat acute models of inflammation. Inflamm Res 2002;51:603–10. [DOI] [PubMed] [Google Scholar]
- 29.Cuzzocrea S, Mazzon E, Serraino I, et al. Celecoxib, a selective cyclo-oxygenase-2 inhibitor reduced the severity of experimental colitis induced dinitrobenzene sulfonic acid in rats. Eur J Pharmacol 2001;431:91–102. [DOI] [PubMed] [Google Scholar]
- 30.Charalambous D, Farmer C, O’Brien PE. Sulindac and indomethacin inhibit formation of aberrant crypt foci in the colons of dimethyl hydrazine treated rats. J Gastroenterol Hepatol 1996;11:88–92. [DOI] [PubMed] [Google Scholar]
- 31.Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cycloxoygenase-2. Nat Rev Cancer 2001;1:11–21. [DOI] [PubMed] [Google Scholar]
- 32.Chan TA. Non-steroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol 2002;3:166–74. [DOI] [PubMed] [Google Scholar]
- 33.Carlton PS, Gopalakrishnan R, Gupta A, et al. Piroxicam is an ineffective inhibitor of N-nitrosomethylbenzylamine-induced tumorigenesis in the rat esophagus. Cancer Res 2002;62:4376–82. [PubMed] [Google Scholar]
- 34.Baron JA, Cole BF, Sandler RS, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med 2003;348:891–9. [DOI] [PubMed] [Google Scholar]
- 35.Karin M, Cao Y, Greten FR, et al. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2002;2:301. [DOI] [PubMed] [Google Scholar]
- 36.Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998;396:77–80. [DOI] [PubMed] [Google Scholar]
- 37.Lai GH, Zhang Z, Sirica AE. Celecoxib acts in a cyclooxygenase-2-independent manner and in synergy with emodin to suppress rat cholangiocarcinoma growth in vitro through a mechanism involving enhanced Akt inactivation and increased activation of caspases-9 and -3. Mol Cancer Ther 2003;2:265–71. [PubMed] [Google Scholar]
- 38.Watanabe T, Tada M, Nagai H, et al. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology 1998;115:642–8. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi S, Fujita T, Yamamoto A. Role of cyclooxygenase-2 in Helicobacter pylori-induced gastritis in Mongolian gerbils. Am J Physiol Gastrointest Liver Physiol 2000;279:G791–8. [DOI] [PubMed] [Google Scholar]