

Abstract
Risk for ischemic stroke is mediated by both environmental and genetic factors. Although several environmental exposures have been implicated, relatively little is known about the genetic basis of predisposition to this disease. Recent studies in Iceland identified risk polymorphisms in two putative candidate genes for ischemic stroke: phosphodiesterase 4D (PDE4D) and 5-lipoxygenase activating protein (ALOX5AP). A collection of North American sibling pairs concordant for ischemic stroke and two cohorts of prospectively ascertained North American ischemic stroke cases and control subjects were used for evaluation of PDE4D and ALOX5AP. Although no evidence supported linkage of ischemic stroke with either of the two candidate genes, single-nucleotide polymorphisms and haplotypic associations were observed between PDE4D and ischemic stroke. There was no evidence of association between variants of ALOX5AP and ischemic stroke. These data suggest that common variants in PDE4D may contribute to the genetic risk for ischemic stroke in multiple populations.
Stroke is a leading cause of death in the Western hemisphere,1 and 85 to 90% of strokes are ischemic.2,3 Risk for ischemic stroke is mediated by both genetic and environmental factors.4 A genome-wide scan was performed on an Icelandic genealogy consisting of 476 patients (clustered into 179 families) and 438 relatives in a search for stroke susceptibility genes.5 Significant evidence for linkage with stroke was found in a 20cM region of human chromosome 5q12 (STRK1). This region included the 5′ end of a candidate gene, PDE4D. The PDE4D protein product was hypothesized to be involved in the cause of stroke risk through an atherosclerotic pathway.6 A risk haplotype, comprising a microsatellite (AC008818-1) and a single-nucleotide polymorphism (SNP45), conferred a 1.5-fold increased risk for ischemic stroke in the Icelandic population. The strongest association with disease was noted in cardiogenic and carotid subtypes of ischemic stroke, suggesting an atherosclerotic cause.
In a separate study in the Icelandic population, a susceptibility locus predisposing to stroke and myocardial infarction was mapped to 13q12-13.7 Within this region, a candidate gene, ALOX5AP, was identified that has been previously implicated in risk for atherosclerosis. A haplotype of ALOX5AP, (Hap A) defined by four SNPs (SG13S25, SG13S114, SG13S89, and SG13S32) that span the first four exons, was most strongly associated with risk for stroke and myocardial infarction. In the Icelandic population, Hap A conferred a 1.8-fold increased risk for myocardial infarction and a 1.67-fold increased risk for stroke. Hap A was also found to be a risk factor for stroke in a Scottish population.8 Hap A was not a risk factor for stroke in a British population, but a different haplotype of ALOX5AP (Hap B) was positively associated with stroke. It was suggested that variants of ALOX5AP are involved in the pathogenesis of myocardial infarction and stroke by increasing production of leukotriene B4, a critical regulator in the 5-lipoxygenase pathway and a marker of inflammation in the arterial wall.
To examine the role of variants in PDE4D and ALOX5AP with stroke risk in a general population, we performed linkage analysis in the Siblings With Ischemic Stroke Study (SWISS; siblings concordant and discordant for ischemic stroke) and association analysis in two cohorts, the Ischemic Stroke Genetics Study (ISGS)9 and the Mayo Stroke Genetics Data Bank (MSGD).10
Subjects and Methods
Study Populations
Institutional review board approval was sought and obtained at all participating institutions. In all instances, subjects were enrolled after providing written, informed consent. Samples were collected under the auspices of three protocols: 249 subjects from 104 families enrolled in SWISS, an ongoing multicenter study of families that assesses genetic risk factors; 544 subjects (329 cases and 215 control subjects) from ISGS, an ongoing, prospective, five-center case-control study; and 96 subjects (48 patients and 48 control subjects) from the MSGD, a single-center controlled repository of DNA collected from patients with ischemic stroke. Protocols for SWISS10 and ISGS9 have been described previously. MSGD inclusion and exclusion criteria for cases and control subjects matched those of ISGS, with the exception that ISGS restricted enrollment to incident cases. Across all DNA collections, stroke was defined according to World Health Organization criteria,11 and index strokes were confirmed to be ischemic by results of computed tomography or magnetic resonance imaging of the head. Index strokes were classified according to the prespecified Trial of Org 10172 in Acute Stroke Treatment (TOAST) criteria12 and were blinded to genotype.
Molecular Genetic Analysis
Polymerase chain reaction (PCR) amplification was performed using 25ng genomic DNA and 10pmol of forward and reverse primers (Table 1) according to the manufacturers’ specifications. TruAllele PCR mix (Applied Biosystems, Foster City, CA) was used for microsatellite markers, Taq with Q solution (Qiagen, Venlo, the Netherlands) was used for all restriction digest assays, and TaqMan Universal PCR Master Mix, No AmpErase was used for all Assay-by-Design experiments (Applied Biosystems). To ensure consistent allele calling and assay accuracy, we included DNA from Centre d’Etude du Polymorphisme Human (CEPH) individual 1347-02 in each amplification. Quality control was assessed by genotyping duplicate DNA from 51 patients.
Table 1.
Polymerase Chain Reaction Amplification Characteristics
| Microsatellite Markers | Primer Sequence | PCR Program | Size Range (bp) |
|---|---|---|---|
| AC008818-1 | F-TGCTTGGTGAAGGAATAGC
R-GTGGGTGGGTATTCATGTG |
57TD52 | 140–160 |
| D13S289 | F-CTGGTTGAGCGGCATT
R-TGCAGCCTGGATGACA |
57TD52 | 245–276 |
| D13S1238 | F-CTCTCAGCAGGCATCCA
R-GCCAACGTAATTGACACCA |
57TD52 | 129–159 |
| Single Nucleotide Polymorphisms | Primer Sequence | PCR Program | Enzyme Digest |
|---|---|---|---|
| SNP32 (rs456009) | F-TAATCTGAGCCTCAGCTTTC
R-TGTAGCTCTGGTATCATAGTGG |
57TD52 | PvuII |
| SNP45 | F-TGGCTGCAGATTACAGTG
R-AGGAGCAGGTTAAAGCAG |
57TD52 | MaeIII |
| SNP56 (rs702553) | F-ACAACAACCCTATAAGGCAG
R-ATTGTCTTGGCTATACAGGC |
57TD52 | AflIII |
| SNP83 (rs966221) | F-GATTTATGTCTTATACTTTC
R-ACGAAAAACTTCTACGTATGAAACA |
57TD52 | MaeIII |
| SNP87 (rs2910829) | F-GATGATGAGTCTGGAGCTG
R-ACTCTAACCAAGTGCTTGC |
60TD50 | SspI |
| SG13S89 | F-TGTGAAGCCCTGGAGAGGTG
R-ACCCTTGTCAGCACAGCAG |
57TD52 | AccII |
| SG13S32 | F-CCTCTCTTCTGTTCCTGG
R-ACAGGTGAACTAAACAGGATAG |
57TD52 | TaqI |
| SG13S25 | F-GACAGCATCAGCTAGTCTCTTTCC
R-GAAATTCATGTTGCTGTGTCCATACA FAM/VIC-CAGCCACTGTTRCCCA |
Assay-by-Design | |
| SG13S114 | F-CAGATGTATGTCCAAGCCTCTCT
R-AGGTAGGTCTATGGTTGCAACATTG FAM/VIC-TTGCAATTCTAWTTAACC |
Assay-by-Design |
PCR = polymerase chain reaction.
Microsatellite markers were analyzed using an ABI Prism 3100 genetic analyzer with the GeneScan 500 LIZ size standard (Applied Biosystems). Resulting data were read using an ABI Prism Genotyper (Applied Biosystems). Genotypes for D13S289 and D13S1238 were corrected to the published sizes for CEPH individual 1347-02 (http://www.cephb.fr/test/cephdb/eng). These data were not available for marker AC008818-1, but all genotypes were corrected to a genotype of 148 and 152, consistently seen for CEPH individual 1347-02. Endonuclease restriction digests were performed according to the manufacturer’s instructions, and resulting fragments were electrophoresed in a 2 to 4% agarose gel stained with ethidium bromide and viewed using ultraviolet transillumination. TaqMan Assays-by-Design SNP Genotyping Services (Applied Biosystems) were used for ALOX5AP markers SG13S25 and SG13S114. Thermal cycling and allelic discrimination were performed on an ABI Prism 7900 HT Sequence Detection System (Applied Biosystems) according to the manufacturer’s instructions. Table 1 shows the microsatellites and SNPs studied. Genotype determination was performed blinded to clinical information.
Statistical Analysis
To describe the study population, we reported categorical data as frequencies and percentages, and continuous data as means with standard deviations. A two-sample test for binomial proportions between cases and control subjects was reported using a χ2 test of independence. The Fisher exact test was used where appropriate. A two-sample t test for independent samples was used to compare age across groups.
Each genetic marker was examined for deviation from Hardy-Weinberg equilibrium proportions. In the SWISS sibships, each marker was also tested for evidence of Mendelian inconsistencies using PedCheck.13 All genotype inconsistencies were converted to “missing.” Linkage analyses were performed using nonparametric single-point and multipoint analysis using the NPL(pairs) statistic within GeneHunter-Plus.14 The one-parameter exponential allele sharing model was computed.15
Association analyses were performed to estimate odds ratios for each SNP with case-control status. A series of generalized estimating equations (GEE1)16 was computed that included relevant covariates (age, sex, race, origin of DNA sample [ISGS or MSGD], hypertension, atrial fibrillation, myocardial infarction, smoking status, family history of stroke). All modeling was performed in a hierarchical manner, with a baseline model that included only the SNP as the predictor. Additional models were tested with age alone; sex alone; age and sex; age, sex, and race; and age, sex, race, and origin of DNA sample. Additional models then were tested by increasing the number of remaining covariates. A final, fully saturated model was also used. The p values were computed using the 2-degree-of-freedom general test of association. To assess the possibility of false-positive results due to study sample stratification, we performed both race-pooled and race-specific (white vs nonwhite) analyses.
Haplotypes were constructed using expectation-maximization algorithm-based methods, and odds ratios and 95% confidence intervals were estimated using GEE1 methods. In PDE4D, four SNPs and one microsatellite marker were used in haplotype estimation, based on the single marker results (SNP83, SNP56, SNP45, AC008818, and SNP32). The most frequent allele in each SNP was assigned “allele 1,” and the minor allele was “allele 2.” For the microsatellite marker, AC008818, the rare alleles (3 alleles accounting for 5 genotypes of 1,204) were coded as missing, thereby providing five alleles for analysis, each with population frequencies greater than 8%. Two and three moving window haplotype analyses across the five PDE4D markers were performed using the software Dandelion.17 These analyses were repeated for the four SNPs that were genotyped in ALOX5AP (SG13S25, SG13S114, SG13S89, SG13S32). Differences in the distribution of PDE4D and ALOX5AP genotypes within cases according to TOAST subtype versus control subjects were determined using χ2 tests. Comparisons were made for cases according to large-vessel, cardioembolic, small-vessel, and combined phenotype (large-vessel and cardioembolic) categories.
Results
Table 2 summarizes the demographic and clinical characteristics of the SWISS subjects analyzed in the linkage component of this study. A total of 93.3% (97/104) of probands identified themselves as white, and 6.7% (7/104) identified themselves as nonwhite. Probands were recruited from Canadian sites in 8.7% (9/104) of cases, and probands were recruited from US sites in 91.3% (95/104) of cases. A total of 50.0% (52/104) of probands had early-onset (<70 years of age) disease, and 39.4% (41/104) of siblings had early-onset disease. Among probands, 8.7% (9/104) of the index strokes were cardioembolic, 27.9% (29/104) were large vessel, 31.7% (33/104) were small vessel, 26.0% (27/104) were of undetermined cause, and 5.8% (6/104) were of other determined cause. Among concordant siblings, 4.8% (5/104) of the index strokes were cardioembolic, 27.9% (29/104) were large vessel, 35.6% (37/104) were small vessel, 28.8% (30/104) were of undetermined cause, and 2.9% (3/104) were of other determined cause.
Table 2.
Demographics and Clinical Characteristics of SWISS Subjects
| Siblings
|
|||
|---|---|---|---|
| Characteristic | Probands (n = 104) | Concordant (n = 104) | Discordant (n = 41) |
| Age, mean ± SD, yr | 68.4 ± 11.0 | 70.3 ± 11.0 | 66.5 ± 11.6 |
| Sex, % male | 54 (51.9) | 56 (53.8) | 9 (22.0) |
| Race, % white | 97 (93.2) | 97 (93.2) | 39 (95.1) |
| Risk factors, n/N, % | |||
| Myocardial infarction | 22/103 (21.4) | 28/102 (27.5) | 6/41 (14.6) |
| Chronic AF | 8/104 (7.7) | 22/101 (21.8) | 3/41 (7.3) |
| Paroxysmal AF | 6/103 (5.8) | 5/102 (4.9) | 5/41 (12.2) |
| Ever smoking | 73/101 (72.3) | 85/101 (84.2) | 25/41 (61.0) |
| Hypertension | 76/104 (73.1) | 75/103 (72.8) | 22/41 (57.3) |
| Hyperlipidemia | 67/104 (64.4) | 69/103 (67.0) | 19/41 (46.3) |
| Diabetes mellitus | 28/104 (26.9) | 29/103 (28.2) | 1/41 (2.4) |
AF = atrial fibrillation; SWISS = Siblings With Ischemic Stroke Study.
Tests of deviation from Hardy-Weinberg equilibrium proportions were performed on the entire sample and within strata defined by ethnicity (white, other) and sex (male, female). Significant (p < 0.002) deviations from expected proportions were observed for SNP56 (rs702553) of the PDE4D gene in the total group, within whites (p = 0.09 in non-whites), and within each sex. Within ALOX5AP, SG13S89 deviated significantly (p < 0.05) by ethnic group and by sex from Hardy-Weinberg expectations for the total sample. Thus, results from these markers should be viewed with caution. All other markers within PDE4D and ALOX5AP did not deviate significantly from Hardy–Weinberg expectations or had sporadic deviations.
To test linkage for ischemic stroke at the PDE4D and ALOX5AP loci, we performed single-point and multipoint analyses (Figs 1 and 2). For PDE4D, single-point linkage analyses produced a maximum logarithm of odds score of 0.25 for D5S2091. For ALOX5AP, single-point linkage analyses produced a maximum logarithm of odds score of 0.14 at marker SG13S114. Multipoint linkage analyses for both PDE4D and ALOX5AP produced no positive logarithm of odds scores. There was no significant difference in allele sharing from expectation under the hypothesis of “no linkage” across markers tested for either ALOX5AP or PDE4D loci. Thus, there was no evidence supporting linkage of either ALOX5AP or PDE4D with ischemic stroke susceptibility in these data.
Fig 1.
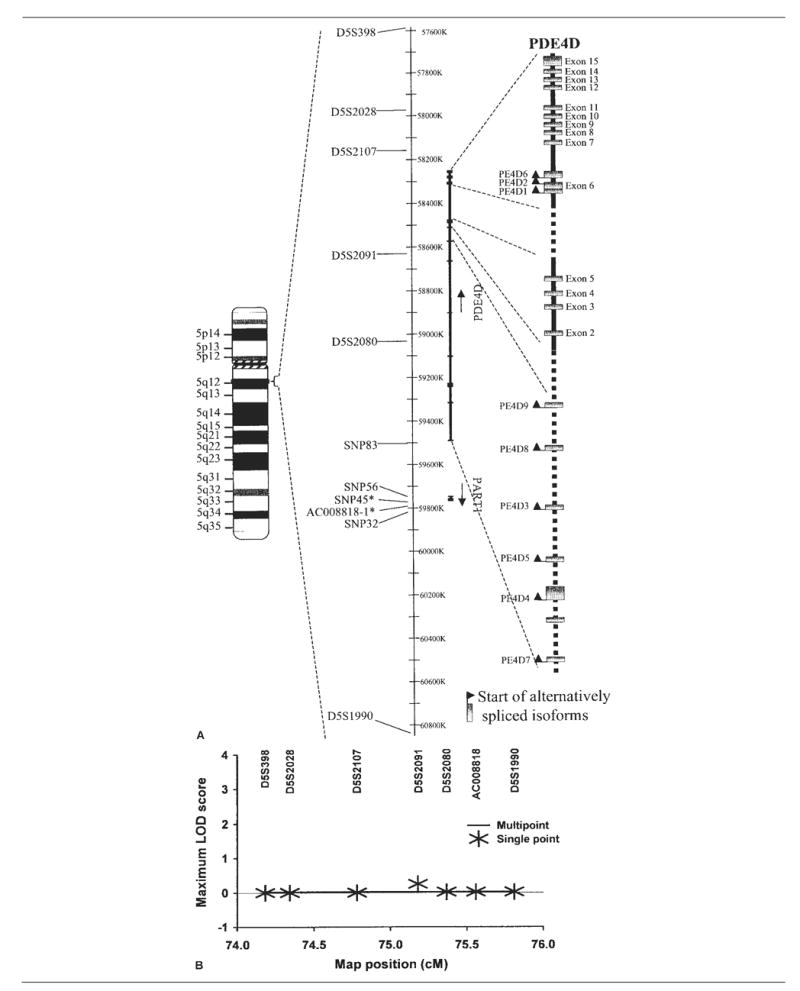
Analysis of PDE4D polymorphisms and risk for ischemic stroke. (A) Ideogram showing the relative locations of the PDE4D and PART1 genes on chromosome 5q12 (left), polymorphisms genotyped (middle), and gene transcripts and alternatively spliced isoforms of PDE4D (right). The single-nucleotide polymorphism indicated by an asterisk and short tandem repeat form the haplotype previously reported. (B) Single-point and multipoint linkage analyses of ischemic stroke risk were performed using GeneHunter-Plus in Siblings With Ischemic Stroke Study (SWISS) affected sibling pairs. The framework map was based on that from deCODE, with AC008818 placed using physical map coordinates and marker order as shown. LOD = logarithm of odds.
Fig 2.
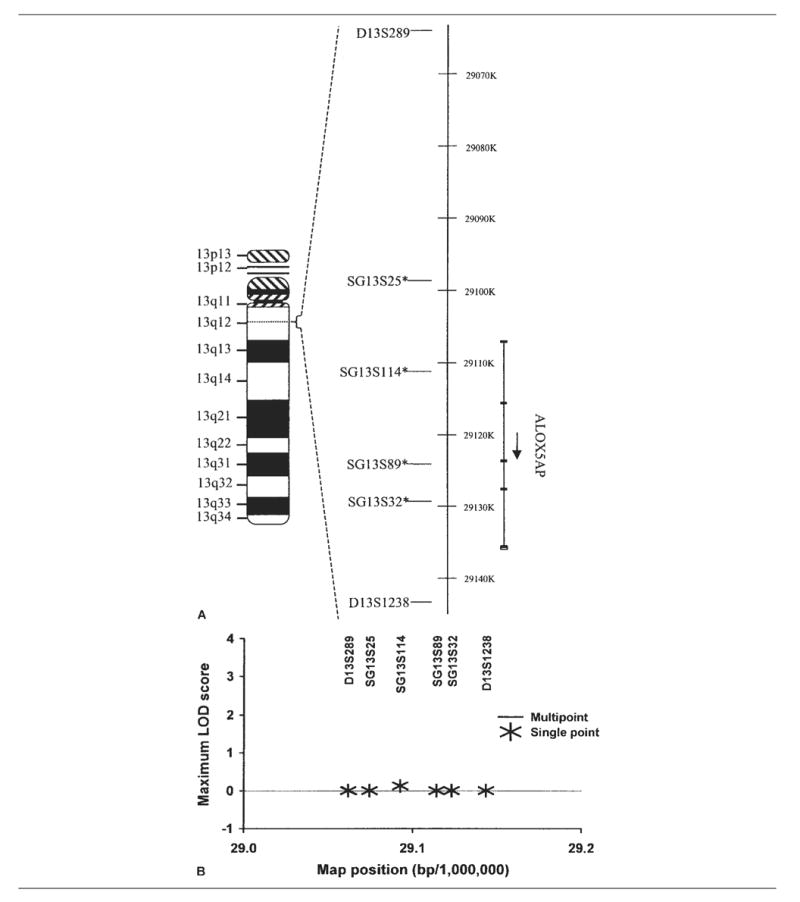
Analysis of 5-lipoxygenase activating protein (ALOX5AP) and risk for ischemic stroke. (A) Ideogram showing the relative locations of the ALOX5AP gene on chromosome 13q12 (left), polymorphisms genotyped (middle), and gene transcripts (right). The single-nucleotide polymorphisms indicated by asterisks form the haplotype previously reported. (B) Single-point and multi-point linkage analyses of ischemic stroke risk were performed using GeneHunter-Plus in Siblings With Ischemic Stroke Study (SWISS) affected sibling pairs. The framework map was based on that from deCODE, with marker order as shown. LOD = logarithm of odds.
Table 3 summarizes the demographic and clinical characteristics of the 640 subjects (377 cases and 263 control subjects) from the ISGS and MSGD studies who were analyzed in the association component of this study. A total of 74.0% (279/377) of cases identified themselves as white, and 26.0% (98/377) identified themselves as nonwhite. A total of 56.8% (214/377) of cases had early-onset (<70 years of age) disease. TOAST subtypes were available for the ISGS participants. A total of 24.9% (82/329) of the index strokes were cardioembolic, 19.5% (64/329) were large vessel, 17.3% (57/329) were small vessel, 34.0% (112/329) were of undetermined cause, and 4.3% (14/329) were of other determined cause.
Table 3.
Demographics and Clinical Characteristics of Subjects in the MSGD and ISGS
| Characteristic | Cases (n = 377) | Controls (n = 263) | p |
|---|---|---|---|
| Age, mean ± SD, yr | 64.8 ± 15.0 | 60.0 ± 14.7 | <0.001 |
| Sex, % male | 202 (53.6) | 100 (38.0) | <0.001 |
| Race, % white | 279 (74.0) | 210 (79.9) | 0.09 |
| Risk factors, n/N, % | |||
| Myocardial infarction | 59/376 (15.7) | 15/263 (5.7) | <0.001 |
| Chronic AF | 27/373 (7.2) | 10/262 (3.8) | 0.07 |
| Paroxysmal AF | 35/372 (9.4) | 9/262 (3.4) | 0.004 |
| Ever smoking | 257/377 (68.2) | 124/263 (47.1) | <0.001 |
| Hypertension | 257/375 (68.5) | 101/260 (38.8) | <0.001 |
| Diabetes mellitus | 90/377 (23.9) | 34/263 (12.9) | <0.001 |
AF = atrial fibrillation; ISGS = Ischemic Stroke Genetics Study; MSGD = Mayo Stroke Genetics Data Bank.
Four SNPs in the ALOX5AP locus (SG13S25, SG13S114, SG13S89, and SG13S32) were evaluated for association with ischemic stroke. After adjustment for age, sex, ethnicity, origin of DNA sample (ISGS or MSGD), family history of stroke, hypertension, atrial fibrillation, myocardial infarction, and smoking status, no significant association with ischemic stroke was observed (Table 4). In addition, using haplotype frequencies estimated from case-control data, we determined that there were no significant haplotype effects on the baseline risk for ischemic stroke. Given the absence of either linkage with stroke or association with stroke for any ALOX5AP SNP, there appears to be little evidence of ALOX5AP in the cause of ischemic stroke risk in this collection.
Table 4.
Association Analyses: ISGS and MSGD Cohorts
| No. of Individuals (case/control) by Genotype
|
|||||||
|---|---|---|---|---|---|---|---|
| Candidate Gene | SNP | Analysis | 1/1a | 1/2 | 2/2 | OR for Risk Genotype (95% CI)b | p |
| PDE4D | rs966221 (SNP83) | Overall | 164/90 | 139/120 | 55/46 | 1.68 (1.00–2.84) | 0.03 |
| White | 129/73 | 102/97 | 33/33 | 2.02 (1.09–3.76) | 0.02 | ||
| Nonwhite | 35/17 | 37/23 | 22/13 | 2.00 (0.63–6.38) | 0.33 | ||
| rs702553 (SNP56) | Overall | 136/97 | 151/90 | 68/67 | 1.63 (1.01–2.62) | 0.04 | |
| White | 100/79 | 112/69 | 52/53 | 1.58 (0.92–2.70) | 0.07 | ||
| Nonwhite | 36/18 | 39/21 | 16/14 | 1.86 (0.61–5.66) | 0.54 | ||
| SNP45 | Overall | 186/113 | 165/120 | 25/29 | 1.73 (0.91–3.28) | 0.13 | |
| White | 138/97 | 124/90 | 17/23 | 1.75 (0.84–3.65) | 0.31 | ||
| Nonwhite | 48/16 | 41/30 | 8/6 | 1.92 (0.43–8.54) | 0.25 | ||
| rs456009 (SNP32) | Overall | 154/112 | 173/109 | 47/42 | 1.21 (0.70–2.07) | 0.14 | |
| White | 107/80 | 135/96 | 35/34 | 1.44 (0.77–2.66) | 0.28 | ||
| Nonwhite | 47/32 | 38/13 | 12/8 | 0.79 (0.22–2.76) | 0.27 | ||
| ALOX5AP | SG13S25 | Overall | 307/201 | 57/50 | 4/1 | 0.23 (0.02–2.29) | 0.20 |
| White | 218/152 | 52/47 | 4/1 | 0.23 (0.02–2.35) | 0.18 | ||
| Nonwhite | 89/49 | 5/3 | 0/0 | 0.59 (0.10–3.29) | — | ||
| SG13S114 | Overall | 140/97 | 144/112 | 83/50 | 1.60 (0.87–2.92) | 0.18 | |
| White | 136/94 | 113/93 | 21/19 | 2.24 (1.04–4.85) | 0.10 | ||
| Nonwhite | 62/31 | 31/19 | 4/3 | 2.39 (0.30–19.15) | 0.58 | ||
| SG13S89 | Overall | 287/221 | 71/34 | 11/6 | 0.76 (0.25–2.30) | 0.15 | |
| White | 215/176 | 53/28 | 8/4 | 0.61 (0.16–2.24) | 0.25 | ||
| Nonwhite | 72/45 | 18/6 | 3/2 | 1.79 (0.20–15.94) | 0.48 | ||
| SG13S32 | Overall | 95/70 | 180/129 | 101/63 | 1.15 (0.70–1.87) | 0.79 | |
| White | 73/58 | 138/106 | 68/45 | 0.99 (0.57–1.73) | 0.98 | ||
| Nonwhite | 22/12 | 42/23 | 33/18 | 2.03 (0.62–6.69) | 0.46 | ||
The 1 allele is the more common allele, and the 2 allele is the less common allele.
Covariates in model include age, sex, race, cohort, family history, hypertension, atrial fibrillation, myocardial infarction, and smoker.
CI = confidence interval; ISGS = Ischemic Stroke Genetics Study; MSGD = Mayo Stroke Genetics Data Bank; SNP = single nucleotide polymorphism; OR = odds ratio.
Four SNPs in PDE4D (rs966221, SNP83; rs702553, SNP56; SNP45; rs456009, SNP32) were tested for association with ischemic stroke in the ISGS-MSGD sample of cases and control subjects (see Table 4). After adjustment for the same set of covariates, the independent effect of PDE4D genotype on risk for ischemic stroke was significantly increased for SNP83 (p = 0.03) and SNP56 (p = 0.04). The two-marker haplotype (SNP83, SNP56) was also significantly associated with risk for ischemic stroke (p = 0.04). The “high-risk” haplotype previously observed in the Icelandic population (SNP45, AC008818) was not significantly associated with increased risk (p = 0.09). When the analyses were restricted to the white population, the association remained significant for SNP83 (p = 0.02) and approached significance for SNP56 (p = 0.07).
The distribution of PDE4D SNP genotypes was examined across TOAST subtypes within cases. The frequency of genotypes for SNP83, SNP56, SNP45, and SNP32 did not differ across subtypes. Within each TOAST subtype, the individual PDE4D and ALOX5AP SNPs were used to predict case status (compared with all 263 control subjects), using the same covariates as applied previously. The risk for ischemic stroke by TOAST subtype did not differ for ALOX5AP SNPs. For PDE4D, there was a significant (p < 0.01) association with SNP83 in the large artery group (64 cases), with risk being associated with an additive model (common risk allele frequency of 0.63) and homozygote carrier risk (odds ratio) of 4.44 and heterozygote risk of 1.82. There was no significant association with SNP56 by TOAST subtype. SNP45 exhibited significant (p = 0.01) association with ischemic stroke risk within the combined phenotype (146 cases with large-vessel or cardioembolic subtypes), consistent with an additive risk model (common risk allele frequency of 0.69) and homozygote carrier risk of 3.90 and heterozygote risk of 2.17. For this SNP, the cardioembolic category (82 cases) had marginal (p = 0.05) association, consistent with an additive risk model; the large-vessel subtype was not associated with ischemic stroke for SNP45. Finally, there was an association of ischemic stroke with SNP32 within the large-vessel subgroup (p = 0.02); however, the observed risk was not consistent with a dominant, additive, or recessive risk model. In summary, the results of association of SNPs within ALOX5AP and PDE4D by TOAST subtype suggest the following: (1) there is no significant association of ischemic stroke with ALOX5AP SNPs; (2) there is differential association of ischemic stroke with PDE4D SNPs by TOAST subtype; (3) PDE4D contributes to ischemic stroke risk most significantly in those with large-vessel disease; and (4) the PDE4D SNPs that are most associated with large-vessel ischemic stroke risk are SNP83 and SNP45, with a common (approximately 65% frequency) risk allele that acts in an additive manner.
Discussion
Our results indicate that variation within the ALOX5AP and PDE4D loci, previously implicated as genetic risk factors for ischemic stroke in Iceland, do not appear to be linked in a North American population. There is strong evidence, however, that specific variants in PDE4D, but not ALOX5AP, are associated with ischemic stroke.
Earlier reports of the role of PDE4D and ALOX5AP in risk for ischemic stroke were based on an initial linkage analysis of a highly characterized set of kindreds from Iceland that were integrated into a large genealogy. The regions of linkage were followed by dense marker mapping and positional candidate analysis. In this research, these loci were evaluated for linkage using a subset of genetic markers implemented in the Icelandic study, but in a set of North American affected sibling pairs. The absence of support for linkage for PDE4D and ALOX5AP would argue against a highly penetrant rare variant (with a substantial genetic risk ratio, λs). Although the number of affected sibling pair families in this study was not large (~100 pairs), the power to confirm PDE4D or ALOX5AP with a modest locus-specific λs (~2) is more than 95% under Lander and Kruglyak’s criteria18 for confirmation of linkage (p = 0.01).
In this set of two case-control collections, the finding of an association with markers of PDE4D, but not with ALOX5AP, suggests that these PDE4D variants may be common, with minor effects on risk in this North American cohort. To focus specifically on the effect of the candidate loci, we included ischemic stroke risk factors as covariates in the analyses. These data indicate that specific variants within PDE4D, singly or in combination as a part of haplotypes, do confer a significantly increased risk for ischemic stroke. In the genetic models tested, the risk allele appears to be common in the population (approximately 60%) and increases the risk for ischemic stroke by approximately 75% over baseline (those without the risk allele). This association study has outstanding power (>95%) to replicate the findings for odds ratios greater than 2.0, which is consistent with those observed in the Icelandic studies. Thus, the power to detect association of individual SNPs or haplotypes in PDE4D remains high (even with a less strong association), suggesting that variants in PDE4D contribute to ischemic stroke risk in the North American population.
Our association study may have yielded false-positive results due to study sample stratification. However, we are reassured that our findings are varied, because a significant association remained for SNP83 among white subjects in the race-specific analysis. The absence of significant associations for PDE4D SNPs in nonwhite subjects may be due to small sample size. Larger studies in nonwhite populations are warranted.
Our results have implications for the interpretation of PDE4D and ALOX5AP or genes proximal to these loci and the risk for ischemic stroke. Several possibilities may explain these differing results. First, these loci could be participating in a large biological pathway that has multiple contributors to genetic risk for stroke. ALOX5AP may contribute substantially to stroke risk in Iceland, but it may play a smaller role in the North American population. Although we have demonstrated that the analyses performed in this study have substantial power to confirm the contribution of PDE4D and ALOX5AP to ischemic stroke risk, these estimates are based on the locus-specific effect sizes reported in Iceland. Should the effect of ALOX5AP be minor in the North American population under study, then our study may not possess sufficient power to detect such a minor effect. A larger association study performed in a Central European population detected a modest association of variants in ALOX5AP and ischemic stroke.19 Finally, the ALOX5AP result obtained by Gretarsdottir and colleagues6 and Helgadottir and colleagues7 may represent false-positive linkage and association; thus, as in our studied population, ALOX5AP may not be involved in genetic risk for ischemic stroke. Nonetheless, the confirmation of PDE4D as an ischemic stroke risk factor may provide a valuable tool for pre-symptomatic diagnosis and prediction of stroke risk and better identification of environmental factors that interact with PDE4D genetic risk in humans.
PDE4D is one of four genes belonging to a complex multigene family that encode several PDE4 isoforms. Genetic variants of PDE4D may effect differential expression of isoforms. PDE4 enzyme activity may modify stroke risk through its effects on inflammation20 atherosclerotic plaque stability,21,22 response to vascular injury,23 pathological angiogenesis,24,25 and risk for and response to low-grade infections such as those caused by Chlamydia pneumoniae.26 PDE4 is the predominant enzyme that metabolizes cyclic adenosine monophosphate in inflammatory cells.27 Inhibitors of PDE4 decrease leukocyte–endothelial cell interactions by reducing adhesion molecule expression in response to proinflammatory stimuli,28,29 and selective PDE4 inhibitors decrease smooth muscle cell activation after vascular injury in an animal model.23 PDE4 may become a novel pharmacological target for stroke prevention.
Appendix
Siblings With Ischemic Stroke Study Centers and Investigators Listed by Proband Enrollment as of February 7, 2005
Mayo Clinic, Rochester, MN (probands enrolled, 52)—Principal investigator (PI): Robert D. Brown, Jr., MD; Coordinator: Colleen S. Albers, RN; Subinvestigators (SI): George W. Petty, MD, Eelco F.M. Wijdicks, MD, Irene Meissner, MD, Bruce A. Evans, MD, Kelly D. Flemming, MD, Edward M. Manno, MD, Jimmy R. Fulgham, MD, David O. Wiebers, MD. University of Florida/Shands Hospital, Jacksonville, FL (39)—PI: Scott Silliman, MD; Coordinators: Barbara Quinn, RN, Cicely Bryant. Mayo Clinic, Jacksonville, FL (36)–PI: Thomas G. Brott, MD; Coordinator: Jacob Rosen-berg, CRC; SI: James F. Meschia, MD, Frank A. Rubino, MD, Benjamin H. Eidelman, MD. University of Virginia, Charlottesville, VA (34)–PI: Bradford Worrall, MD, MSc; Coordinator: Martha Davis, RN; SI: E. Clarke Haley, Jr., MD, Karen Johnston, MD, MSc, Jaclyn van Wingerden. University of Cincinnati, Cincinnati, OH (32)–PI: Brett Kissela, MD; Coordinator: Kathleen Alwell, RN; SI: Joseph Broderick, MD, Daniel Woo, MD, Daniel Kanter, MD, Dawn Kleindorfer, Alexander Schneider, MD, Matthew Flaherty, MD. Neurological Associates, Inc, Richmond, VA (28)–PI: Francis McGee, Jr., MD; Coordinators: Sharon McQueen-Goss, RN, Janet McGee, CCRC; SI: Stephen Thurston, MD, Thomas Smith, MD, Robert White, MD, Philip Davenport, MD, John Brush, MD, Susanna Mathe, MD, Robert Cohen, MD, J. Kim Harris, MD, John O’Bannon III, MD, John Blevins, MD. Mercy General Hospital, Sacramento, CA (16)–PI: Paul Akins, MD, PhD; Coordinator: Deidre Wentworth, RN. Maine Line Health Stroke Program, Bryn Mawr, PA (16)–PI: Gary Friday, MD; Coordinator: Angela Whittington-Smith, RN. Centre Hospitalier Affilie Universitaire de Québec, Québec City, PQ (13)–PI: Denis Simard, MD; Coordinators: Annette Hache, RN, Sophie Dube, RN; SI: Ariane Mackey, MD. Luther-Midelfort Clinic, Eau Claire, WI (13)—PI: Felix Chukwudelunzu, MD; Coordinators: Tonya Kunz, RN, Karen Snobl, RN; SI: James Bounds, MD, Rae Hanson, MD, David Nye, MD, Donn Dexter, MD. Wake Forest University School of Medicine, Winston-Salem, NC (11)—PI: David Lefkowitz, MD; Coordinators: Jean Satterfield, RN, Elizabeth Westerberg, CCRC; SI: Charles Tegeler IV, MD, Patrick Reynolds, MD. Maine Medical Center, Portland, ME (11)—PI: John Belden, MD; Coordinator: Diane Diconzo-Fanning, RN; SI: Paul Muscat, MD. Mayo Clinic, Scottsdale, AZ (11)—PI: David W. Dodick, MD; Coordinators: Erica Boyd, RN, Rebecca Rush, RN, Gail LeBrun, RN, Nadine Lendzion, RN, Barbara Cleary, RN; SI: Bart M. Demaerschalk, MD. Kaleida Stroke Center, Millard Fillmore Hospital, Buffalo, NY (11)—PI: F.E. Munschauer, MD, Coordinator: Kathleen Wrest, MLS; SI: Peterkin Lee-Kwen, MD. University of South Alabama, Mobile, AL (10)—PI: Richard Zweifler, MD; Coordinators: Robin Yunker, RNC, MSN, Mel Parnell, RN, BSN; SI: Ivan Lopez, MD, M. Asim Mahmood, MD. University of Pennsylvania Medical Center, Philadelphia, PA (10)—PI: Scott Kasner, MD; Coordinator: Jessica Clarke, RN; SI: David S. Liebeskind, MD, Brett L. Cucchiara, MD, Michael L. Mc-Garvey, MD, Steven R. Messe, MD, Robert A. Taylor, MD. Stroke Prevention and Atherosclerosis Research Centre (SPARC), London, ON (10)—PI: David Spence, MD; Coordinator: Rose Freitas; SI: Claudio Munoz, MD. Cleveland Clinic Florida, Weston, FL (9)—PI: Virgilio Salanga, MD; Coordinators: Anupama Podichetty, MD, Jose Alvarez, MD; SI: Eduardo Locatelli, MD, Nestor Galvez-Jimenez, MD, Efrain Salgado, MD. Hospital Charles Le Moyne, Greenfield Park, PQ (9)—PI: Leo Berger, MD; Coordinators: Martine Maineville, Denise Racicot. University of Iowa Hospital, Iowa City, IA (9)—PI: Patricia Davis, MD; Coordinator: Jeri Sieren, RN; SI: Harold P. Adams, Jr., MD. Metro Health Medical Center, Cleveland, OH (9)—PI: Joseph Hanna, MD; Coordinators: Alice Liskay, RN, Joan Kappler, RN, Dana Simcox, RN; SI: Marc Winkelman, MD; Nimish Thakore, MD, DM. University of Maryland, Baltimore, MD (9)—PI: Steven Kittner, MD; Coordinator: Mary J. Sparks; SI: John Cole, MD. Washington University School of Medicine, St. Louis, MO (8)—PI: Jin-Moo Lee, MD, PhD; Coordinator: Denise Shearrer, RMA, BS; SI: Abdullah Nassief, MD. University of Texas Southwestern Medical Center at Dallas, Dallas, TX (8)—PI: D. Hal Unwin, MD; Coordinator: J. Greggory Wright, BS; SI: Dion Graybeal, MD, Mark Johnson, MD, Mounzer Kassab, MD. Emory University School of Medicine, Atlanta, GA (7)—PI: Barney Stern, MD; Coordinator: Betty Jo Shipp, RN; SI: Michael Frankel, MD, Marc Chimowitz, MD, Owen Samuels, MD. University of California, Davis School of Medicine, Sacramento, CA (7)—PI: Piero Verro, MD; Coordinator: Shari Nichols. University of Wisconsin, Madison, WI (7)—PI: Robert Dempsey, MD; Coordinator: Pam Winne; SI: George Newman, MD, Douglas Dulli, MD, Madeleine Geraghty, MD. Indiana University School of Medicine, Indianapolis, IN (7)—PI: Linda Williams, MD; SI: Askiel Bruno, MD; William Jones, MD. Inova Fairfax Hospital, Church Falls, VA (7)—PI: Paul Nyquist, MD; Coordinator: Barbara Farmer, RN, MSN. Marshfield Clinic, Marshfield, WI (7)—PI: Percy Karanjia, MD; Coordinator: Kathy Mancl, CCRC; SI: Kenneth Madden, MD. East Bay Region Associates in Neurology, Berkeley, CA (6)—PI: Brian Richardson, MD; Coordinator: Lauren McCormick. Ohio State University, Columbus, OH (6)—PI: Andrew Slivka, MD; Coordinator: Peggy Notestine, CCRC; SI: Yousef Mohammad, MD. Mercy Ruan Center for Neurologic Research Des Moines, IA (5)—PI: Michael Jacoby, MD; Coordinator: Judi Greene, RN; SI: Bruce Hughes, MD, Randall Hamilton, MD, Paul Babikian, MD, Mark Puricelli, DO. Helen Hayes Hospital, West Haverstraw, NY (5)—PI: Laura Lennihan, MD; Coordinator: Laura Tenteromano, RN. Thomas Jefferson University Hospital, Philadelphia, PA (5)—PI: Rodney Bell, MD; Coordinator: Lisa Bowman, MNS, CRNP, CNRN; SI: David G. Brock, MD, Carissa Pineda, MD. Florida Neurovascular Institute, Tampa, FL (5)—PI: Erfan Albakri, MD; Coordinators: Taryn Chauncey, RN, Judy Jackson, Mary Katherine Taylor, ARNP. University of Kentucky, Lexington, KY (4)—PI: L. Creed Pettigrew, MD; Coordinator: DeborahTaylor, MS; SI: Stephen Ryan, MD, Anand G. Vaishnav, MD. Scripps Clinic, La Jolla, CA (4)—PI: Mary Kalafut, MD; Coordinator: Carmen James, RN; Joy Reyes. University of California Los Angeles Stroke Center, Los Angeles, CA (4)—PI: Jeffery Saver, MD; Coordinator: Gina Paek, BA; SI: Bruce Ovbiagele, MD, Scott Selco, MD, Venkatakrishna Rajajee, MD. Field Neurosciences Institute, Saginaw, MI (3)—PI: Faith Abbott, MD; Coordinator: Richard Herm, RN, BSN, CEN; SI: Malcolm Field, MD, Debasish Mridha, MD. Johns Hopkins Bayview Medical Center, Baltimore, MD (3)—PI: Rafael Llinas, MD; Coordinator: Janice Alt; SI: Christopher Earley, MD. Medical University of South Carolina, Charleston, SC (3)—PI: Timothy Carter, MD. Royal University Hospital, Saskatoon, SK (3)—PI: Ali Rajput, MD; Coordinator: Theresa Shirley, RN; SI: Alexander Rajput, MD. Rush-Presbyterian-St Luke’s Medical Center, Chicago, IL (3)—PI: Sean Ruland, DOMD; Coordinator: Karen Whited, RN; SI: Michael Schneck, MD; Michael Sloan, MD, Phillip Gorelick, MD, MPH. University of California San Diego Stroke Center, San Diego, CA (3)—PI: Patrick Lyden, MD; Coordinators: Nancy Kelly, RN, Janet Werner, RN; SI: Christy Jackson, MD, Thomas Hemmen, MD, Brett Meyer, MD. University of Illinois at Chicago, Chicago, IL (3)—PI: Cathy Helgason, MD; Coordinator: Joan N. Martellotto, RN, PhD. Yale University School of Medicine, New Haven, CT (3)—PI: Mark Gorman, MD; Coordinator: Janet Halliday, RN, BS. Chattanooga Neurology Associates, Chattanooga, TN (3)—PI: Thomas Devlin, MD; Coordinators: Patty Wade-Hardie, RN, Tammy Owens, RN; SI: Adele Ackell, MD, Sharon Farber, MD, Ravi Chander, MD, G. Hagan Jackson, MD, Kadrie Hytham, MD; Bruce Kaplan, MD, David Rankine, MD. Charles R. Drew University of Medicine and Science, Los Angeles, CA (1)—PI: George Locke, MD; Coordinators: Marcia Montenegro, RN, Derek Knight; SI: Lowell Nelson, PhD. Rhode Island Hospital, Providence, RI (1)—Janet Wilterdink, MD; Coordinator: Carol Cirillo, RN. Syracuse VA Medical Center, Syracuse, NY (1)—PI: Antonio Culebras, MD; Coordinator: Therese Dean, RN.
Ischemic Stroke Genetics Study Centers and Invesgators as of October 6, 2005
Mayo Clinic, Jacksonville, Florida (subjects enrolled, 188): Principal Investigator (PI): James F. Meschia, MD; coordinators: Alexa Richie, Dale Gamble; subinvestigators (SI): Thomas G. Brott, MD, Benjamin H Eidelman, MD, Pablo R. Castillo, MD, Frank A. Rubino, MD. University of Florida/Shands Hospital, Jacksonville, Florida (216): PI: Scott Silliman, MD; coordinators: Barbara Quinn, RN, Yvonne Douglas, Marc Lojacono, CCRC; SI: Nader Antonios, MD. Emory University School of Medicine, Atlanta, Georgia (228): PI: Michael R. Frankel, MD; coordinator, Sharion Smith, RN. University of Virginia, Charlottesville, Virginia (212): PI: Bradford B. Worrall, MD, MSc; coordinators: Martha Davis, RN, Helen Roehl, RN. Mayo Clinic, Rochester, Minnesota (228): PI: Robert D. Brown, Jr, MD; coordinators: Colleen S. Albers, RN, Debra E. Herzig, RN.
The Mayo Stroke Genetics Databank investigators are James F. Meschia, MD (Principal Investigator); Thomas G. Brott, MD; Frank A. Rubino, MD; Marc Lojacono; Elizabeth J. Atkinson, MS; Richard Crook; John Hardy, PhD.
Footnotes
Members of the SWISS, ISGS and MSGD Study Groups are listed in the Appendix on page 359–360.
This study was supported by the NIH (National Institute of Neurological Disorders and Stroke, R01-NS39987; R01-NS42733, J.F.M., Swiss, ISGS) and the Mayo Foundation for Medical Education and Research (Protocol Institutional Review Board No. 0-1806-00, M.S.G.D.).
References
- 1.Murray CJL, Lopez AD. Mortality by cause for eight regions of the world: Global Burden of Disease Study. Lancet. 1997;349:1269–1276. doi: 10.1016/S0140-6736(96)07493-4. [DOI] [PubMed] [Google Scholar]
- 2.Rothwell PM, Coull AJ, Giles MF, et al. for the Oxford Vascular Study. Change in stroke incidence, mortality, case-fatality, severity, and risk factors in Oxfordshire, UK from 1981 to 2004 (Oxford Vascular Study) Lancet. 2004;363:1925–1933. doi: 10.1016/S0140-6736(04)16405-2. [DOI] [PubMed] [Google Scholar]
- 3.Brown RD, Whisnant JP, Sicks JD, et al. Stroke incidence, prevalence, and survival: secular trends in Rochester, Minnesota, through 1989. Stroke. 1996;27:373–380. [PubMed] [Google Scholar]
- 4.Flossmann E, Schulz UG, Rothwell PM. Systematic review of methods and results of studies of the genetic epidemiology of ischemic stroke. Stroke. 2004;35:212–227. doi: 10.1161/01.STR.0000107187.84390.AA. [DOI] [PubMed] [Google Scholar]
- 5.Gretarsdottir S, Sveinbjornsdottir S, Jonsson HH, et al. Localization of a susceptibility gene for common forms of stroke to 5q12. Am J Hum Genet. 2002;70:593–603. doi: 10.1086/339252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gretarsdottir S, Thorleifsson G, Reynisdottir ST, et al. The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet. 2003;35:131–138. doi: 10.1038/ng1245. [DOI] [PubMed] [Google Scholar]
- 7.Helgadottir A, Manolescu A, Thorleifsson G, et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet. 2004;36:233–239. doi: 10.1038/ng1311. [DOI] [PubMed] [Google Scholar]
- 8.Helgadottir A, Gretarsdottir S, St Clair D, et al. Association between the gene encoding 5-lipoxygenase activating protein and stroke replicated in a Scottish population. Am J Hum Genet. 2005;76:505–509. doi: 10.1086/428066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meschia JF, Brott TG, Brown RD, Jr, et al. The Ischemic Stroke Genetics Study (ISGS) protocol. BMC Neurol. 2003;3:4. doi: 10.1186/1471-2377-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meschia JF, Brown RD, Jr, Brott TG, et al. The Siblings With Ischemic Stroke Study (SWISS) protocol. BMC Med Genet. 2002;3:1. doi: 10.1186/1471-2350-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.WHO MONICA Project Principal Investigators. The World Health Organization MONICA Project (monitoring trends and determinants in cardiovascular disease): a major international collaboration. J Clin Epidemiol. 1988;41:105–114. doi: 10.1016/0895-4356(88)90084-4. [DOI] [PubMed] [Google Scholar]
- 12.Adams HP, Jr, Bendixen BH, Kappelle LJ, et al. Classification of subtype of acute ischemic stroke: definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke. 1993;24:35–41. doi: 10.1161/01.str.24.1.35. [DOI] [PubMed] [Google Scholar]
- 13.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 15.Kong A, Cox NJ. Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang K-Y, Zeger SL. Longitudinal data analysis using generalized linear models. Biometrika. 1986;73:13–22. [Google Scholar]
- 17.Green LE, Lange EM, Langefeld CD. Power comparison of phase-known versus phase-unknown haplotype analyses for case-control designs. Am J Hum Genet. 2001;69(suppl):1948. (Abstract) [Google Scholar]
- 18.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 19.Lõhmussaar E, Gschwendtner A, Mueller JC, et al. ALOX5AP gene and the PDE4D gene in a Central European population of stroke patients. Stroke. 2005;36:731–736. doi: 10.1161/01.STR.0000157587.59821.87. [DOI] [PubMed] [Google Scholar]
- 20.Ariga M, Neitzert B, Nakae S, et al. Nonredundant function of phosphodiesterases 4D and 4B in neutrophil recruitment to the site of inflammation. J Immunol. 2004;173:7531–7538. doi: 10.4049/jimmunol.173.12.7531. [DOI] [PubMed] [Google Scholar]
- 21.Kohyama T, Liu X, Wen F-Q, et al. PDE4 inhibitors attenuate fibroblast chemotaxis and contraction of native collagen gels. Am J Respir Cell Mol Biol. 2002;26:694–701. doi: 10.1165/ajrcmb.26.6.4743. [DOI] [PubMed] [Google Scholar]
- 22.Banner KH, Trevethick MA. PDE4 inhibition: a novel approach for the treatment of inflammatory bowel disease. Trends Pharmacol Sci. 2004;25:430–436. doi: 10.1016/j.tips.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 23.Kondo K, Umemura K, Miyaji M, Nakashima M. Milrinone, a phosphodiesterase inhibitor, suppresses intimal thickening after photochemically induced endothelial injury in the mouse femoral artery. Atherosclerosis. 1999;142:133–138. doi: 10.1016/s0021-9150(98)00203-2. [DOI] [PubMed] [Google Scholar]
- 24.Favot L, Keravis T, Holl V, et al. VEGF-induced HUVEC migration and proliferation are decreased by PDE2 and PDE4 inhibitors. Thromb Hemost. 2003;90:334–343. doi: 10.1160/TH03-02-0084. [DOI] [PubMed] [Google Scholar]
- 25.Favot L, Keravis T, Lugnier C. Modulation of VEGF-induced endothelial cell cycle protein expression through cyclic AMP hydrolysis by PDE2 and PDE4. Thromb Haemost. 2004;92:634–645. doi: 10.1160/TH03-12-0768. [DOI] [PubMed] [Google Scholar]
- 26.Haralambieva IH, Iankov ID, Ivanova PV, et al. Chlamydophila pneumoniae induces p44/p42 mitogen-activated protein kinase activation in human fibroblasts through Toll-like receptor 4. J Med Microbiol. 2004;53:1187–1193. doi: 10.1099/jmm.0.45758-0. [DOI] [PubMed] [Google Scholar]
- 27.Jacob C, Martin-Chouly C, Lagente V. Type 4 phosphodiesterase-dependent pathways: role in inflammatory processes. Therapie. 2002;57:163–168. [PubMed] [Google Scholar]
- 28.Sanz MJ, Alvarez A, Piqueras L, et al. Rolipram inhibits leukocyte-endothelial cell interactions in vivo through P- and E-selectin downregulation. Br J Pharmacol. 2002;135:1872–1881. doi: 10.1038/sj.bjp.0704644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blease K, Burke-Gaffney A, Hellewell PG. Modulation of cell adhesion molecule expression and function on human lung microvascular endothelial cells by inhibition of phosphodiesterases 3 and 4. Br J Pharmacol. 1998;124:229–237. doi: 10.1038/sj.bjp.0701833. [DOI] [PMC free article] [PubMed] [Google Scholar]