

Abstract
Male-killing bacteria are maternally inherited endosymbionts that selectively kill male offspring of their arthropod hosts. Using both analytical techniques and computer simulations, we studied the impact of these bacteria on the population genetics of their hosts. In particular, we derived and corroborated formulas for the fixation probability of mutant alleles, mean times to fixation and fixation or extinction, and heterozygosity for varying male-killer prevalence. Our results demonstrate that infections with male-killing bacteria impede the spread of beneficial alleles, facilitate the spread of deleterious alleles, and reduce genetic variation. The reason for this lies in the strongly reduced fitness of infected females combined with no or very limited gene flow from infected females to uninfected individuals. These two properties of male-killer-infected populations reduce the population size relevant for the initial emergence and spread of mutations. In contrast, use of Wright's equation relating sex ratio to effective population size produces misleading predictions. We discuss the relationship to the similar effect of background selection, the impact of other sex-ratio-distorting endosymbionts, and how our results affect the interpretation of empirical data on genetic variation in male-killer-infected populations.
MALE-KILLING bacteria are maternally inherited endosymbionts that selectively kill male hosts early in their development. They have been reported in many insect species from all major orders and also in pseudoscorpions (e.g., Williamson and Poulson 1979; Werren et al. 1986; Hurst et al. 1999; Fialho and Stevens 2000; Montenegro et al. 2000; Zeh et al. 2005). Male-killers are taxonomically diverse and include members of the bacterial genera Wolbachia, Rickettsia, Spiroplasma, and Arsenophonus (reviewed in Hurst et al. 2003). The prevalence of male-killing bacteria varies considerably both between and within species, with values ranging from close to 0% to 99%. Because of the maternal mode of transmission, male-killing is not selectively disfavored in the bacteria, and it can even be a selectively advantageous behavior if sisters benefit from the death of their brothers (Hurst and Majerus 1993; Jaenike et al. 2003).
It has been suggested that male-killing bacteria play an important role in shaping the ecology and evolution of their hosts (Hurst and McVean 1998; Jiggins et al. 2000; Randerson et al. 2000; Normark 2004; Engelstädter and Hurst 2006). One aspect that has attracted attention recently is the impact of male-killing infections on the hosts' population genetics. However, almost all studies conducted so far have focused on the population genetics of the mitochondrial genome, because of the obvious impact of disequilibrium with any other maternally inherited agents (Johnstone and Hurst 1996; Schulenburg et al. 2002; Jiggins 2003; Dyer and Jaenike 2004; Hurst and Jiggins 2005; Jiggins and Tinsley 2005). To our knowledge, there is only one study on the impact of male-killing bacteria on the population genetics of nuclear genes (Telschow et al. 2006). In this theoretical investigation, the process of host adaptation was analyzed in a two-island model, and it was demonstrated that adaptation can be impeded when one population is infected with male-killing bacteria and the other is not infected.
Here, we investigate the interplay between selection and drift acting on nuclear alleles in populations infected with male-killing bacteria. We present both analytic approximations and simulation results on the probability of fixation of mutant alleles, the rate of evolution, and expected times until fixation or extinction of alleles. We demonstrate that with respect to these quantities, a male-killer-infected population behaves approximately as it would if it consisted of uninfected individuals only. This means, for example, that the probability of fixation of deleterious alleles is increased in infected compared to uninfected populations, while beneficial alleles have a lower chance of fixation in infected populations. This effect is not due to the distorted population sex ratio, but stems from the peculiar inheritance pattern of nuclear alleles in male-killer-infected populations, in which gene flow from infected to uninfected individuals is constrained. The effects are observed even when the male-killer is inefficiently transmitted.
THE MODEL
We assume a host population of diploid individuals that reproduce sexually in discrete, nonoverlapping generations. Each individual is characterized by its sex, its genotype, and its infection state (infected or not infected with male-killing bacteria). For the genotype, we consider one autosomal locus with two alleles, the wild type and a mutant allele. Starting with N adult individuals, we assume a life cycle that consists of the following steps:
Random mating: Genotype–cytotype frequencies of the zygotes in the next generation are obtained. We assume fair meiosis, i.e., the genotype frequencies in the gametes are the same as in the parent population. The primary sex ratio is assumed to be 1:1. Transmission of the male-killing bacteria is strictly maternal. Uninfected females produce uninfected eggs only, while infected females produce a proportion α of infected eggs and 1 − α uninfected eggs.
Male-killing: All infected male zygotes are killed by the male-killing bacteria.
Fitness compensation: Following Hurst (1991), the fitness of siblings of killed males is increased by a factor [1 + bα/(2 − α)]. Here, we assume that the resources freed by the death of males are equally distributed among the surviving siblings and that fitness increases linearly with the amount of resources. The parameter b gives the fraction of resources freed by the death of males that is reallocated to increase the surviving siblings' fitness, and the transmission rate α gives the proportion of male zygotes that are killed.
Selection according to genotype: We assume that individuals that are homozygous for the mutant allele have their fitness altered by the factor 1 + s, and heterozygous individuals have their fitness altered by 1 + hs. s can take both negative and positive values, reflecting deleterious and beneficial mutations, respectively.
Random survival: On the basis of the genotype–cytotype frequencies obtained in the above steps of the life cycle, N new mature adult individuals are randomly drawn. We thus assume that the prevalence of the male-killing bacteria does not affect total population size.
The parameters of the model are summarized in Table 1, and the variables that we use to describe properties of the host population are given in Table 2.
TABLE 1.
Parameters of the model
| Parameter | Description |
|---|---|
| N | Population size at the stage of reproducing adults (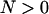 ) ) |
| s | Selection coefficient of the mutant allele (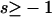 ) ) |
| h | Dominance level: heterozygotes have fitness of 1 + hs (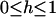 ) ) |
| α | Transmission rate of the male-killing bacteria (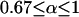 ) ) |
| b | Fitness compensation parameter: gives the amount of resources that are reallocated from dead males to their surviving siblings (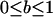 ) ) |
| μ | Mutation rate per allele per generation |
TABLE 2.
Variables of the model
| Variable | Description |
|---|---|
| y, y* | Actual and equilibrium prevalence of the male-killing bacteria (i.e., fraction of infected females among all females in the population) |
| u0, uMK | Fixation probability of newly arisen mutations in uninfected and male-killer-infected populations, respectively |
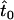 , , 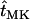
|
Expected times until fixation of new mutations, provided fixation occurs |
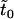 , , 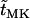
|
Expected times until either fixation or extinction of new mutations |
| FMK, HMK | Expected homozygosity and heterozygosity in a male-killer-infected population |
Note that y is used as a parameter in the first part of our model (when transmission is perfect), but is a variable in the second part (when transmission is imperfect).
PERFECT TRANSMISSION
Throughout this section we assume that all offspring of infected females are again infected (i.e., 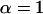 ) and that daughters from infected mothers do not receive any fitness benefit through the death of their brothers (i.e., b = 0). The assumption of perfect transmission not only is convenient for the derivation of analytic approximations, but also is a situation found in some natural populations infected with male-killing bacteria (e.g., Jiggins et al. 2002; Dyson and Hurst 2004). Moreover, we assume that the prevalence of the male-killing bacteria and the population sex ratio remain constant over the time frame under consideration.
) and that daughters from infected mothers do not receive any fitness benefit through the death of their brothers (i.e., b = 0). The assumption of perfect transmission not only is convenient for the derivation of analytic approximations, but also is a situation found in some natural populations infected with male-killing bacteria (e.g., Jiggins et al. 2002; Dyson and Hurst 2004). Moreover, we assume that the prevalence of the male-killing bacteria and the population sex ratio remain constant over the time frame under consideration.
The probability of fixation of a mutant allele:
Following Kimura (1962) the fixation probability for a newly arisen mutant allele in an uninfected population with even sex ratio can be approximated by
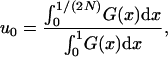 |
(1) |
with
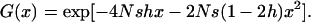 |
We now consider the case where the population is infected with a male-killing parasite with perfect maternal transmission. We assume that the prevalence of the male-killer is fixed at a proportion y of infected females among all females and the sex ratio is distorted accordingly. The relative proportions of infected females, uninfected females, and uninfected males in the adult population are then given by 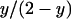 ,
, 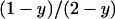 , and
, and 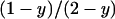 , respectively.
, respectively.
A newly arisen nuclear mutant allele will be found either in an infected female [with probability 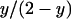 ] or in an uninfected individual [with probability
] or in an uninfected individual [with probability 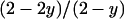 ]. However, if the mutant allele emerges in an infected female, the chances of quick extinction are extremely high unless the allele confers a very large fitness advantage to its carrier in the heterozygous state. This is simply because nuclear genes in infected females have a priori a fitness that is only about half that of the same allele in uninfected females (see Sullivan and Jaenike 2006 for a similar argument).
]. However, if the mutant allele emerges in an infected female, the chances of quick extinction are extremely high unless the allele confers a very large fitness advantage to its carrier in the heterozygous state. This is simply because nuclear genes in infected females have a priori a fitness that is only about half that of the same allele in uninfected females (see Sullivan and Jaenike 2006 for a similar argument).
We can thus assume that neutral alleles, detrimental alleles, and slightly or moderately beneficial alleles will invariably become extinct when they arise in infected females. Therefore, only alleles that arise in the uninfected proportion of the population have a chance of becoming fixed in the population. Since alleles can spread only from uninfected males to infected females, but not from infected females to uninfected individuals, the dynamics of new alleles are completely determined by the uninfected individuals in the population. Thus, building upon Equation 1, we can approximate the fixation probability of a newly arisen allele by
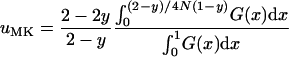 |
(2) |
with
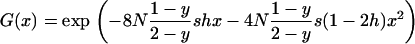 |
In this approximation, we take into account the probability that a mutation occurs in an uninfected individual and replace the population size N of Equation 1 with the number of uninfected individuals in the population, 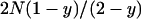 . Note that the sex ratio within the uninfected part of the population is even, so that no sex-ratio distortion needs to be taken into account.
. Note that the sex ratio within the uninfected part of the population is even, so that no sex-ratio distortion needs to be taken into account.
Unfortunately, in general Equation 2 cannot be simplified. However, we can shed light on the fixation probability of nuclear alleles in populations infected with male-killers by making two specific assumptions about the selection and dominance coefficients s and h.
Neutral alleles:
With s = 0, the function G(x) greatly simplifies to 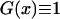 , yielding
, yielding 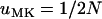 . As expected from neutral theory, this is the same fixation probability as for a neutral allele in an uninfected population.
. As expected from neutral theory, this is the same fixation probability as for a neutral allele in an uninfected population.
Semidominance (h = 0.5):
For a semidominant allele, the fixation probability in an infected population simplifies to
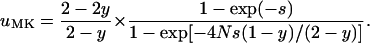 |
It can be proved that uMK < u0 always for s > 0, and uMK > u0 when s < 0 (see appendix). Thus, male-killing bacteria hinder the spread of beneficial and foster the spread of deleterious alleles. Moreover, for large values of Ns, 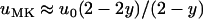 holds; i.e., the probability that a beneficial allele becomes fixed in a sufficiently large population is approximately the fraction of uninfected individuals multiplied by the probability of fixation of the same allele in an uninfected population of the same size.
holds; i.e., the probability that a beneficial allele becomes fixed in a sufficiently large population is approximately the fraction of uninfected individuals multiplied by the probability of fixation of the same allele in an uninfected population of the same size.
Figure 1 shows estimates of the fixation probability in populations carrying a male-killer relative to fixation probabilities in uninfected populations (i.e., 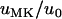 ) for different slices of the parameter space. Also shown are the average fixation rates observed in computer simulations, in which we ran at least 106 simulations per data point. As can be seen, the simulation results are in very good agreement with our analytic predictions. Finally, Figure 1 gives the predictions for the fixation probability derived from the application of Wright's equation relating effective population size to sex ratio,
) for different slices of the parameter space. Also shown are the average fixation rates observed in computer simulations, in which we ran at least 106 simulations per data point. As can be seen, the simulation results are in very good agreement with our analytic predictions. Finally, Figure 1 gives the predictions for the fixation probability derived from the application of Wright's equation relating effective population size to sex ratio, 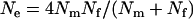 (Wright 1931). Here, the resulting formula in terms of the actual population size N and the male-killer prevalence y,
(Wright 1931). Here, the resulting formula in terms of the actual population size N and the male-killer prevalence y, 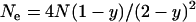 , was inserted into the function G(x) instead of N in Equation 1. As can be seen, the prediction based on the skewed sex ratio is not appropriate for male-killer-infected populations.
, was inserted into the function G(x) instead of N in Equation 1. As can be seen, the prediction based on the skewed sex ratio is not appropriate for male-killer-infected populations.
Figure 1.—

Fixation probabilities of nuclear mutations in male-killer-infected populations, relative to fixation probabilities in uninfected populations. The solid curve gives the analytic approximation, and open triangles represent fixation rates averaged over (A) 107 or (B–D) 106 simulations per datum. The dotted lines give fixation probability estimates based on Wright's approximation for effective population size in populations with biased sex ratio. Unless varied, parameters take the values N = 1000, s = 0.01, h = 0.5, y = 0.5.
For semidominant alleles we have demonstrated analytically that a male-killer infection increases the fixation probability of deleterious, but decreases the fixation probability of beneficial alleles (see above and appendix). To test whether this result also holds for arbitrary dominance levels, we conducted further computer simulations with randomly chosen parameters s, h, N, and y. The results shown in Figure 2 confirm that the male-killer infection makes the fixation of deleterious alleles more likely, and the fixation of beneficial alleles less so, across all parameter values.
Figure 2.—

Relative fixation probabilities of nuclear alleles in male-killer-infected populations (i.e., uMK/u0). Each datum in the plot is based on fixation rates from 106 computer simulations with an infected population and 106 simulations with an uninfected population. Parameters for the 1000 sets of simulations conducted were chosen randomly, with selection coefficients s as shown, dominance levels h between 0 and 1, population sizes N between 100 and 500, and male-killer infection frequencies y between 0.2 and 0.99. Note that for 30 of 1000 parameter combinations, simulations results could not be included in the plot because the alleles never became fixed in the uninfected population during the 106 simulations conducted.
Expected time until fixation:
The expected time until fixation for a mutant allele in a population of size N was estimated by Kimura and Ohta (1969). As has become apparent in the previous section, for a mutant allele to spread and become fixed in a male-killer-infected population, it is necessary that the mutant arises and spreads in the uninfected individuals of the population. Fixation within the uninfected individuals will be followed quickly by fixation of the allele within infected females, because of the strong influx of the allele each generation through males. Hence, we can estimate the expected time until fixation in an infected population simply as that found in an uninfected population with size equal to the uninfected fraction of the infected population. Denoting by 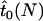 the expected time until fixation in an uninfected population of size N and by
the expected time until fixation in an uninfected population of size N and by 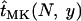 the expected time until fixation in a population of size N and with male-killer prevalence y, we have
the expected time until fixation in a population of size N and with male-killer prevalence y, we have
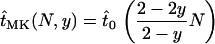 |
(3) |
For a selectively neutral allele, the expected time until fixation in an uninfected population is ∼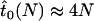 (Kimura and Ohta 1969). Accordingly, in an infected population we have
(Kimura and Ohta 1969). Accordingly, in an infected population we have 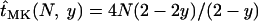 for neutral alleles. Thus, the expected time until fixation of neutral alleles is decreased in infected compared to uninfected populations.
for neutral alleles. Thus, the expected time until fixation of neutral alleles is decreased in infected compared to uninfected populations.
Expected time until either fixation or extinction:
Aside from this conditional time until fixation, we can also ask what the expected time is for a mutant allele to either become fixed or be lost. Let us denote by 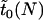 the expected time until fixation or extinction of an allele in an uninfected population of size N. An approximate formula for
the expected time until fixation or extinction of an allele in an uninfected population of size N. An approximate formula for 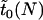 is readily derived from Kimura and Ohta (1969).
is readily derived from Kimura and Ohta (1969).
To derive an approximation for the unconditioned time a mutant allele stays polymorphic in a male-killer-infected population, it is again useful to consider the different individuals the mutant allele might arise in. With probability (2 − 2y)/(2 − y), the mutant arises in an uninfected individual, in which case the expected time until fixation or loss can be estimated by 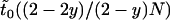 . To get an estimate for the expected time until fixation or loss when the mutant arises in an infected female, we do not attempt an approximation based on the diffusion equation, but adopt a more direct approach. We assume that the fitness effects of the allele itself are negligible compared to the deleterious effect that the male-killing bacteria have on the allele's fitness, namely a fitness reduction by one-half. The probability distribution of the frequency of the mutant allele in infected females is described by a Markov chain with transition matrix
. To get an estimate for the expected time until fixation or loss when the mutant arises in an infected female, we do not attempt an approximation based on the diffusion equation, but adopt a more direct approach. We assume that the fitness effects of the allele itself are negligible compared to the deleterious effect that the male-killing bacteria have on the allele's fitness, namely a fitness reduction by one-half. The probability distribution of the frequency of the mutant allele in infected females is described by a Markov chain with transition matrix
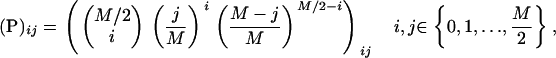 |
(4) |
where 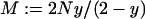 is the number of alleles present in the infected females.
is the number of alleles present in the infected females. 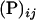 is the probability that i mutant alleles are present in the infected females provided that j mutant alleles were present in the previous generation. Note that the maximum number of mutant alleles present in infected females is M/2 only, because every infected female inherits one wild-type allele from her father. The probability that the allele is extinct at generation k after the mutation event is given by
is the probability that i mutant alleles are present in the infected females provided that j mutant alleles were present in the previous generation. Note that the maximum number of mutant alleles present in infected females is M/2 only, because every infected female inherits one wild-type allele from her father. The probability that the allele is extinct at generation k after the mutation event is given by 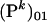 . In our approximation, we consider only the first two generations, as the probability that the mutant allele survives longer than that is very low.
. In our approximation, we consider only the first two generations, as the probability that the mutant allele survives longer than that is very low.
Putting all these considerations together, we get as an estimate for the expected time until fixation or loss of a mutant allele in a population infected with male-killers:
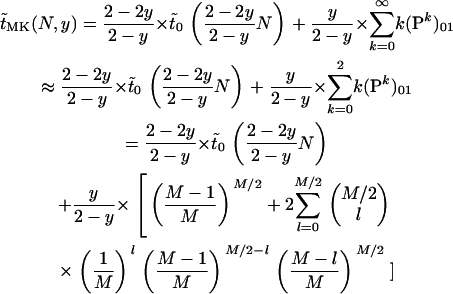 |
(5) |
Estimates for both the conditional expected time until fixation and the expected time until fixation or extinction of mutant alleles in male-killer-infected populations are shown in Figure 3, again relative to the respective expected times in uninfected populations (i.e., 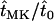 and
and 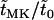 ). Both expected times are lower in infected than in uninfected populations and decrease with increasing frequency of the male-killer. Also shown in Figure 3 are simulation results for the relative expected times until fixation and fixation or extinction, which are in good agreement with the analytical predictions.
). Both expected times are lower in infected than in uninfected populations and decrease with increasing frequency of the male-killer. Also shown in Figure 3 are simulation results for the relative expected times until fixation and fixation or extinction, which are in good agreement with the analytical predictions.
Figure 3.—

Times until fixation (for alleles that fix) and times until fixation or extinction in a population infected with a male-killer infected relative to an uninfected population. The solid curve gives the approximation for the relative time until fixation, and the boldface curve gives the approximation for the relative time until fixation or extinction. Triangles and circles give the respective simulation results for these quantities. Simulation results are based on the same data and parameters take the same values as in Figure 1.
Genetic variation:
One measure of genetic variation in a population is the level of heterozygosity H, the proportion of heterozygous individuals in a population. This quantity is equal to 1 − F, where F is the proportion of homozygous individuals in the population. Consider a locus where mutations occur at rate μ, and each mutation leads to a new allele not previously present in the population (“infinite-alleles model,” Kimura and Crow 1964).
Assuming that the actual population size equals the effective population size and that all alleles are selectively neutral, the expected equilibrium homozygosity has then been estimated (Kimura and Crow 1964) to be 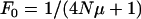 . Applying this fundamental result to a population infected with male-killing bacteria, we first note that the equilibrium homozygosity in the uninfected part of the population will be equal to the equilibrium homozygosity of an uninfected population that has the same size as the uninfected part of the infected population. This is simply because there is no gene flow from infected females to uninfected individuals. Thus,
. Applying this fundamental result to a population infected with male-killing bacteria, we first note that the equilibrium homozygosity in the uninfected part of the population will be equal to the equilibrium homozygosity of an uninfected population that has the same size as the uninfected part of the infected population. This is simply because there is no gene flow from infected females to uninfected individuals. Thus,
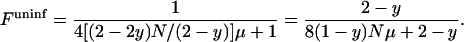 |
(6) |
Because newly arisen mutations in infected females will quickly disappear from the population, the genetic composition within infected females will be approximately the same as that within uninfected females. Therefore, we predict that the total homozygosity in an infected population will be approximately the same as the homozygosity in the uninfected fraction of that population, as given in Equation 6.
Given this reasoning, the expected equilibrium heterozygosity in a male-killer-infected population is approximately
 |
(7) |
Clearly, the expected genetic variability in a male-killer-infected population as measured by heterozygosity is smaller than that in an uninfected population of the same size. Figure 4 shows how the expected equilibrium heterozygosity in male-killer-infected populations declines with increasing prevalence of the male-killers. The decline is most notable for prevalence values >0.6; here marked reductions in heterozygosity are observed. Also shown are the results of computer simulations that confirm our analytical approximations.
Figure 4.—

Predicted heterozygosity (lines) derived from Equation 7 and corresponding simulation results (symbols), depending on the prevalence of the male-killer infection. In the simulations, we initiated the population with one allele fixed and took values of heterozygosity every 1000 generations. Each datum is an average of 300 values of heterozygosity. In all simulations, N = 100. The mutation rate μ takes values of 0.0001 (shaded circles), 0.0005 (crosses), 0.001 (solid diamonds), and 0.003 (open squares).
IMPERFECT TRANSMISSION
In the previous section, we assumed perfect transmission of the bacteria. However, in many cases, infected females also produce some uninfected offspring. Transmission rates ranging from 80 to 100% have been reported, although they are usually in the range of 95–100% (Hurst and Majerus 1993; Jiggins et al. 2002; Dyer and Jaenike 2004). Thus, let us now assume that only a proportion α 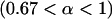 of the offspring of infected females inherit the infection, and (1 − α) of the offspring are uninfected (α > 0.67 is required for male-killer invasion). We also assume that surviving siblings of infected females receive a fitness compensation benefit b due to the death of their brothers and relax our previous assumption of a constant prevalence of the male-killing bacteria. We do not attempt to derive approximations for the case of imperfect male-killer transmission, but rather determine by computer simulations the extent to which fixation probabilities and times until fixation and/or extinction deviate from the approximations derived for perfect transmission.
of the offspring of infected females inherit the infection, and (1 − α) of the offspring are uninfected (α > 0.67 is required for male-killer invasion). We also assume that surviving siblings of infected females receive a fitness compensation benefit b due to the death of their brothers and relax our previous assumption of a constant prevalence of the male-killing bacteria. We do not attempt to derive approximations for the case of imperfect male-killer transmission, but rather determine by computer simulations the extent to which fixation probabilities and times until fixation and/or extinction deviate from the approximations derived for perfect transmission.
In a deterministic model, the polymorphic equilibrium frequency of the male-killing bacteria (proportion of infected females among all females) is given by
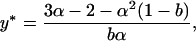 |
(8) |
and this equilibrium is always stable when it exists (Hurst 1991). This equilibrium corresponds to the expected frequency of the male-killing bacteria in our stochastic model, as long as the male-killer does not become extinct and the population does not go extinct due to lack of males.
How can we expect inefficient transmission to affect the population genetics of host alleles? First, given a certain frequency of the male-killing bacteria within females, decreasing transmission rates will both increase the fitness of infected females and lead to gene flux from infected females to uninfected individuals. This will result in fixation probabilities and expected times to fixation and/or extinction that lie in between the predictions for an uninfected population and an infected population with perfect male-killer transmission. Second, we also need to take into account that the male-killing bacteria themselves will be subject to random genetic drift. For a given equilibrium male-killer frequency, drift will become stronger with increasing transmission rate and correspondingly decreasing fitness compensation benefit b. The impact of male-killer drift on the drift of the nuclear alleles is not straightforward to predict.
We obtained simulation estimates for fixation probabilities of alleles with different selection coefficients s and dominance levels h for varying transmission efficiencies, α (Figure 5). In these simulations, the expected frequency of the male-killing bacteria 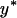 was kept at a constant value with varying α by adjusting the fitness compensation parameter b according to Equation 8. Each simulation was initiated with the male-killing bacteria at their expected frequency.
was kept at a constant value with varying α by adjusting the fitness compensation parameter b according to Equation 8. Each simulation was initiated with the male-killing bacteria at their expected frequency.
Figure 5.—

The effect of relaxing the assumption of perfect vertical transmission. Simulation results are given with varying rates of imperfect transmission. The plots show simulation estimates for relative fixation rates of alleles, depending on the transmission rate of the male-killing bacteria α. Three different allele classes have been assumed in these simulations: a beneficial semidominant allele with s = 0.01 and h = 0.5 (open squares), a neutral allele (solid circles), and a deleterious recessive allele with s = −0.001 and h = 0 (shaded triangles). Each datum represents an average of 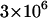 simulations. Solid lines show the analytical predictions based on perfect transmission. Other parameters take the values N = 1000 and
simulations. Solid lines show the analytical predictions based on perfect transmission. Other parameters take the values N = 1000 and 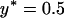 ; the fitness compensation parameter b was varied according to transmission rate to keep equilibrium male-killer frequency constant.
; the fitness compensation parameter b was varied according to transmission rate to keep equilibrium male-killer frequency constant.
As can be seen in Figure 5, the predictions based on perfect transmission give fairly good approximations for a wide range of transmission rates <1. For low transmission rates, estimates for the fixation probabilities 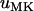 are slightly closer to
are slightly closer to 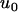 than predicted by the estimates for perfect transmission and fixed male-killer frequency. This is in accord with the above reasoning. We also compared the average times until fixation and until fixation or extinction of alleles to the predictions based on perfect transmission (results not shown). Again, the deviations of the simulation results from the predictions based on perfect transmission are not severe, and the direction of the deviations is toward the expected times in an uninfected population.
than predicted by the estimates for perfect transmission and fixed male-killer frequency. This is in accord with the above reasoning. We also compared the average times until fixation and until fixation or extinction of alleles to the predictions based on perfect transmission (results not shown). Again, the deviations of the simulation results from the predictions based on perfect transmission are not severe, and the direction of the deviations is toward the expected times in an uninfected population.
To determine regions of the parameter space where deviations in fixation probabilities from the analytical predictions are highest, we performed simulations with randomly chosen parameters N, s, α, and b. Plotting relative fixation probabilities against the selection coefficient s yields a graph very similar to that in Figure 2 (not shown), confirming that imperfect transmission does not qualitatively alter our fundamental result of increased and decreased fixation probabilities of deleterious and beneficial alleles, respectively.
Figure 6 shows relative deviations of fixation rates yielded from these simulations from the analytical predictions with perfect transmission. For beneficial alleles (Figure 6A), most deviations are relatively small and tend to be positive, at least for low transmission rates. This means that the actual fixation rates are higher than the predictions based on the assumption of perfect transmission and thus closer to fixation probabilities in uninfected populations. However, for very high transmission rates, deviations become negative, meaning that fixation rates are even lower than predicted. The strongest negative deviations are observed with high transmission rate and high equilibrium frequency (i.e., when both α and b are high). This may be explained by the surmise that with male-killing bacteria drifting at high frequencies, the frequencies above the equilibrium will have a much stronger impact than frequencies below the equilibrium. The reasoning behind this conjecture is an analogy with populations of fluctuating size, in which it has been demonstrated that the effective population size is given by the harmonic mean of population sizes and, thus, is smaller than the arithmetic mean (Kimura and Crow 1963).
Figure 6.—

Relative deviations of fixation rates derived from simulations with imperfect transmission from predictions based on the assumption of perfect transmission. Each datum in the two plots represents an average of 106 simulations. For each of the 1000 points per plot, parameters were chosen randomly, with transmission rate 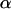 between 0.95 and 1, equilibrium frequency
between 0.95 and 1, equilibrium frequency 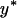 between 0 and 1, population size between 100 and 500, and selection coefficients s between (A) 0 and 0.02 (beneficial alleles) and (B) −0.001 and 0 (deleterious alleles). The fitness compensation parameter b was chosen according to formula (9), but only simulations with biologically reasonable values of
between 0 and 1, population size between 100 and 500, and selection coefficients s between (A) 0 and 0.02 (beneficial alleles) and (B) −0.001 and 0 (deleterious alleles). The fitness compensation parameter b was chosen according to formula (9), but only simulations with biologically reasonable values of 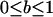 are included in the plots. Semidominance (h = 0.5) was assumed in all simulations.
are included in the plots. Semidominance (h = 0.5) was assumed in all simulations.
For deleterious alleles (Figure 6B), deviations of fixation rates from the predictions tend to be higher than those for beneficial alleles, as can be expected from the fact that such fixation events are very rare and thus the variance of fixation rates in a finite set of simulations is much higher than that for beneficial alleles. Most deviations are negative; i.e., fixations occurred less often than predicted. Again, this is explained by the fact that with imperfect transmission, there is allele flux from infected females to uninfected individuals and thus more efficient purging of deleterious mutations.
DISCUSSION
We have studied basic aspects of the population genetics of populations infected with male-killing bacteria. Our results suggest that when maternal transmission of the bacteria is perfect and their prevalence in the population stable, the population behaves approximately as if it consisted of the uninfected part of the population only. This includes increased fixation probabilities for deleterious alleles and decreased fixation probabilities for beneficial alleles, decreased times to fixation and fixation or extinction, and reduced genetic variation. When transmission of the male-killing bacteria is imperfect, the effect is somewhat mitigated, but still strong.
In contrast to what might be expected at first glance, the distorted sex ratio in populations infected with male-killing bacteria is not primarily connected to these results. Rather, the reason for the effects observed is that alleles arisen in an infected female cannot spread, because (1) infected females have only about half the fitness of uninfected females, and (2) the nucleotype of infected females is tightly linked to the male-killing cytotype. Thus, although reproducing, the infected females in the population can be regarded as “living deads” [to use a metaphor framed for individuals of inferior genotype in asexual populations (Rice 2002)], and their number has almost no impact on population genetics (see also Sullivan and Jaenike 2006). Our results can also be understood in terms of reduction of effective population size due to different genetic contributions of different classes within the population (e.g., Caballero 1994; Woolliams et al. 1999). In fact, a male-killer-infected population can be regarded as an extreme case of class structure: a potentially large fraction of the population, although reproducing, contributes either very little or nothing genetically in the long term, and this class structure is stable over a long time period, as long as infection persists.
Selection for or against alleles is not completely determined by the phenotype that the allele induces, but also by linkage to other alleles. For example, recurrently arising deleterious mutations can reduce genetic variability at linked neutral loci, a process known as “background selection” (Charlesworth et al. 1993). The effect of male-killing endosymbionts is similar to background selection, as both stem from linkage to deleterious genetic elements. However, provided that their prevalence is sufficiently high, male-killing bacteria can be expected to have a stronger impact on genetic variability than deleterious nuclear alleles. This is because first, linkage of all newly arisen alleles in infected females to the male-killing element is complete or at least very high. Linkage here corresponds to the vertical transmission rate of the male-killing bacteria, for which quantity values in the range of 0.8–1 have been reported in natural and laboratory populations of various insect species (Hurst and Majerus 1993 and references therein; Jiggins et al. 2002; Dyer and Jaenike 2004). Second, despite their being strongly deleterious to nuclear alleles, male-killing bacteria carry a drive and can be stably maintained in populations at high frequencies (Dyer and Jaenike 2004; Dyson and Hurst 2004). This is in contrast to deleterious nuclear alleles, which are constantly purged and usually kept at low frequencies. Combined, these two aspects are expected to result in very strong selection even against beneficial alleles when they arise in infected females and correspondingly in a strong reduction in neutral genetic variation and adaptability.
One assumption in our model is that changes in the prevalence do not affect the total population size. In contrast, there may be either a positive or a negative impact of male-killer prevalence on population size, depending on the type of density-dependent population dynamics (P. Kirkby and G. D. D. Hurst, unpublished results; see also Hatcher et al. 1999). Two different types of impacts of change in population size as a consequence of a male-killer infection need to be distinguished. First, the population size when the male-killer is at equilibrium frequency in the host population may be different from that if the population is uninfected. This is important when one is interested in how the infection of a particular population with male-killing bacteria changed the population genetics of that population. On the other hand, if one is interested in how the population genetics of a male-killer-infected population deviate from the expectations if it were not known that the population is infected, it is irrelevant how the equilibrium male-killer frequency affects the population size. (The former case is a comparison prior and, after infection, the latter a comparison of an infected and an uninfected population of the same size.) Second, and independent of the type of question asked, fluctuations in population size as a result of fluctuations in the male-killer prevalence need to be taken into account. Fluctuations of this kind are likely to make the effects found in our model more severe, as the effective population size will be decreased below the population size where the male-killer is at equilibrium frequency.
A question we have not resolved is the degree to which the processes predicted will be observable in currently infected species. Some of the processes occur in the medium term (e.g., the accumulation of deleterious mutations) and may be hard to detect in currently infected species. Processes that do not completely depend upon mutation events (e.g., the reduction in heterozygosity at high male-killer prevalence) should be observable in the short term, as should impeded adaptation. We note, additionally, that some infections appear to be long lived (e.g., Dyer and Jaenike 2004), and, in these cases, the hallmark of male-killer infection on the host genome will be most apparent.
We can also examine the extent to which the results revealed for male-killing infections are likely to be true for other factors distorting the sex ratio. Feminization is another strategy of reproductive parasitism that is found in arthropods. The best-studied example is the woodlouse Armadillidium vulgare, which is infected by feminizing Wolbachia (reviewed in Rigaud 1997). In some populations, the ancestral sex determination of female heterogamety has been replaced by a system where all infected individuals are females and all uninfected individuals are males; the W chromosome has become extinct, so that all individuals are “genetic males” with a ZZ constitution. Thus, females produce offspring in a female-biased sex ratio that corresponds to the transmission rate of Wolbachia. Correspondingly, as with male-killer infections, the population sex ratio is female biased. However, in contrast to a male-killer infection, there is unrestricted gene flow between infected and uninfected individuals (i.e., females and males). Therefore, the effect of selection and drift on nuclear alleles will be the same as that in any uninfected population with the same population sex ratio, and applying Wright's (1931) formula for the effective population size should be adequate. Similarly, Wright's equations should be sufficient for describing cases of X chromosome meiotic drive.
Our results imply that caution needs to be taken when estimating population genetic properties such as fixation rates or genetic variability in arthropod populations with biased sex ratio. Clearly, it is not sufficient to apply Wright's formula for the effective population size without determining the reason for the distorted sex ratio. In studies comparing mitochondrial with nuclear DNA variation in male-killer-infected populations (e.g., Jiggins 2003; Dyer and Jaenike 2004), this may lead to overestimates of mitochondrial genetic variability relative to nuclear variability and, correspondingly, the age of a male-killer infection may be overestimated.
Moreover, we caution against disregarding the peculiar population genetics of male-killer-infected populations in ESS models (e.g., Hurst and McVean 1998). Whether a beneficial gene will spread or not will largely depend on its fitness impact in uninfected individuals. Taking the average of the fitness effect in infected and uninfected individuals will often produce misleading conclusions. Aside from genes that suppress male-killing (Hornett et al. 2006) we can expect adaptations beneficial solely in infected females to be strongly impeded if genes conferring these adaptations are maladaptive in uninfected individuals. By contrast, the evolution of traits that are adaptive for uninfected and infected individuals alike (e.g., alterations in mate choice and mating behavior, as suggested by Randerson et al. 2000) will not be impeded beyond the impediment due to decreased effective population size as reported in this article.
Acknowledgments
We thank Kelly Dyer, Kirsten Hilgenbröcker, Matthias Flor, and three anonymous reviewers for helpful comments on the manuscript. We acknowledge support of a University College London Graduate School Research Scholarship to J.E. and funding from the National Environment Research Council to G.H.
APPENDIX
In what follows we demonstrate that in the case of semidominance (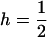 ) and perfect transmission of the male-killer, the fixation probability of a positively selected mutant allele is always lower in an infected than in an uninfected population; i.e.,
) and perfect transmission of the male-killer, the fixation probability of a positively selected mutant allele is always lower in an infected than in an uninfected population; i.e., 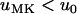 . We have
. We have
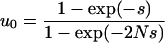 |
(A1) |
resulting from Equation 1 and
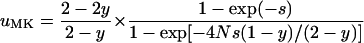 |
(A2) |
from Equation 2. Let s > 0 and 0 < y < 1 be true, and let 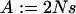 (
(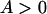 ) and
) and 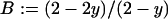 (
(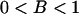 ). Then
). Then 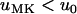 is equivalent to
is equivalent to
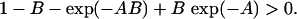 |
(A3) |
The left side of this equality is zero for 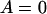 . Also, the derivative of the left side with respect to
. Also, the derivative of the left side with respect to 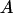 ,
, 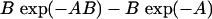 , is always >0. From this it follows that inequality (A3) is true for all
, is always >0. From this it follows that inequality (A3) is true for all 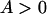 and
and 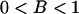 , which proves the assertion. Analogously, it can be demonstrated that
, which proves the assertion. Analogously, it can be demonstrated that 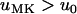 holds for
holds for 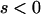 .
.
References
- Caballero, A., 1994. Developments in the prediction of effective population size. Heredity 73: 657–679. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., M. T. Morgan and D. Charlesworth, 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer, K. A., and J. Jaenike, 2004. Evolutionarily stable infection by a male-killing endosymbiont in Drosophila innubila. Genetics 168: 1443–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson, E. A., and G. D. D. Hurst, 2004. Persistence of an extreme sex-ratio bias in a natural population. Proc. Natl. Acad. Sci. USA 101: 6520–6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelstädter, J., and G. D. D. Hurst, 2006. Can maternally transmitted endosymbionts facilitate the evolution of haplodiploidy? J. Evol. Biol. 19: 194–202. [DOI] [PubMed] [Google Scholar]
- Fialho, R. F., and L. Stevens, 2000. Male-killing Wolbachia in a flour beetle. Proc. R. Soc. Lond. Ser. B Biol. Sci. 267: 1469–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher, M. J., D. E. Taneyhill, A. M. Dunn and C. Tofts, 1999. Population dynamics under parasitic sex ratio distortion. Theor. Popul. Biol. 56: 11–28. [DOI] [PubMed] [Google Scholar]
- Hornett, E. A., S. Charlat, A. M. R. Duplouy, N. Davies, G. K. Roderick et al., 2006. Evolution of male killer suppression in a natural population. PLoS Biol. 4: e283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst, G. D. D., and F. M. Jiggins, 2005. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc. R. Soc. Lond. Ser. B Biol. Sci. 272: 1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst, G. D. D., and M. E. N. Majerus, 1993. Why do maternally inherited microorganisms kill males? Heredity 71: 81–95. [Google Scholar]
- Hurst, G. D. D., and G. T. McVean, 1998. Parasitic male-killing bacteria and the evolution of clutch-size. Ecol. Entomol. 23: 350–353. [Google Scholar]
- Hurst, G. D. D., F. M. Jiggins, J. H. G. V. D. Schulenburg, D. Bertrand, S. A. West et al., 1999. Male-killing Wolbachia in two species of insect. Proc. R. Soc. Lond. Ser. B Biol. Sci. 266: 735–740. [Google Scholar]
- Hurst, G. D. D., F. M. Jiggins and M. E. N. Majerus, 2003. Inherited microorganisms that selectively kill male hosts: The hidden players of insect evolution?, pp. 177–198 in Insect Symbiosis, edited by K. Bourtzis and T. A. Miller. CRC Press, Boca Raton, FL.
- Hurst, L. D., 1991. The incidences and evolution of cytoplasmic male killers. Proc. R. Soc. Lond. Ser. B Biol. Sci. 244: 91–99. [Google Scholar]
- Jaenike, J., K. A. Dyer and L. K. Reed, 2003. Within-population structure of competition and the dynamics of male-killing Wolbachia. Evol. Ecol. Res. 5: 1023–1036. [Google Scholar]
- Jiggins, F. M., 2003. Male-killing Wolbachia and mitochondrial DNA: selective sweeps, hybird introgression and parasite population dynamics. Genetics 164: 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiggins, F. M., and M. C. Tinsley, 2005. An ancient mitochondrial polymorphism in Adalis bipunctata linked to a sex-ratio-distorting bacterium. Genetics 71: 1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiggins, F. M., G. D. D. Hurst and M. E. N. Majerus, 2000. Sex-ratio-distorting Wolbachia causes sex-role reversal in its butterfly host. Proc. R. Soc. Lond. Ser. B Biol. Sci. 267: 69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiggins, F. M., J. P. Randerson, G. D. D. Hurst and M. E. N. Majerus, 2002. How can sex ratio distorters reach extreme prevalences? Male-killing Wolbachia are not suppressed and have near-perfect vertical transmission efficiency in Acraea encedon. Evolution 56: 2290–2295. [DOI] [PubMed] [Google Scholar]
- Johnstone, R. A., and G. D. D. Hurst, 1996. Maternally inherited male-killing microorganisms may confound interpretation of mitochondrial DNA variability. Biol. J. Linn. Soc. 58: 453–470. [Google Scholar]
- Kimura, M., 1962. On the probability of fixation of mutant genes in a population. Genetics 47: 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., and J. F. Crow, 1963. The measurement of effective population number. Evolution 17: 279–288. [Google Scholar]
- Kimura, M., and J. F. Crow, 1964. The number of alleles that can be maintained in a finite population. Genetics 49: 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., and T. Ohta, 1969. Average number of generations until fixation of a mutant allele in a finite population. Genetics 61: 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenegro, H., W. N. Souza, D. D. Leite and L. B. Klaczko, 2000. Male-killing selfish cytoplasmic element causes sex-ratio distortion in Drosophila melanogaster. Heredity 85: 465–470. [DOI] [PubMed] [Google Scholar]
- Normark, B. B., 2004. Haplodiploidy as an outcome of coevolution between male-killing cytoplasmic elements and their hosts. Evolution 58: 790–798. [DOI] [PubMed] [Google Scholar]
- Randerson, J. P., F. M. Jiggins and L. D. Hurst, 2000. Male killing can select for male mate choice: a novel solution to the paradox of the lek. Proc. R. Soc. Lond. Ser. B Biol. Sci. 267: 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, W. R., 2002. Experimental test for the adaptive significance of sexual recombination. Nat. Rev. Genet. 3: 241–251. [DOI] [PubMed] [Google Scholar]
- Rigaud, T., 1997. Inherited microorganisms and sex determination of arthropod hosts, pp. 81–102 in Influential Passengers: Inherited Microorganisms and Invertebrate Reproduction, edited by S. L. O'Neill, A. A. Hoffmann and J. H. Werren. Oxford University Press, New York.
- Schulenburg, J. H. G. V. D., G. D. D. Hurst, D. Tetzlaff, G. E. Booth, I. A. Zakharov et al., 2002. History of infection with different male-killing bacteria in the two-spot ladybird beetle Adalia bipunctata revealed through mitochondrial DNA sequence analysis. Genetics 160: 1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, J., and J. Jaenike, 2006. Male-killing Wolbachia and male mate choice: a test with Drosophila innubila. Evol. Ecol. Res. 8: 91–102. [Google Scholar]
- Telschow, A., J. Engelstädter, N. Yamamura, P. Hammerstein and G. D. D. Hurst, 2006. Asymmetric gene flow and constraints on adaptation caused by sex ratio distorters. J. Evol. Biol. 19: 869–878. [DOI] [PubMed] [Google Scholar]
- Werren, J. H., S. W. Skinner and A. M. Huger, 1986. Male-killing bacteria in a parasitic wasp. Science 231: 990–992. [DOI] [PubMed] [Google Scholar]
- Williamson, D. L., and D. F. Poulson, 1979. Sex ratio organisms (Spiroplasmas) of Drosophila, pp. 175–208 in The Mycoplasmas, edited by R. F. Whitcomb and J. G. Tully. Academic Press, New York.
- Woolliams, J. A., P. Bijma and B. Villanueva, 1999. Expected genetic contributions and their impact on gene flow and genetic gain. Genetics 153: 1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16: 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeh, D. W., J. A. Zeh and M. Bonilla, 2005. Wolbachia, sex ratio bias and apparent male killing in the harlequin beetle riding pseudoscorpion. Heredity 95: 41–49. [DOI] [PubMed] [Google Scholar]