

Abstract
Composite interval mapping (CIM) is the most commonly used method for mapping quantitative trait loci (QTL) with populations derived from biparental crosses. However, the algorithm implemented in the popular QTL Cartographer software may not completely ensure all its advantageous properties. In addition, different background marker selection methods may give very different mapping results, and the nature of the preferred method is not clear. A modified algorithm called inclusive composite interval mapping (ICIM) is proposed in this article. In ICIM, marker selection is conducted only once through stepwise regression by considering all marker information simultaneously, and the phenotypic values are then adjusted by all markers retained in the regression equation except the two markers flanking the current mapping interval. The adjusted phenotypic values are finally used in interval mapping (IM). The modified algorithm has a simpler form than that used in CIM, but a faster convergence speed. ICIM retains all advantages of CIM over IM and avoids the possible increase of sampling variance and the complicated background marker selection process in CIM. Extensive simulations using two genomes and various genetic models indicated that ICIM has increased detection power, a reduced false detection rate, and less biased estimates of QTL effects.
THE rapid increase in availability of fine-scale genetic marker maps has led to the intensive use of QTL mapping in the genetic study of quantitative traits (Falconer and Mackay 1996; Doerge et al. 1997; Lynch and Walsh 1998; Kearsey 2002; Steinmetz et al. 2002; Wu and Lin 2006). A number of statistical methods have been developed for QTL detection and effect estimation (Lander and Botstein 1989; Haley and Knott 1992; Jansen 1994; Wright and Mowers 1994; Zeng 1994; Satagopan et al. 1996; Whittaker et al. 1996; Piepho and Gauch 2001; Sen and Churchill 2001; Broman and Speed 2002; van den Oord and Sullivan 2003; Xu 2003; Bogdan et al. 2004).
From a statistical perspective, methods for QTL mapping are based on three broad classes: regression (Haley and Knott 1992; Whittaker et al. 1996), maximum-likelihood (Doerge et al. 1997), and Bayesian models (Sillanpää and Corander 2002). The simplest single-marker analysis identifies QTL on the basis of the difference between the mean phenotypes of different marker groups, but cannot separate the estimates of recombination fraction and QTL effect (Soller et al. 1976; Doerge et al. 1997). Interval mapping (IM) is based on maximum-likelihood parameter estimation and provides a likelihood-ratio test for QTL position (Lander and Botstein 1989). Regression interval mapping was proposed to approximate maximum-likelihood interval mapping to save computation time at one or multiple genomic positions (Haley and Knott 1992; Martinez and Curnow 1992). The major disadvantage of IM is that the estimates of locations and effects of QTL may be biased when QTL are linked (Haley and Knott 1992; Martinez and Curnow 1992; Zeng 1994). Composite interval mapping (CIM) (Jansen 1994; Zeng 1994) combines IM with multiple-marker regression analysis, which controls the effects of QTL on other intervals or chromosomes onto the QTL that is being tested and thus increases the precision of QTL detection. More recently, the use of Bayesian models has been widely explored for QTL mapping (Satagopan et al. 1996; Uimari and Hoeschele 1997; Sen and Churchill 2001; Xu 2003; Bogdan et al. 2004; Wang et al. 2005a). However, methods based on Bayesian models have not been widely used in practice, partially due to the difficulty of choosing prior distributions, complexity of computation, and lack of user-friendly software.
Due to the accessibility of the freely available software QTL Cartographer (Wang et al. 2005b) CIM is now the most commonly used method for QTL mapping with populations derived from biparental crosses. However, in Zeng's algorithm, QTL effect at the current testing position and regression coefficients of the marker variables used to control genetic background were estimated simultaneously in an expectation and conditional maximization (ECM) algorithm. Thus, the same marker variable may have different coefficient estimates as the testing position changes along the chromosomes. The algorithm used in CIM cannot completely ensure that the effect of QTL at the current testing interval is not absorbed by the background marker variables and may result in biased estimation of the QTL effect (see Table 4 and Figure 1 in Zeng 1994).
TABLE 4.
Deviations of QTL position and effect estimates based on 100 simulation runs
| QZ1 | QZ2 | QZ3 | QZ4 | QZ5 | QZ6 | QZ7 | QZ8 | QZ9 | QZ10 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Position estimated from all peaks in the confidence interval | |||||||||||
| ADD, H = 0.8 | CIM | 0.77 (2.80) | −0.23 (2.85) | −0.18 (2.97) | −1.53 (2.36) | 0.94 (2.76) | −1.12 (2.32) | −0.75 (2.62) | 1.42 (2.00) | 0.66 (2.55) | −0.13 (3.03) |
| ICIM | 1.09 (3.34) | 0.94 (2.78) | 0.92 (2.65) | −0.83 (2.25) | −0.67 (2.19) | 0.65 (2.45) | −1.19 (2.71) | 0.69 (1.66) | 0.83 (2.01) | 0.05 (3.12) | |
| ADD, H = 0.5 | CIM | 1.48 (2.89) | 0.16 (2.94) | 0.81 (2.61) | −1.91 (2.11) | −0.33 (2.84) | −0.31 (2.98) | −1.06 (2.71) | 0.78 (2.40) | 0.82 (2.49) | 0.56 (3.02) |
| ICIM | 2.40 (2.75) | 1.09 (2.67) | 1.60 (1.92) | −1.51 (2.12) | −0.70 (3.07) | 0.34 (3.11) | −1.37 (2.73) | 1.01 (2.30) | 0.57 (1.97) | 0.89 (3.34) | |
| ADD + EPI, H = 0.8 | CIM | 1.08 (3.08) | −0.61 (2.81) | 0.26 (2.82) | −1.66 (2.06) | 0.42 (2.96) | 0.17 (2.88) | −1.34 (2.63) | 1.30 (2.21) | 0.05 (2.78) | 0.42 (2.96) |
| ICIM | 1.77 (3.07) | 0.85 (2.49) | 1.12 (2.46) | −1.23 (2.20) | −1.03 (2.68) | 1.03 (2.80) | −2.23 (1.97) | 1.13 (2.13) | 0.51 (2.22) | 0.73 (3.37) | |
| ADD + EPI, H = 0.5 | CIM | 1.13 (3.13) | −0.39 (3.35) | 0.98 (2.45) | −1.58 (2.45) | 0.21 (3.20) | 0.08 (3.00) | −1.06 (3.13) | 0.42 (2.54) | −0.27 (2.33) | 1.31 (3.23) |
| ICIM | 1.17 (3.59) | 0.40 (2.67) | 1.47 (1.88) | −1.58 (2.45) | −0.91 (3.02) | 0.54 (3.05) | −1.93 (2.33) | 0.89 (2.23) | 0.46 (1.89) | 1.89 (3.30) | |
| Position estimated from peaks that have LOD scores >2.5 in the confidence interval | |||||||||||
| ADD, H = 0.8 | CIM | 0.14 (2.62) | −0.51 (2.70) | −0.66 (3.17) | −1.04 (2.65) | 1.00 (2.75) | −1.10 (2.33) | 0.56 (2.67) | 1.42 (2.00) | 0.67 (2.57) | 0.11 (2.88) |
| ICIM | 0.95 (3.35) | 0.95 (2.80) | 0.83 (2.87) | −0.83 (2.25) | −0.67 (2.19) | 0.65 (2.45) | −0.78 (2.65) | 0.69 (1.66) | 0.83 (2.01) | −0.05 (3.07) | |
| ADD, H = 0.5 | CIM | 0.19 (2.58) | −0.21 (2.87) | −0.09 (1.98) | −1.77 (2.06) | 1.00 (2.66) | −0.40 (2.94) | 1.17 (1.07) | 0.74 (2.39) | 0.23 (2.67) | 0.28 (2.98) |
| ICIM | 1.00 (3.39) | 1.36 (2.46) | 1.47 (2.00) | −1.29 (2.15) | −0.60 (3.13) | 0.33 (3.12) | −0.17 (3.27) | 1.05 (2.22) | 0.66 (2.00) | 0.25 (3.12) | |
| ADD + EPI, H = 0.8 | CIM | 0.40 (2.92) | −1.00 (2.70) | −0.26 (2.92) | −1.16 (2.11) | 0.21 (2.95) | 0.04 (2.87) | −1.50 (2.60) | 1.31 (2.23) | 0.10 (2.74) | 0.70 (2.72) |
| ICIM | −0.29 (3.10) | 0.69 (2.78) | 1.20 (2.68) | −1.19 (2.26) | −0.91 (2.66) | 0.96 (2.84) | −2.00 (2.52) | 1.14 (2.14) | 0.47 (2.25) | 0.89 (3.25) | |
| ADD + EPI, H = 0.5 | CIM | 2.00 (2.14) | −1.04 (3.52) | 0.40 (2.80) | −0.93 (2.02) | 0.58 (3.05) | −0.62 (3.10) | 0.48 (2.56) | −0.50 (2.13) | 0.17 (3.53) | |
| ICIM | 1.27 (3.31) | −0.30 (3.27) | 0.92 (2.27) | −0.87 (2.67) | −0.62 (3.49) | 0.19 (3.23) | 1.40 (3.72) | 0.75 (2.23) | 0.33 (2.07) | 1.83 (3.29) | |
| Effect estimated from all peaks in the confidence interval | |||||||||||
| ADD, H = 0.8 | CIM | 0.24 (0.37) | 0.34 (0.40) | 0.03 (0.21) | −0.53 (0.39) | 0.34 (0.51) | −0.41 (0.36) | 0.11 (0.32) | −0.19 (0.26) | 0.04 (0.25) | −0.02 (0.25) |
| ICIM | 0.13 (0.30) | 0.02 (0.30) | 0.01 (0.31) | −0.08 (0.26) | 0.11 (0.33) | −0.03 (0.30) | 0.01 (0.31) | −0.06 (0.28) | 0.09 (0.25) | 0.01 (0.28) | |
| ADD, H = 0.5 | CIM | 0.22 (0.52) | 0.20 (0.49) | 0.08 (0.37) | −0.42 (0.40) | 0.20 (0.75) | −0.35 (0.41) | 0.02 (0.55) | −0.14 (0.46) | 0.10 (0.42) | −0.08 (0.40) |
| ICIM | 0.09 (0.55) | 0.16 (0.51) | 0.00 (0.46) | −0.17 (0.39) | 0.06 (0.52) | −0.03 (0.54) | −0.08 (0.49) | −0.08 (0.49) | 0.16 (0.55) | 0.06 (0.53) | |
| ADD + EPI, H = 0.8 | CIM | 0.26 (0.46) | 0.36 (0.48) | 0.07 (0.46) | −0.35 (0.44) | 0.20 (0.63) | −0.36 (0.54) | 0.10 (0.45) | −0.21 (0.46) | 0.08 (0.37) | −0.06 (0.34) |
| ICIM | 0.09 (0.55) | 0.04 (0.51) | 0.01 (0.52) | −0.02 (0.39) | 0.06 (0.50) | −0.05 (0.46) | 0.05 (0.54) | −0.19 (0.47) | 0.01 (0.52) | 0.02 (0.55) | |
| ADD + EPI, H = 0.5 | CIM | 0.35 (0.67) | 0.29 (0.59) | 0.12 (0.54) | −0.30 (0.60) | 0.11 (0.83) | −0.34 (0.70) | 0.33 (0.65) | −0.20 (0.59) | 0.09 (0.54) | 0.00 (0.43) |
| ICIM | 0.26 (0.87) | 0.00 (0.67) | 0.14 (0.72) | −0.17 (0.62) | 0.04 (0.89) | −0.23 (0.87) | 0.24 (0.58) | −0.24 (0.69) | 0.15 (0.62) | 0.00 (0.58) | |
| Effect estimated from peaks that have LOD scores >2.5 in the confidence interval | |||||||||||
| ADD, H = 0.8 | CIM | 0.67 (0.24) | 0.46 (0.22) | 0.17 (0.14) | −0.27 (0.29) | 0.24 (0.49) | −0.42 (0.34) | −0.35 (0.14) | −0.19 (0.26) | 0.05 (0.24) | 0.08 (0.20) |
| ICIM | 0.27 (0.20) | 0.05 (0.26) | 0.12 (0.20) | −0.08 (0.26) | 0.11 (0.33) | −0.03 (0.30) | −0.23 (0.20) | −0.06 (0.28) | 0.09 (0.25) | 0.05 (0.23) | |
| ADD, H = 0.5 | CIM | 0.95 (0.22) | 0.54 (0.25) | 0.59 (0.26) | 0.05 (0.19) | −0.55 (0.60) | −0.41 (0.34) | −0.99 (0.28) | −0.08 (0.42) | 0.36 (0.29) | 0.37 (0.21) |
| ICIM | 0.86 (0.29) | 0.45 (0.31) | 0.52 (0.28) | 0.03 (0.23) | −0.06 (0.43) | −0.09 (0.49) | −0.67 (0.23) | −0.04 (0.45) | 0.38 (0.43) | 0.42 (0.29) | |
| ADD + EPI, H = 0.8 | CIM | 0.72 (0.16) | 0.57 (0.26) | 0.63 (0.19) | 0.09 (0.32) | −0.33 (0.56) | −0.49 (0.43) | −0.74 (0.10) | −0.17 (0.43) | 0.28 (0.29) | 0.31 (0.14) |
| ICIM | 0.64 (0.23) | 0.35 (0.29) | 0.46 (0.33) | 0.10 (0.29) | −0.03 (0.42) | −0.09 (0.42) | −0.74 (0.33) | −0.17 (0.46) | 0.25 (0.37) | 0.32 (0.46) | |
| ADD + EPI, H = 0.5 | CIM | 1.29 (0.28) | 0.84 (0.26) | 0.91 (0.16) | 0.60 (0.37) | −0.86 (0.53) | −0.73 (0.46) | 0.04 (0.41) | 0.62 (0.23) | 0.71 (0.20) | |
| ICIM | 0.90 (1.03) | 0.72 (0.22) | 1.07 (0.56) | 0.45 (0.32) | −0.59 (0.60) | −0.61 (0.61) | −1.00 (0.18) | 0.02 (0.47) | 0.59 (0.40) | 0.74 (0.41) | |
Figure 1.—

Average LOD profiles of CIM and ICIM across 100 simulation runs for different genetic models and heritability levels under genome 1. Arrow size and direction represent the approximate effect size and direction of the pointed QTL, respectively. (A–D) Mean LOD profile across 100 runs: (A) additive genetic model, H = 0.8; (B) additive genetic model, H = 0.5; (C) additive and epistasis model, H = 0.8; and (D) additive and epistasis model, H = 0.5.
In this article, we propose a modified algorithm to render CIM more inclusive of all marker data [inclusive composite interval mapping (ICIM)] and then compare ICIM with CIM through extensive simulations.
MATERIALS AND METHODS
The linear regression model and its properties in QTL mapping:
For simplicity, it is supposed that two inbred parents P1 and P2 differ in m QTL, being distributed in m intervals flanked by m + 1 markers. The parental QTL genotype is assumed to be Q1Q1Q2Q2…QmQm for P1 and q1q1q2q2…qmqm for P2. We consider a backcross population where P1 is the recurrent parent. For an individual in a backcross population X = (x1, x2,…, xm, xm+1) represents marker variables that are 1 and −1, standing for the two marker types (homozygote and heterozygote), respectively, and G = (g1, g2,…, gm) represents the QTL variables that are 1 and −1, standing for the two QTL types (homozygote and heterozygote), respectively. Additive effects of QTL are represented by a1, a2,…, and am. Under the assumption of additivity of QTL effects, the genetic value G of an individual under an additive genetic model can be written in the following form:
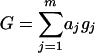 |
(1) |
The expectation of QTL genotype 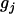 is dependent on the position of the jth QTL on the chromosomal interval flanked by the jth and (j + 1)th markers and the length of the interval (Zeng 1993; Wright and Mowers 1994; Whittaker et al. 1996); i.e.,
is dependent on the position of the jth QTL on the chromosomal interval flanked by the jth and (j + 1)th markers and the length of the interval (Zeng 1993; Wright and Mowers 1994; Whittaker et al. 1996); i.e.,
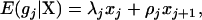 |
(2) |
where 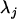 and
and 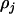 are functions of the three recombination fractions between the jth marker and jth QTL, between the jth QTL and (j + 1)th marker, and between the jth and (j + 1)th markers. Therefore, the expectation of the genotypic value G conditional on the known marker types can be written as a linear function of marker variables; i.e.,
are functions of the three recombination fractions between the jth marker and jth QTL, between the jth QTL and (j + 1)th marker, and between the jth and (j + 1)th markers. Therefore, the expectation of the genotypic value G conditional on the known marker types can be written as a linear function of marker variables; i.e.,
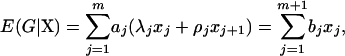 |
(3) |
where 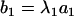 ,
, 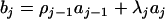 (j = 2,…, m), and
(j = 2,…, m), and 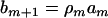 . The coefficient of the jth marker is affected by QTL only on intervals (j − 1, j) and (j, j + 1). If there are no QTL in the neighboring intervals of the current interval (j, j + 1), corresponding to the assumption of isolated QTL according to Whittaker et al. (1996), the two coefficients
. The coefficient of the jth marker is affected by QTL only on intervals (j − 1, j) and (j, j + 1). If there are no QTL in the neighboring intervals of the current interval (j, j + 1), corresponding to the assumption of isolated QTL according to Whittaker et al. (1996), the two coefficients 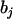 and
and 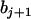 contain all the position and additive effect information of the QTL in the interval (j, j + 1), which provides the theoretical basis for mapping additive QTL in CIM (Zeng 1994) and other regression mapping methods (Wright and Mowers 1994; Whittaker et al. 1996).
contain all the position and additive effect information of the QTL in the interval (j, j + 1), which provides the theoretical basis for mapping additive QTL in CIM (Zeng 1994) and other regression mapping methods (Wright and Mowers 1994; Whittaker et al. 1996).
Suppose that we have a sample of n individuals from a backcross population with observations on a quantitative trait of interest and m + 1 ordered markers. The following linear regression model based on Equation 3 can be used in mapping additive QTL; i.e.,
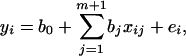 |
(4) |
where 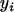 is the trait value of the ith individual in the mapping population;
is the trait value of the ith individual in the mapping population; 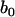 is the overall mean of the model;
is the overall mean of the model; 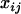 is a dummy variable for the genotype of the ith individual at the jth marker, taking value 1 for homozygote of marker type and −1 for heterozygote;
is a dummy variable for the genotype of the ith individual at the jth marker, taking value 1 for homozygote of marker type and −1 for heterozygote; 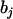 is the regression coefficient of the phenotype on the jth marker conditional on all other markers; and
is the regression coefficient of the phenotype on the jth marker conditional on all other markers; and 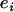 is the residual random error that is assumed to be normally distributed.
is the residual random error that is assumed to be normally distributed.
According to Zeng (1994), the two major properties of CIM were:
Property 1: In the multiple-regression analysis, assuming additivity of QTL effects between loci (i.e., ignoring epistasis), the expected partial regression coefficient of the trait on a marker depends only on those QTL that are located on the interval bracketed by the two neighboring markers and is unaffected by the effects of QTL located on other intervals.
Property 2: Conditioning on unlinked markers in the multiple-regression analysis will reduce the sampling variance of the test statistic by controlling some residual genetic variation and thus will increase the power of QTL mapping.
Both properties come from the regression properties of regression model (4). In Zeng's algorithm, both QTL effect at the current testing interval and regression coefficients of the background markers were estimated simultaneously by an ECM algorithm. However, this algorithm may not completely ensure the two properties.
A modified CIM algorithm:
The basic idea behind the modified algorithm is to use all marker information when building model (4), so that properties 1 and 2 in Zeng (1993, 1994) can be completely guaranteed, and then the interval mapping approach of Lander and Botstein (1989) is applied on the adjusted phenotypic data. Considering that the number of QTL is always much lower than the number of markers, stepwise regression can be used to select the most important marker variables and therefore select the significant QTL. The coefficients of unselected markers through stepwise regression are set to 0 in model (4). When scanning for QTL along the chromosomes, the parameters in model (4) are estimated only once. For a testing position in interval (k, k + 1), the observation values in model (4) can be adjusted by
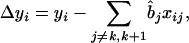 |
(5) |
where 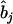 is the estimate of
is the estimate of 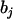 in model (4). As shown in model (3), the two estimates
in model (4). As shown in model (3), the two estimates 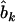 and
and 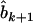 contain all the position- and additive-effect information of the QTL located on the current interval (k, k + 1) under the condition of no QTL in its neighboring intervals and the condition of large samples. Therefore, the use of
contain all the position- and additive-effect information of the QTL located on the current interval (k, k + 1) under the condition of no QTL in its neighboring intervals and the condition of large samples. Therefore, the use of 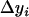 in the subsequent interval mapping does not lose any information of the QTL at the current mapping interval, but the effects of QTL located on other intervals and chromosomes are controlled through the introduction of other coefficients in Equation 5. The adjusted observation
in the subsequent interval mapping does not lose any information of the QTL at the current mapping interval, but the effects of QTL located on other intervals and chromosomes are controlled through the introduction of other coefficients in Equation 5. The adjusted observation 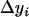 does not change until the testing position moves into a new interval. Please note that the only assumption we made here is that the QTL on the same linkage group or chromosome are isolated by at least one empty interval (isolated QTL according to Whittaker et al. 1996).
does not change until the testing position moves into a new interval. Please note that the only assumption we made here is that the QTL on the same linkage group or chromosome are isolated by at least one empty interval (isolated QTL according to Whittaker et al. 1996).
For a testing position in an interval, all individuals in the backcross population can be classified into four groups on the basis of the two flanking markers (Table 1). If there is one QTL (with the two alleles denoted as Q and q) at the testing position, individuals in all the four groups have QTL genotypes QQ or Qq and hence follow a mixture distribution consisting of components 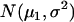 and
and 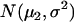 (Table 1) (McLachlan and Basford 1988). The distribution proportions in each mixture distribution depend on the recombination frequencies between QTL and the two flanking markers (Table 1). The existence of QTL at the current mapping position can be tested by the following hypotheses:
(Table 1) (McLachlan and Basford 1988). The distribution proportions in each mixture distribution depend on the recombination frequencies between QTL and the two flanking markers (Table 1). The existence of QTL at the current mapping position can be tested by the following hypotheses:
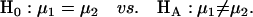 |
TABLE 1.
Marker types on the current mapping interval and their QTL distributions in a backcross population
| Marker genotype
|
Frequency of QTL genotype
|
||||||
|---|---|---|---|---|---|---|---|
| Group | Sample size | Frequency | j | j + 1 | Distribution of 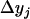
|
||
| 1 | n1 | 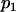 |
+ | + | 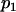 |
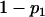 |
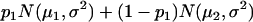 |
| 2 | n2 | 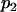 |
+ | − | 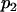 |
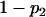 |
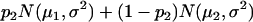 |
| 3 | n3 | 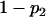 |
− | + | 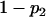 |
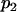 |
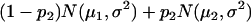 |
| 4 | n4 | 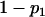 |
− | − | 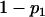 |
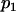 |
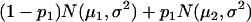 |
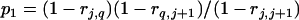 and
and 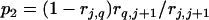 , where
, where 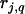 ,
, 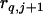 , and
, and 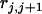 are the recombination frequencies between marker j and the QTL, between the QTL and marker j + 1, and between markers j and j + 1, respectively. “+” denotes homozygote for the marker genotype and “−” denotes heterozygote.
are the recombination frequencies between marker j and the QTL, between the QTL and marker j + 1, and between markers j and j + 1, respectively. “+” denotes homozygote for the marker genotype and “−” denotes heterozygote. 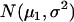 and
and 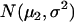 represent the distributions for the two QTL genotypes QQ and Qq, respectively.
represent the distributions for the two QTL genotypes QQ and Qq, respectively.
Supposing that all the n individuals have been sorted on the basis of their marker types, the log-likelihood function under the alternative hypothesis HA is
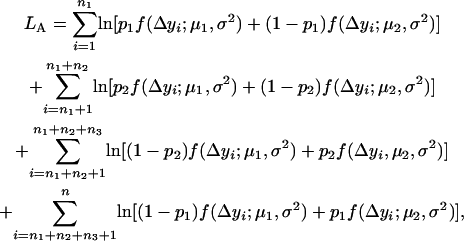 |
(6) |
where 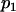 and
and 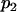 are the proportions of individuals with QQ genotype in group 1 and group 2 or the proportions of individuals with Qq genotype in group 4 and group 3, respectively.
are the proportions of individuals with QQ genotype in group 1 and group 2 or the proportions of individuals with Qq genotype in group 4 and group 3, respectively. 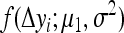 and
and 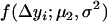 represent the probability densities of the two normal distributions of
represent the probability densities of the two normal distributions of 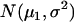 and
and 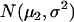 , corresponding to the two QTL genotypes QQ and Qq, respectively (Table 1).
, corresponding to the two QTL genotypes QQ and Qq, respectively (Table 1).
The expectation and maximization (EM) algorithm (Dempster et al. 1977; McLachlan and Basford 1988) is used to estimate the two means and one variance in Equation 6. The initial values of the three unknown parameters can be defined from groups 1 and 4 (Table 1) as
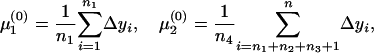 |
and
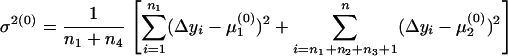 |
In the E-step, the posterior probabilities of an individual being QQ at the QTL in groups 1–4 are
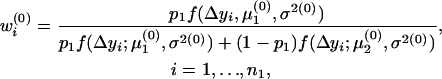 |
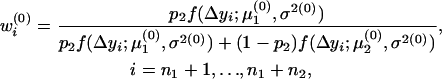 |
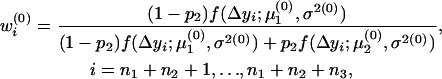 |
and
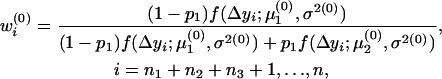 |
respectively.
In the M-step, the three parameters were updated as
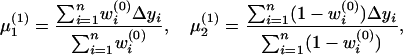 |
and
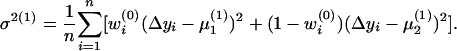 |
The EM algorithm continues until the difference in likelihood function between two consecutive iterations reaches a preassigned precision criterion. The maximum-likelihood estimates thus obtained are represented as 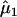 ,
, 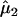 , and
, and 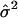 , from which the additive effect of the putative QTL can be estimated.
, from which the additive effect of the putative QTL can be estimated.
Under the null hypothesis, H0, all 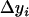 defined by Equation 5 follow a normal distribution denoted as
defined by Equation 5 follow a normal distribution denoted as 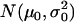 . The mean and variance of this distribution can be estimated as
. The mean and variance of this distribution can be estimated as
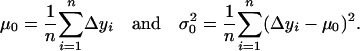 |
Thus, the log-likelihood function under the null hypothesis H0 is
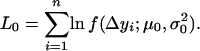 |
The LOD score at the testing position can be calculated from the log-likelihoods under the two hypotheses.
Genetic models used in simulation studies:
Two hypothetical genomes were used in simulation. One genome consisted of six chromosomes, each of 150 cM in length and with 16 evenly distributed markers. Ten QTL (represented by QZ1–QZ10; Table 2) were assumed to contribute to the trait of interest. Three QTL were located on each of the first three chromosomes and one QTL on the fourth chromosome. There was no QTL on chromosomes 5 and 6. The locations and effects of these QTL were similar to the scenario used by Zeng (1994). Both coupling and repulsive linkages and unequal QTL effects were considered in this scenario and therefore should have a wide applicability. To investigate the effect of epistasis on mapping additive QTL, two genetic models were simulated for this genome, one consisting of only additive genetic effects and the other consisting of both additive effects and digenic interactions (Table 2). The additive effects in the epistasis model were the same as those in the additive model, and the interaction effect was drawn from a Gamma distribution implemented by QTL Cartographer (Wang et al. 2005b). Under the QTL distribution in Table 2, the theoretical additive variance was 4.0, and the theoretical epistasis variance was 2.0 (estimated by QTL Cartographer). Two heritability (in the broad sense) levels were considered: H = 0.8 (representing high heritability traits) and H = 0.5 (representing medium heritability traits). One hundred backcross populations of 200 individuals were simulated for each model by heritability combination using QTL Cartographer.
TABLE 2.
Chromosomal position and additive and additive-by-additive epistatic effects of 10 QTL
| Chromosome
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1 | 2 | 2 | 2 | 3 | 3 | 3 | 4 | ||
| Position (cM): | 16 | 48 | 108 | 3 | 43 | 77 | 33 | 68 | 129 | 26 | |
| QTL symbol: | QZ1 | QZ2 | QZ3 | QZ4 | QZ5 | QZ6 | QZ7 | QZ8 | QZ9 | QZ10 | |
| QZ1 | 0.54 | ||||||||||
| QZ2 | 0.16 | 0.95 | |||||||||
| QZ3 | 0.45 | 0.17 | 0.73 | ||||||||
| QZ4 | 0.92 | 1.29 | |||||||||
| QZ5 | 0.61 | −1.57 | |||||||||
| QZL6 | −1.16 | 0.46 | 0.21 | −1.61 | |||||||
| QZ7 | 0.17 | 1.30 | −0.59 | ||||||||
| QZ8 | 1.18 | 2.91 | 0.36 | 2.05 | |||||||
| QZ9 | 0.30 | −1.12 | −0.30 | −1.72 | 1.12 | ||||||
| QZ10 | −0.44 | −0.96 | 2.96 | 0.94 | |||||||
| PVE of additive genetic model | |||||||||||
| H = 0.8 | 1.46 | 4.51 | 2.66 | 8.32 | 12.30 | 12.96 | 1.74 | 21.01 | 6.27 | 4.42 | |
| H = 0.5 | 0.91 | 2.82 | 1.67 | 5.20 | 7.70 | 8.10 | 1.09 | 13.13 | 3.92 | 2.76 | |
| PVE of additive and epistasis genetic model | |||||||||||
| H = 0.8 | 0.97 | 3.01 | 1.78 | 5.55 | 8.22 | 8.64 | 1.16 | 14.01 | 4.18 | 2.95 | |
| H = 0.5 | 0.61 | 1.88 | 1.11 | 3.47 | 5.14 | 5.40 | 0.73 | 8.76 | 2.61 | 1.84 | |
The additive variance (VA) was 4.0, and the interaction variance (VI) was half of the additive variance. The interaction effect was drawn from a Gamma distribution 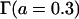 . The error variance (Ve) was calculated by
. The error variance (Ve) was calculated by 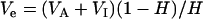 , where H is the heritability in the broad sense. When interaction was not included, the error variances were 1.0 and 4.0 for H = 0.8 and H = 0.5, respectively. When interaction was included, the error variances were 1.5 and 6.0 for H = 0.8 and H = 0.5, respectively. PVE, percentage of variance explained by individual QTL.
, where H is the heritability in the broad sense. When interaction was not included, the error variances were 1.0 and 4.0 for H = 0.8 and H = 0.5, respectively. When interaction was included, the error variances were 1.5 and 6.0 for H = 0.8 and H = 0.5, respectively. PVE, percentage of variance explained by individual QTL.
The other genome consisted of four chromosomes, each with 100 cM in length and 21 markers evenly distributed. Eight large-effect QTL (represented by QY1–QY8) and 16 small-effect QTL contributed to the expression of a quantitative trait of interest (for details see Table 1 in Yi et al. 2003). To compare CIM and ICIM with the Bayesian mapping methods of Yi et al. (2003), 100 backcross populations each of 300 individuals were generated, and the residual variance 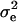 was adjusted to 1. The population size and the residual variance were the same as those used in Yi et al. (2003).
was adjusted to 1. The population size and the residual variance were the same as those used in Yi et al. (2003).
For CIM, we applied different background marker selection methods available in QTL Cartographer. The model using stepwise regression to select control markers was the best in terms of the estimates of QTL positions and effects, so other models for CIM were not considered in power analysis. For ICIM, the stepwise regression was used to select markers and estimate the parameters in model (4), in which the largest P-value for entering variables was set at 0.05, and the smallest P-value for removing variables was 0.10. CIM was implemented by QTL Cartographer, and IM (where applicable) and ICIM were implemented by an in-house computer program called IciMapping (available from http://www.isbreeding.net/software.html). On the basis of a permutation test, a LOD threshold of 2.5 was used to declare the presence of a QTL.
Power calculation and position and effect estimation:
QTL mapping based on an interval test is not a point estimation, which makes it complicated to calculate power through simulation. Especially, when QTL are closely linked, it is difficult to determine which putative QTL the LOD peak belongs to. We adopted two methodologies to calculate power. First, a power was calculated for each interval defined by markers. This power calculation allows monitoring of QTL locations if not on the predefined intervals. Second, each predefined QTL was assigned to a 10-cM interval centered at the true QTL location, and then the power was estimated for the so-defined confidence interval. QTL identified in other intervals were viewed as false positives.
We also adopted two methodologies to calculate the mean QTL position and effect (Zeng 1994). One was calculated from all peaks in the confidence interval across 100 runs, and the other from the peaks having a LOD score over the predefined threshold of 2.5.
SIMULATION RESULTS
LOD score distribution of ICIM:
Permutation tests (Churchill and Doerge 1994) were conducted to find the LOD score distributions of ICIM under the null hypothesis. These distributions were different for different genetic models and heritability levels in genome 1 (Table 3). For a population size of 200, the probabilities that the LOD score was >2.5 were 0.0630, 0.0311, 0.0462, and 0.0204 (calculated from Table 3) for the four combinations of two genetic models and two heritability levels, respectively. For a population size of 300, these probabilities were 0.1393, 0.0899, 0.0932, and 0.0474, respectively. Results in Table 3 indicate that different LOD thresholds should be applied for different data sets to ensure the same level of false-positive rates. For simplicity, we applied the LOD threshold of 2.5 in the simulation study. This threshold value may not result in a false discovery rate <0.05, but will have little effect on the comparison of different mapping methods. Moreover, we compared ICIM with CIM not only in terms of mapping power but also in terms of the number of false positives.
TABLE 3.
Accumulated probability of LOD score, P(LOD < x), from a permutation test
| Population size 200
|
Population size 300
|
|||||||
|---|---|---|---|---|---|---|---|---|
| ADD
|
ADD + EPI
|
ADD
|
ADD + EPI
|
|||||
| x | H = 0.8 | H = 0.5 | H = 0.8 | H = 0.5 | H = 0.8 | H = 0.5 | H = 0.8 | H = 0.5 |
| 2.0 | 0.9018 | 0.9434 | 0.9255 | 0.9610 | 0.8250 | 0.8773 | 0.8771 | 0.9214 |
| 2.5 | 0.9370 | 0.9689 | 0.9538 | 0.9796 | 0.8607 | 0.9101 | 0.9068 | 0.9526 |
| 3.0 | 0.9609 | 0.9823 | 0.9724 | 0.9893 | 0.8891 | 0.9351 | 0.9261 | 0.9720 |
| 3.5 | 0.9757 | 0.9900 | 0.9836 | 0.9943 | 0.9131 | 0.9543 | 0.9401 | 0.9837 |
| 4.0 | 0.9857 | 0.9936 | 0.9902 | 0.9973 | 0.9328 | 0.9691 | 0.9519 | 0.9913 |
| 4.5 | 0.9921 | 0.9966 | 0.9950 | 0.9987 | 0.9477 | 0.9800 | 0.9607 | 0.9953 |
| 5.0 | 0.9955 | 0.9979 | 0.9975 | 0.9993 | 0.9597 | 0.9875 | 0.9685 | 0.9977 |
| 5.5 | 0.9973 | 0.9991 | 0.9988 | 0.9997 | 0.9696 | 0.9921 | 0.9743 | 0.9986 |
| 6.0 | 0.9987 | 0.9996 | 0.9994 | 0.9999 | 0.9772 | 0.9950 | 0.9791 | 0.9993 |
Power analysis of CIM and ICIM from genome 1:
On the average LOD profiles of ICIM displayed clear peaks around most of the predefined QTL, but this was not the case for CIM especially on chromosomes where there were multiple QTL (Figure 1). The three QTL on chromosome 2, i.e., QZ4, QZ5, and QZ6 (explaining 8.32, 12.30, and 12.96% of the phenotypic variance under the additive genetic model and H = 0.8, respectively; Table 2), had similar effects. QZ4 was linked with QZ5 in repulsive phase, and QZ5 was linked with QZ6 in coupling phase. Three clear peaks were observed on the average LOD profiles of ICIM, but it was hard to distinguish QZ5 and QZ6 on the average LOD profiles of CIM (Figure 1). The average LOD profiles were very low on chromosomes 5 and 6 on which there were no QTL, indicating that both CIM and ICIM are less likely to locate a QTL on one chromosome to other chromosomes.
When powers were calculated for all marker intervals along the six chromosomes, the probability that QTL were mapped onto the two devoid chromosomes (i.e., 5 and 6) was rather low for both CIM and ICIM (Figure 2), as has been seen from the average LOD profiles in Figure 1. The advantage of ICIM over CIM was not significant for chromosome 3 (Figure 2), for which QZ7 has a very small effect (explaining 1.74% of phenotypic variance under the additive model and H = 0.8; Table 2), and QZ8 and QZ9 are far apart (61 cM apart on chromosome 3; Table 2). The advantage was not significant for chromosome 4 either, on which there was only one QTL (Figure 2). However, the advantage of ICIM was significant for chromosome 2 for all genetic models and heritability levels, on which there were three QTL of similar effects (QZ4, QZ5, and QZ6), and the distances between QZ4 and QZ5 and QZ5 and QZ6 were 40 and 34 cM, respectively (Table 2). ICIM had higher powers to map QZ4 and QZ5 in the right intervals and lower probability to assign them to incorrect intervals than CIM (Figures 2 and 3).
Figure 2.—

Power of QTL detection of CIM and ICIM for two genetic models and heritability levels under genome 1. Power was calculated as the proportion of runs that detected the presence of QTL for each of the 90 intervals defined by the 96 markers evenly distributed on six chromosomes. Arrow size and direction represent the approximate effect size and direction of the pointed QTL, respectively. (A) Additive genetic model, H = 0.8. (B) Additive genetic model, H = 0.5. (C) Additive and epistasis model, H = 0.8. (D) Additive and epistasis model, H = 0.5.
Figure 3.—

Power of QTL detection of CIM and ICIM for two genetic models and heritability levels under genome 1. Power was calculated as the proportion of runs that detected QTL within the interval defined as 5 cM from each side of the predefined QTL. The QTL were rearranged in ascending order by the percentage of variance explained. (A) Additive genetic model, H = 0.8. (B) Additive genetic model, H = 0.5. (C) Additive and epistasis model, H = 0.8. (D) Additive and epistasis model, H = 0.5.
The advantage of ICIM over CIM was much clearer when power was calculated on the basis of an interval of 10 cM centered on the predefined QTL (Figure 3). For QZ1, CIM had higher powers than ICIM for the additive genetic model under heritability 0.5 and the additive and epistasis model under heritability 0.8. For QZ2 and QZ6, CIM also had higher powers in most cases. For QZ8, the powers of CIM and ICIM for the additive genetic model under heritability 0.5 and the additive and epistasis model under heritability 0.5 were the same, and for QZ9 those powers for the additive genetic model under heritability 0.5 were the same. But for all other cases, ICIM had higher powers. On average, CIM identified 6.11, 3.63, 3.97, and 2.35 QTL for the two genetic models and two heritability levels in each run, respectively, while ICIM identified 7.52, 4.40, 4.84, and 2.72 QTL, respectively, and all of them were higher than those observed from CIM (Figure 4). QTL identified in intervals other than the defined intervals were viewed as false positives. On average the false QTL numbers in each run were 6.58, 4.82, 4.85, and 3.36 for CIM, but 3.67, 3.71, 3.31, and 2.73 for ICIM, respectively (Figure 4). Thus, the proportions of true to false positives were 0.93, 0.75, 0.82, and 0.70 for CIM, but 2.05, 1.19, 1.46, and 1.00 for ICIM for the two genetic models and two heritability levels, respectively. As shown in Figure 2, many false positives were located in the neighboring intervals on the chromosomes, and the proportion of true QTL vs. false positives was generally dependent on the width of confidence intervals.
Figure 4.—

Average number of QTL identified on the 10-cM intervals of the 10 predefined QTL (A) and other chromosome regions (B) for two genetic models and heritability levels under genome 1.
Lower heritability and the inclusion of epistasis reduced the power for mapping additive QTL using CIM and ICIM (Figures 1–4). The mapping power under the additive and epistasis model and heritability 0.8 was similar to that under the additive genetic model and heritability 0.5. When mapping was conducted assuming additivity of QTL, the epistatic effect should enter into the sampling error. The effect of epistasis on additive QTL mapping was equivalent to additional random errors on phenotypic data. For the additive and epistasis genetic model under heritability 0.8 (Table 2), the proportion of additive variance to the phenotypic variance was 4/(4 + 2 + 1.5) ≈ 53%, which was similar to the additive genetic model of heritability 0.5 (Table 2). This suggests that CIM and ICIM are still effective for locating the QTL and estimating their additive effects when epistasis is present if the heritability in the narrow sense is not too low.
Estimation of QTL positions and effects from genome 1:
The estimates of QTL positions can be on the left or right of the true position. The deviation of the average position estimates across the 100 simulations ranged from −2.23 to 2.40 cM (Table 4). There is a tendency that QTL were mapped toward to their closest markers. In other words, a QTL closer to its left flanking marker had a negative deviation (i.e., QZ4 at 3 cM and QZ5 at 43 cM on chromosome 2 and QZ7 at 33 cM on chromosome 3), and a QTL closer to its right flanking marker had a positive deviation (i.e., QZ2 at 48 cM and QZ3 at 108 cM on chromosome 1, QZ6 at 77 cM on chromosome 2, and QZ8 at 68 cM and QZ9 at 129 cM on chromosome 3). This is understandable by looking into the two coefficients in model (2). If a QTL is located in the middle of a flanking interval, its effect will be evenly absorbed by the two flanking markers. Otherwise, the marker closer to the QTL will absorb most of the QTL variation and is more likely retained through stepwise regression. Therefore, a QTL is more likely to be mapped onto the marker closer to it. Due to the same reason, the highest power of ICIM was not reached on interval (120, 130) on chromosome 3 where QZ9 was located (i.e., 129 cM), but was achieved on interval (130, 140), as shown in Figure 2.
When calculated from all peaks, the estimate of QTL effect was almost unbiased. In comparison, the QTL effect was generally overestimated when calculated from the significant peaks only, which was expected as small-effect estimates in some simulation runs were not counted. Therefore, in any simulation studies, it is not likely to achieve an unbiased estimation of QTL effect if only significant QTL are counted. Compared with CIM, ICIM tends to have smaller bias in effect estimation in most cases (Table 4).
Simulation results from genome 2:
The advantage of ICIM over CIM is also significant for genome 2 (Figures 5 and 6). The power given in Figure 5A was calculated for each chromosomal interval of 5 cM as defined by two neighboring markers. For ICIM, the highest powers were achieved in the intervals where QY1, QY3, QY4, QY5, QY7, and QY8 were located, which were 0.56, 0.69, 0.67, 0.72, 0.71, and 0.78, respectively. The highest powers were achieved in the neighboring intervals for QY2 and QY6 (Figure 5A). The result of CIM was comparable to that of ICIM for chromosome 4, for which the two QTL were linked in coupling with a distance of 35 cM. The worst performance of CIM was for chromosome 2, for which the two QTL were linked in repulsion with a distance of 21 cM (Figure 5A).
Figure 5.—

Power of QTL detection (A), average LOD (B), and additive effect (C) profiles across the 100 simulation runs of CIM and ICIM based on genome 2. Power was calculated as the proportion of runs that detected the presence of QTL for each of the 80 intervals defined by 84 markers evenly distributed on four chromosomes. Arrow size and direction represent the approximate effect size and direction of the pointed QTL, respectively.
Figure 6.—

Power (A) and deviation of position estimation (B) in genome 2 from 100 simulation runs. Each predefined QTL was assigned to a 10-cM interval centered at the true QTL location.
The mean LOD profile of ICIM across the 100 simulations had eight clear peaks corresponding to the eight major predefined QTL, while that of CIM only had two clear peaks on chromosome 4 (Figure 5B). The mean estimated effects from ICIM across the 100 simulation runs were close to the true QTL effects for all the eight predefined major QTL. But CIM tended to overestimate QTL effects on chromosomes 1, 3, and 4 where the linked QTL were in the coupling phase, but tended to underestimate QTL effects on chromosome 2, where the linked QTL were in the repulsive phase (Figure 5C).
When a confidence interval of 10 cM with the predefined QTL at the center was considered, ICIM had a power >0.87 to map the eight major QTL (Figure 6A). The powers of CIM were >0.76 for QTL on chromosomes 1, 3, and 4, for which linked QTL were in the coupling phase, but were only 0.09 for QY3 and 0.47 for QY4 on chromosome 2, where the two QTL were linked in repulsion (Figure 6A). In addition, ICIM also resulted in less false positives (total false positives in intervals other than the eight confidence intervals divided by 8) than CIM (Figure 6A). Across the 100 simulations, the estimates of QTL effects were almost unbiased for ICIM (Figure 6B), while the effect estimates of QY3 and QY4 on chromosome 2 were about twice the true QTL effects for CIM.
DISCUSSION
Theoretical justification of ICIM:
For both ICIM and CIM, Mendelian segregation and recombination laws (Table 1) and quantitative genetic theories [models (1)–(3)] provide the theoretical basis (Falconer and Mackay 1996; Lynch and Walsh 1998; Hartl and Jones 2005), and regression and maximum-likelihood principles [Table 1, model (4), and Equation 6] provide the statistical basis. The statistical assumption made in CIM and ICIM is that the residual errors in model (4) are normally distributed. The genetic assumptions are that (i) the genotypic value of an individual is the summation of effects from all loci affecting the trait of interest and (ii) linked QTL are separated by at least one blank interval. These well-established genetic and statistical theories ensure that these mapping methods are valid under these assumptions. However, simulations are useful if one wants to investigate their sensitivities to the violation of the underlying assumptions, such as nonisolated QTL and epistasis.
ICIM makes the background marker selection process easier:
Various methods for selecting background markers are available in QTL Cartographer implementing CIM (Wang et al. 2005b), and different methods may result in different, sometime controversial, mapping results. A mapping population from the first simulation run using the additive genetic model and H = 0.8 in genome 1 was used to demonstrate this point. Three cofactor selection methods used in CIM, i.e., unlinked marker control, all marker control, and the standard model using stepwise regression (window size 10 cM), gave rather different LOD profiles (Figure 7A). The method using unlinked markers as a control was similar to IM (Figure 7A), which should not be recommended. The method using all markers as a control had clear peaks where major QTL were located, but the LOD score was generally lower than that from IM (Figure 7A). Moreover, power analysis showed that this method resulted in a large number of false positives on the two devoid chromosomes (results not shown), and this method cannot be applied when markers outnumber the mapping population size.
Figure 7.—

LOD profiles of (A) IM, CIM (with three background marker selection methods), and (B) ICIM (with three probability levels). The first simulated backcross population of 200 individuals was used, where the 10 QTL were additive, and H = 0.8. For clarity, 20, 40, and 60 were added to the LOD scores of CIM with unlinked marker control, of CIM with all marker control, and of CIM standard model control, respectively. Similarly, 20, 40, and 60 were added to the LOD scores of ICIM with PIN = 0.10 and POUT = 0.20, of ICIM with PIN = 0.05 and POUT = 0.10, and of ICIM with PIN = 0.01 and POUT = 0.02, respectively.
In ICIM, the background markers were selected only once using the standard stepwise regression, and thus the difficulty in choosing the background markers associated with CIM can be avoided. To investigate the influence of marker inclusion and exclusion criteria in stepwise regression on mapping results, different P-values for entering variables (PIN) and removing variables (POUT) were applied to the population used above for comparing marker selection methods in CIM. The LOD profiles from three levels of PIN and POUT showed very little difference (Figure 7B). All three probability levels identified the 8 largest QTL (Figure 7B). When PIN = 0.10 and POUT = 0.20 were used, the average power to detect the 10 predefined QTL from the additive genetic model and H = 0.8 was slightly lower than that from PIN = 0.05 and POUT = 0.10, i.e., 73.7 vs. 75.2 (calculated from Table 5), but the false positive was much higher, i.e., 511 vs. 367 (Table 5). Similar results were also observed for other genetic models. Therefore, ICIM is robust to the choice of probability levels, and in practice a lower probability level should be applied to further reduce the false positives without sufficiently changing the detection power (Figure7B and Table 5).
TABLE 5.
Effect of marker inclusion and exclusion probabilities in the stepwise regression of ICIM
| Genetic model | PIN | POUT | QZ1 | QZ2 | QZ3 | QZ4 | QZ5 | QZ6 | QZ7 | QZ8 | QZ9 | QZ10 | False QTLa |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ADD, H = 0.8 | 0.01 | 0.02 | 35 | 77 | 57 | 94 | 94 | 95 | 22 | 99 | 82 | 71 | 267 |
| 0.05 | 0.10 | 41 | 76 | 59 | 96 | 95 | 92 | 32 | 99 | 86 | 76 | 367 | |
| 0.10 | 0.20 | 40 | 72 | 56 | 94 | 94 | 92 | 35 | 99 | 82 | 73 | 511 | |
| ADD, H = 0.5 | 0.01 | 0.02 | 13 | 32 | 11 | 51 | 50 | 71 | 8 | 79 | 41 | 24 | 286 |
| 0.05 | 0.10 | 12 | 36 | 15 | 63 | 63 | 72 | 18 | 82 | 47 | 32 | 371 | |
| 0.10 | 0.20 | 18 | 33 | 24 | 68 | 68 | 71 | 26 | 83 | 49 | 33 | 531 | |
| ADD + EPI, H = 0.8 | 0.01 | 0.02 | 10 | 44 | 24 | 61 | 56 | 73 | 7 | 91 | 47 | 26 | 250 |
| 0.05 | 0.10 | 14 | 35 | 25 | 74 | 69 | 77 | 11 | 92 | 51 | 36 | 331 | |
| 0.10 | 0.20 | 15 | 34 | 28 | 73 | 68 | 77 | 14 | 92 | 54 | 38 | 481 | |
| ADD + EPI, H = 0.5 | 0.01 | 0.02 | 9 | 20 | 9 | 17 | 26 | 50 | 2 | 54 | 23 | 7 | 194 |
| 0.05 | 0.10 | 11 | 20 | 13 | 31 | 37 | 52 | 5 | 61 | 30 | 12 | 273 | |
| 0.10 | 0.20 | 11 | 22 | 15 | 36 | 44 | 52 | 8 | 68 | 33 | 15 | 459 |
ADD, additive genetic model as defined in Table 2; ADD + EPI, additive and epistasis genetic model as defined in Table 2; H, heritability in the broad sense; PIN, the largest P-value for entering variables; POUT, the smallest P-value for removing variables.
All significant positives not located in the 10 predefined QTL intervals.
ICIM does not increase sampling variance compared to IM:
According to property 3 proposed by Zeng (1994), conditioning on linked markers in the multiple-regression analysis will reduce the influence of interference caused by possible multiple linked QTL on hypothesis testing and parameter estimation, but with a possible increase of sampling variance. The increased sampling variance can be seen from the lower LOD scores (compared with IM) that resulted from CIM using all markers as a control (Figure 7A). But this is not the case for ICIM. At most peak positions, ICIM has higher LOD scores than IM (Figure 7, A and B). So for ICIM, properties 2 and 3 of CIM (Zeng 1994) can be merged as “Conditioning on both linked and unlinked markers in the multiple regression analysis will reduce the sampling variance of the test statistic by controlling some residual genetic variation and thus will increase the power of QTL mapping” (Zeng 1994, p. 1460).
CIM and ICIM are valid and simple methods for mapping with populations derived from biparental crosses:
CIM, when implemented properly, represents the best single-interval mapping method based on linear model and maximum-likelihood principles. Recently, Bayesian models have gained some popularity among theoreticians. In a sense and in the context of QTL mapping using populations derived from biparental crosses, both frequentist statistics and Bayesian statistics deal with the maximization of likelihood function. The major difference is that a prior distribution has to be considered in any Bayesian model, and the choice of the prior in the case of QTL mapping is rather arbitrary and a tedious process (Xu 2003). On the basis of the prior, Bayesian statistics derive the posterior and then conduct inference on the basis of the posterior distribution. The conventional maximum likelihood can be viewed as a special case of Bayesian models where a uniform density is used as the prior distribution. It should be noted that the effect and advantage of prior distribution diminish as the sample size increases (Gelman et al. 2004). The population size for a QTL mapping population is normally hundreds, which can be reasonably regarded as a large sample in statistics. Theoretically, Bayesian mapping has no flaw in terms of models. However, Bayesian statistics may not have significant advantages over frequentist statistics in QTL mapping for some standard mapping populations derived from biparental crosses, considering the difficulty in choosing prior distributions and the complexity in computing posterior distributions.
Genome 2 used in our simulation has been used by Yi et al. (2003) to demonstrate their Bayesian mapping method. The results of ICIM shown in Figure 5 were comparable with the probability profile from the Bayesian method (Figure 1 in Yi et al. 2003). The mean effects for the eight identified large-effect QTL were 1.02, 0.96, −0.83, 0.81, −0.78, −0.87, 1.00, and 1.03, respectively (Figure 5C), which were close to the true additive effects. But ICIM is much simpler in principle and faster in computation. It required <5 min for ICIM to complete the 100 simulation runs in a personal computer. In addition, we also compared ICIM with the Bayesian mapping method proposed by Sillanpää and Arjas (1998), and the results of ICIM were very similar to those from Bayesian models (results not shown).
Since the number of QTL is always much lower than the number of markers, QTL mapping can be viewed as an issue of model selection (Broman and Speed 2002; Sillanpää and Corander 2002). A number of statistical methods are available to search through the space of models and various criteria can be used to select the best model (Miller 1990; Piepho and Gauch 2001). However, there is no conclusion in statistics as to which model selection method is the best (Miller 1990). On the basis of our simulation results, the performance of stepwise regression is satisfactory. However, we do not exclude the possibility that other model selection methods may achieve similar performance as the stepwise regression used in ICIM.
The calculation of probability that a QTL is in a given interval is viewed as a major advantage of Bayesian models (Ball 2001). In any likelihood-ratio test-based mapping methods such as IM, CIM, and ICIM, the QTL position is estimated as the peak of the LOD profile with a LOD score over a specified threshold value. The LOD score is actually a likelihood-ratio test (LRT) [LRT = 2 log(10)LOD ≈ 4.61 LOD]. In the case of mixture models (McLachlan and Basford 1988; Goffinet et al. 1992), the asymptotic distribution of LRT may not exactly follow a 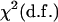 , where d.f. is the difference in the number of dependent variables under the two hypotheses. However, Zeng (1994) showed when the sample size was large and the number of markers fitted to the model was relatively small, the LRT statistic was still approximately distributed as
, where d.f. is the difference in the number of dependent variables under the two hypotheses. However, Zeng (1994) showed when the sample size was large and the number of markers fitted to the model was relatively small, the LRT statistic was still approximately distributed as 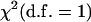 . Another way to find the LOD threshold is to use permutation tests (Churchill and Doerge 1994). A probability may be calculated at any testing position if required. In any mapping methods based on the likelihood-ratio test, the LOD score actually indicates the likelihood of a QTL at the testing position. The similarity between the mean LOD score profile shown in Figure 5B and the probability profile shown in Figure 1 in Yi et al. (2003) is consequently not unexpected.
. Another way to find the LOD threshold is to use permutation tests (Churchill and Doerge 1994). A probability may be calculated at any testing position if required. In any mapping methods based on the likelihood-ratio test, the LOD score actually indicates the likelihood of a QTL at the testing position. The similarity between the mean LOD score profile shown in Figure 5B and the probability profile shown in Figure 1 in Yi et al. (2003) is consequently not unexpected.
Multiple interval mapping (MIM) was proposed to map multiple QTL simultaneously (Kao et al. 1999; Zeng et al. 1999). MIM may have avoided the complicated background selection process associated with CIM, but introduced various model selection methods (Zeng et al. 1999; Wang et al. 2005b). We applied two model selection methods available in QTL Cartographer to the same mapping population previously used for comparing different cofactor selection methods in CIM. Eleven QTL were identified when the forward and backward selection on markers was used, while only 6 QTL were identified when the MIM forward search method was used (results not shown). Again, different MIM model selection methods resulted in different mapping results.
Further considerations of QTL mapping:
An important assumption of most QTL mapping methods is that the QTL are separated by at least one blank interval. It is expected that this assumption is more satisfactory for narrow than for wide marker spacing. Similarly, it is a better assumption for independent or loosely linked than for tightly linked QTL. With the rapid development of molecular technology, high-density linkage maps are becoming available for more and more species. The treatment of tightly linked QTL is more an issue of biology than of statistical methodology. Two linked QTL can be separated only if recombinants are sampled in the mapping population (Kearsey 2002). Therefore, the mapping resolution is limited by the practicable mapping population size. Populations of size ≥500 are rarely seen in practice for mapping using primary populations such as backcross, F2, and recombination inbred lines. Therefore, QTL mapping using primary populations can give only a rough position and effect estimation due to the limited population size and errors in both genotyping and phenotyping. Once the QTL interval has been identified, some secondary mapping populations should be built from the preliminary mapping population and fine mapping needs to be conducted (Kearsey 2002). As whole-genome genotyping is not requested in the secondary population, and selective phenotyping may be implemented, a larger population can be used. At the same time, new markers in the candidate intervals may be discovered and added to the linkage map. By then we may determine whether the identified QTL contain one gene or multiple genes.
It is now common that the number of markers exceeds the sample size of the mapping population. The performance of ICIM and other recommended mapping methods needs to be investigated under this situation. We have used a backcross population to illustrate our method in this article. However, the extension of ICIM to F2, doubled haploids, and recombination inbred lines is straightforward. CIM is not extendable to epistasis (Zeng et al. 1999). As can be seen, models (1)–(3) can be easily extended to include epistasis. For instance, by inclusion of marker-pair multiplications in model (4) digenic epistasis can be modeled. This work is currently under development.
Conclusions:
The problem with the current CIM method is the arbitrariness of choosing the cofactors. Different methods of cofactor selection will generate different, sometime controversial results. In ICIM, significant cofactors are selected and their corresponding effects are estimated by using stepwise regression analysis prior to interval mapping. The effects of the cofactors are then fixed when used in the genome-scanning process. This eliminates the arbitrariness of cofactor selection associated with CIM. ICIM has a simpler form and faster convergence speed (EM algorithm converges after three to five iterations), without losing the optimal properties of CIM. ICIM gives clearly high LOD scores at chromosomal regions with QTL but rather low LOD scores where no QTL is located and results in less biased estimates of QTL effects, thereby improving the mapping power and precision. Extensive simulations showed that ICIM improved the performance of QTL mapping over the existing CIM method.
Acknowledgments
The authors thank two anonymous reviewers for their useful comments on earlier versions of the manuscript. This work was supported by the National 973 Projects of China (no. 2006CB101700) and the Generation Challenge Program of the Consultative Group for International Agricultural Research.
References
- Ball, R. D., 2001. Bayesian methods for quantitative trait locus mapping based on model selection: approximate analysis using the Bayesian information criterion. Genetics 159: 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan, M., J. K. Ghosh and R. W. Doerge, 2004. Modifying the Schwarz Bayesian information criterion to locate multiple interacting quantitative trait loci. Genetics 167: 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., and T. P. Speed, 2002. A model selection approach for the identification of quantitative trait loci in experimental crosses. J. R. Stat. Soc. B 64: 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138: 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster, A., N. Laird and D. Rubin, 1977. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. B 39: 1–38. [Google Scholar]
- Doerge, R. W., Z-B. Zeng and B. S. Weir, 1997. Statistical issues in the search for genes affecting quantitative traits in experimental populations. Stat. Sci. 12: 195–219. [Google Scholar]
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics, Ed. 4. Longman, Essex, England.
- Gelman, A., J. B. Carlin, H. S. Stern and D. B. Rubin, 2004. Bayesian Data Analysis. Chapman & Hall/CRC Press, London.
- Goffinet, B., P. Loisel and B. Lawrent, 1992. Testing in normal mixture models when the proportions are known. Biometrika 79: 842–846. [Google Scholar]
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative loci in line crosses using flanking markers. Heredity 69: 315–324. [DOI] [PubMed] [Google Scholar]
- Hartl, D., and E. Jones, 2005. Genetics: Analysis of Genes and Genomes, Ed. 6. Jones & Bartlett, Sudbury, MA.
- Jansen, R. C., 1994. High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136: 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C.-H., Z-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152: 1203–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearsey, M. J., 2002. QTL analysis: problems and (possible) solutions, pp. 45–58 in Quantitative Genetics, Genomics and Plant Breeding, edited by M. S. Kang. CABI Publishing, Wallingford, UK.
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Martinez, O., and R. N. Curnow, 1992. Estimating the locations and the sizes of the effects of quantitative trait loci using flanking markers. Theor. Appl. Genet. 85: 480–488. [DOI] [PubMed] [Google Scholar]
- McLachlan, G. J., and K. E. Basford, 1988. Mixture Models: Inference and Applications to Clustering. Marcel Dekker, New York.
- Miller, A. J., 1990. Subset Selection in Regression (Monographs on Statistics and Applied Probability 40). Chapman & Hall, London.
- Piepho, H.-P., and H. G. Gauch, 2001. Marker pair selection for mapping quantitative trait loci. Genetics 157: 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satagopan, J. M., B. S. Yandell, M. A. Newton and T. C. Osborn, 1996. A Bayesian approach to detect quantitative trait loci using Markov chain Monte Carlo. Genetics 144: 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen, S., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159: 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpää, M. J., and E. Arjas, 1998. Bayesian mapping of multiple quantitative trait loci from incomplete inbred line cross data. Genetics 148: 1373–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpää, M. J., and J. Corander, 2002. Model choice in gene mapping: what and why. Trends Genet. 18: 302–307. [DOI] [PubMed] [Google Scholar]
- Soller, M., T. Brody and A. Genizi, 1976. On the power of experimental design for detection of linkage between marker loci and quantitative loci in crosses between inbred lines. Theor. Appl. Genet. 47: 35–39. [DOI] [PubMed] [Google Scholar]
- Steinmetz, L. M., H. Sinha, D. R. Richards, J. I. Spiegelman, P. J. Oefner et al., 2002. Dissecting the architecture of a quantitative trait locus in yeast. Nature 416: 326–330. [DOI] [PubMed] [Google Scholar]
- Van Den Oord, E. J. C. G., and P. F. Sullivan, 2003. False discoveries and models for gene discovery. Trends Genet. 19: 537–542. [DOI] [PubMed] [Google Scholar]
- Uimari, P, and I. Hoeschele, 1997. Mapping-linked quantitative trait loci using Bayesian analysis and Markov chain Monte Carlo algorithms. Genetics 146: 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., Y.-M. Zhang, X. Li, G. L. Masinde, S. Mohan et al., 2005. a Bayesian shrinkage estimation of quantitative trait loci parameters. Genetics 170: 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S., C. J. Basten and Z-B. Zeng, 2005. b Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC.
- Whittaker, J. C., R. Thompson and P. M. Visscher, 1996. On the mapping of QTL by regression of phenotype on marker-type. Heredity 77: 23–32. [Google Scholar]
- Wright, A. J., and R. P. Mowers, 1994. Multiple regression for molecular-marker, quantitative trait data from large F2 populations. Theor. Appl. Genet. 89: 305–312. [DOI] [PubMed] [Google Scholar]
- Wu, R., and M. Lin, 2006. Functional mapping—how to map and study the genetic architecture of dynamic complex traits. Nat. Rev. Genet. 7: 229–237. [DOI] [PubMed] [Google Scholar]
- Xu, S., 2003. Estimating polygenic effects using markers of the entire genome. Genetics 163: 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., V. George and D. B. Allison, 2003. Stochastic search variable selection for identifying multiple quantitative trait loci. Genetics 164: 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z-B., 1993. Theoretical basis for separation of multiple linked gene effects in mapping of quantitative trait loci. Proc. Natl. Acad. Sci. USA 90: 10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z-B., 1994. Precision mapping of quantitative trait loci. Genetics 136: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z-B., C.-H. Kao and C. J. Basten, 1999. Estimating the genetic architecture of quantitative traits. Genet. Res. 74: 279–289. [DOI] [PubMed] [Google Scholar]