

Abstract
During a search for causative genes in patients with concurrent multiple primary colon tumours, we found a novel case with a germline mutation of the p53 gene, from GCC (Ala) to GTC (Val) at codon 189. Of the six primary colon tumours that this patient had, one large advanced carcinoma exhibited a somatic p53 mutation and a somatic APC mutation, in addition to the germline p53 mutation. Two early carcinomas and three adenomas had somatic APC mutations but no somatic p53 mutation or loss of the p53 allele. K-ras-2 mutations were detected in an advanced carcinoma and an early carcinoma. The present results suggest that a patient with a certain type of germline p53 mutation is predisposed to concurrent multiple colon tumours. It is also suggested that in such a patient, a somatic APC mutation is involved in tumour formation and that an additional somatic p53 mutation contributes to tumour progression.
Keywords: multiple colon tumour, germline p53 mutation, somatic p53 mutation, somatic APC mutation
There are two well known hereditary diseases predisposed to colorectal carcinomas: familial adenomatous polyposis (FAP), associated with a germline mutation of the APC gene, 1,2 and hereditary non-polyposis colorectal cancer (HNPCC), associated with a germline mutation of the DNA mismatch repair genes.3,4 Typical FAP patients develop hundreds and thousands of colorectal adenomas which convert to carcinomas, while attenuated-type FAP patients produce a small number of adenomas and carcinomas.5 Many HNPCC patients produce multiple colorectal carcinomas that show microsatellite instability.6,7 However, when no germline APC mutation is found, and when no microsatellite instability or germline mutation of the DNA mismatch repair gene is detected, it is difficult to diagnose whether a patient with multiple colorectal carcinomas and/or adenomas is a sporadic case or a hereditary disease patient. Li-Fraumeni (LF) syndrome, which is associated with germline mutations of the p53 gene,8,9 is characterised by early onset of sarcoma, breast cancer, and other malignant tumours.10–12 Although LF patients sometimes produce colorectal carcinomas, the frequency is extremely low, and the characteristics of colorectal carcinomas in this syndrome are still unclear. To clarify whether the p53 gene is causative for concurrent multiple colon tumours, we examined 15 patients without germline mutations of the APC and mismatch repair genes. We found a novel case exhibiting a germline mutation of the p53 gene, and further examined somatic genetic changes in multiple colon tumours in this case.
CASE REPORT
Fifteen patients without germline mutations of the APC and mismatch repair genes were examined after approval by the Komagome Hospital Review Committee. Each of the 15 patients had both colorectal carcinomas and colorectal polyps. The number of carcinomas ranged from 1 to 5 and polyps from 1 to 7. All tumours exhibited no microsatellite instability. Of these patients, a 73 year old man (COK169) was referred to the Tokyo Metropolitan Komagome Hospital because of lower right abdominal pain. Barium enema examination showed the existence of a large tumour between the caecum and ascending colon, and polyps in the ascending colon. No personal history or obvious family history of tumours was known. Neither of his parents nor any of his three brothers were affected by tumours. A right hemicolectomy was performed. Although colon carcinoma did not recur, he died six years later as a result of squamous cell carcinoma of the lung. He was a non-smoker. Pathological diagnosis of the resected colon showed that the large colon tumour, located at the caecum, was an advanced carcinoma with an ulcerative carcinomatous lesion (Ca in fig 1 ▶). The histological type of this carcinoma was a well to moderately differentiated adenocarcinoma invading the subserosa. Dukes' classification for this carcinoma was B. Moreover, there were five polyps, including two early carcinomas and three adenomas of the ascending colon. Both of the early carcinomas (P1 and P2 in fig 1 ▶) were diagnosed as well differentiated intramucosal adenocarcinomas. The three adenomas (P3, P4, and P5 in fig 1 ▶) were diagnosed as tubular adenomas, with severe atypia. Neoplastic cells comprised approximately 70% of the total cells in each tumour sample, which was assessed by haematoxylin-eosin staining of formalin fixed paraffin embedded sections. Appropriate areas of tumour tissues were frozen at −80°C until they were used for mutation analysis.
Figure 1.
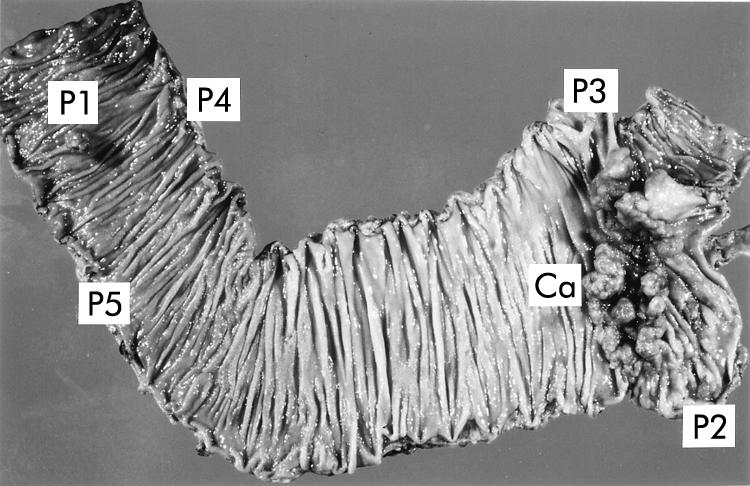
Resected colon from patient COK169. There was one advanced carcinoma (Ca) and five polyps (P1–P5).
DNA was extracted from these tumours and normal mucosa. Mutations of the p53, APC, and K-ras-2 genes in DNA samples were analysed by polymerase chain reaction-single strand conformation polymorphism (PCR-SSCP) using [α-32P]dCTP, as previously described.13 When abnormal bands were detected, single stranded DNA fragments were eluted from the corresponding bands on PCR-SSCP gels. DNA fragments were amplified through the asymmetrical PCR, and the amplified DNA samples were subjected to direct sequencing using a dideoxy chain termination reaction. Analysis of the p53 gene revealed a germline mutation from GCC (Ala) to GTC (Val) at codon 189 (fig 2 ▶). This germline mutation was confirmed to be heterozygous from the fact that PCR-SSCP of normal mucosa DNA from patient COK169 exhibited equal densities of the normal band (which corresponded to the normal allele) and the mutant band (which corresponded to the allele with a germline mutation). In addition to the germline p53 mutation, the large advanced carcinoma (Ca) exhibited a somatic p53 mutation from GGC (Gly) to AGC (Ser) at codon 245, without loss of the p53 allele.
Figure 2.

Sequence of the germline p53 mutation in patient COK169.
To clarify whether germline and somatic p53 mutations existed on different alleles of the p53 gene, we extracted RNA from COK169Ca and performed reverse transcription-PCR-SSCP analysis using a primer set which amplifies the region, including both germline (at codon 189) and somatic (at codon 245) mutations. SSCP showed multiple mutant bands, and direct sequencing of DNA fragments from these bands revealed no mutant band having these two mutations—that is, all mutant bands analysed showed only one of the two mutations (data not shown). These results support the idea that germline and somatic mutations occur in different alleles of the p53 gene. Other tumours (P1–P5) also had a germline p53 mutation but exhibited no somatic p53 mutation or loss of the p53 allele. Somatic APC mutations were detected in advanced carcinoma (Ca), two early carcinomas (P1, P2), and three adenomas (P3, P4, P5). All APC mutations were located at the central region of the APC gene and resulted in stop codons. K-ras-2 mutations were detected in the advanced carcinoma (Ca) and in one of the two early carcinomas (P1). Microsatellite instability was analysed by PCR using BAT26, BAT40, D5S346, D2S123, and TP53 as primers. Instability was not observed in any tumour. These data are summarised in table 1 ▶.
Table 1.
Genetic changes detected in concurrent multiple colon tumours from patient COK169
| Germline p53 mutation | Somatic p53 mutation | Somatic APC mutation | Somatic K-ras mutation | |||||||
| Tumour | Size (mm) | Pathological diagnosis | Codon | Mutation | Codon | Mutation | Codon | Mutation | Codon | Mutation |
| COK169 Ca | 82×40 | Advanced carcinoma | 189 | GCC→GTC | 245 | GGC→AGC | 1487–8 | T deletion | 13 | GGC→GAC |
| 1545 | TCA→TGA | |||||||||
| COK169 P1 | 12×10×6 | Early carcinoma | 189 | GCC→GTC | — | 1378 | CAG→TAG | 12 | GGT→GTT | |
| COK169 P2 | 11×7×3 | Early carcinoma | 189 | GCC→GTC | — | 1449–75 | 80 bp repeat | — | ||
| COK169 P3 | 6×3×2 | Adenoma with severe atypia | 189 | GCC→GTC | — | 1450 | CGA→TGA | — | ||
| Loss of the other allele | ||||||||||
| COK169 P4 | 3×2×1 | Adenoma with severe atypia | 189 | GCC→GTC | — | 1367 | CAG→TAG | — | ||
| COK169 P5 | 3×2×1 | Adenoma with severe atypia | 189 | GCC→GTC | — | 1464 | GAG→TAG | — | ||
—, Mutation was not detected
DISCUSSION
We found a novel case with concurrent multiple primary colon tumours who had a germline p53 mutation from GCC (Ala) to GTC (Val) at codon 189. A somatic p53 mutation has been detected at high frequency (nearly 50%) in colon carcinomas from sporadic and FAP patients13,14 but a germline p53 mutation has not been described in association with colon carcinomas. A germline p53 mutation is usually associated with LF syndrome which is characterised by a family history of various malignant tumours. Patients with typical LF syndrome develop sarcomas at a young age, often in childhood, breast cancers and brain tumours at younger than 44 years, and often form various multiple tumours.8,10 However, colorectal cancer is rare in this syndrome, and onset is at a rather later age (older than 44 years).8 Although the present case was old (aged 73 years), he may be an LF-like patient as he had multiple colon tumours and died later of cancer in another organ. He may carry a new germline mutation as no obvious family history of cancer was observed. The reason why colon carcinomas occurred at such a late age is difficult to understand. Missense mutant-type p53 protein is assumed to form a complex with the wild-type p53 protein resulting in inactivation of the wild-type protein (dominant-negative effect).15 One explanation for the occurrence of colon carcinomas at a late age may be that such a dominant-negative effect of the mutation at codon 189 is weak compared with other typical germline p53 mutations. The effect of an unknown modifier gene(s)16 is also possible.
It is important to determine whether the germline mutation is pathogenic or a rare polymorphism. More than 140 germline p53 mutations have been reported,17 and the p53 gene has polymorphisms at more than six codons.18,19 Germline mutation at codon 189, GCC (Ala) to GTC (Val), has not previously been described, and this mutation may not be a polymorphism as it was not detected in 155 individuals without colorectal cancer in our examination. Codon 189 is located within the L2 loop, which is necessary for binding of p53 protein to the minor groove of DNA.20 Mutation at codon 189 is assumed to perturb DNA binding ability of p53 protein, and from this aspect this mutation is assumed to be pathogenic. A somatic mutation of the same codon has previously been reported in a colon carcinoma,21 although the direction of mutation was different, GCC (Ala) to ACC (Thr). By analogy with this somatic mutation, the germline mutation at codon 189 of our patient may also be pathogenic.
With respect to colon carcinogenesis in LF syndrome, the mechanism is still unclear because genetic alterations in colon tumours from this syndrome have not been reported. The present data suggest a contribution of a somatic APC mutation to tumour formation and an additional somatic p53 mutation, possibly in the wild-type allele, to progression from early to advanced carcinoma. Loss of heterozygosity at the p53 locus detected in various tumours from LF patients has been described to be nearly 50%22; therefore, some of the remaining cases are assumed to have a somatic p53 mutation in the wild-type allele, similar to the present advanced colon carcinoma. The somatic K-ras-2 mutation is also involved in some colon tumours. The genetic events observed in the colon tumours in the present LF-like patient are those occurring in the adenoma-carcinoma sequence.23,24 Later onset of colon cancer in LF syndrome may be due to such a mechanism of carcinogenesis which includes contribution of a biallelic somatic APC mutation.
The present results suggest that in searching for causative genes in patients with multiple colon cancer, it is necessary to examine not only APC and mismatch repair genes but also the p53 gene.
Acknowledgments
This work was supported in part by a Showa University Grant-in-Aid for Innovative Collaborative Research Projects and Special Research Grant-in-Aid for Development of Characteristic Education from the Japanese Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Abbreviations
FAP, familial adenomatous polyposis
HNPCC, hereditary non-polyposis colorectal cancer
LF, Li-Fraumeni syndrome
PCR-SSCP, polymerase chain reaction-single strand conformation polymorphism
REFERENCES
- 1.Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991;66:589–600. [DOI] [PubMed] [Google Scholar]
- 2.Kinzler KW, Nilbert MC, Su L-K, et al. Identification of FAP locus genes from chromosome 5q21. Science 1991;253:661–5. [DOI] [PubMed] [Google Scholar]
- 3.Lynch HT, Kimberling WJ, Albano WA, et al. Hereditary nonpolyposis colorectal cancer (Lynch syndrome I and II). Cancer 1985;56:934–8. [DOI] [PubMed] [Google Scholar]
- 4.Fishel R, Lescoe MK, Rao MSR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993;75:1027–38. [DOI] [PubMed] [Google Scholar]
- 5.Spirio L, Olschwang S, Groden J, et al. Allele of the APC gene: an attenuated form of familial polyposis. Cell 1993;75:951–7. [DOI] [PubMed] [Google Scholar]
- 6.Ionov Y, Peinado MA, Malkhosyan S, et al. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993;363:558–61. [DOI] [PubMed] [Google Scholar]
- 7.Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–16. [DOI] [PubMed] [Google Scholar]
- 8.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990;250:1233–8. [DOI] [PubMed] [Google Scholar]
- 9.Srivestava S, Zou Z, Pirollo K, et al. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 1990;348:747–9. [DOI] [PubMed] [Google Scholar]
- 10.Li FP, Fraumeni Jr JF, Mulvihill JJ, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res 1988;48:5358–62. [PubMed] [Google Scholar]
- 11.Malkin D. p53 and the Li-Fraumeni syndrome. Biochim Biophys Acta 1994;1198:197–213. [DOI] [PubMed] [Google Scholar]
- 12.Varley JM, Evans DGR, Birch JM. Li-Fraumeni syndrome—a molecular and clinical review. Br J Cancer 1997;76:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kikuchi-Yanoshita R, Konishi M, Ito S, et al. Genetic changes of both p53 alleles associated with the conversion from colorectal adenoma to early carcinoma in familial adenomatous polyposis and non-familial adenomatous polyposis patients. Cancer Res 1992;52:3965–71. [PubMed] [Google Scholar]
- 14.Baker SJ, Fearon ER, Nigro JM, et al. Chromosome 17 deletions and p53 mutations in colorectal carcinomas. Science 1989;244:217–21. [DOI] [PubMed] [Google Scholar]
- 15.Kern SE, Pietenpol JA, Thiagalingam S, et al. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science 1992;256:827–30. [DOI] [PubMed] [Google Scholar]
- 16.Moser AR, Dove WF, Roth KA, et al. The min (multiple intestinal neoplasia) mutation: Its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol 1992;116:1517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Database of Germline p53 Mutation. http://www.lf2.cuni.cz/
- 18.Harris N, Brill E, Shohat O, et al. Molecular basis for heterogeneity of the human p53 protein. Mol Cell Biol 1986;6:4650–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toguchida J, Yamaguchi T, Dayton SH, et al. Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med 1992;326:1301–8. [DOI] [PubMed] [Google Scholar]
- 20.Cho Y, Gorina S, Jeffrey PD, et al. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 1994;265:346–55. [DOI] [PubMed] [Google Scholar]
- 21.Slebos RJC, Baas IO, Clement M, et al. Clinical and pathological associations with p53 tumour-suppressor gene mutations and expression of p21WAF1/Cip1 in colorectal carcinoma. Brit J Cancer 1996;74:165–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varley JM, Thorncroft M, McGown G, et al. A detailed study of loss of heterozygosity on chromosome 17 in tumors from Li-Fraumeni patients carrying a mutation to the TP53 gene. Oncogene 1997;14:865–71. [DOI] [PubMed] [Google Scholar]
- 23.Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med 1988;319:525–32. [DOI] [PubMed] [Google Scholar]
- 24.Miyaki M, Seki M, Okamoto M, et al. Genetic changes and histopathological types in colorectal tumors from patients with familial adenomatous polyposis. Cancer Res 1990;50:7166–73. [PubMed] [Google Scholar]