

Abstract
Ribosomal (r)DNA undergoes concerted evolution, the mechanisms of which are unequal crossing over and gene conversion. Despite the fundamental importance of these mechanisms to the evolution of rDNA, their rates have been estimated only in a few model species. We estimated recombination rate in rDNA by quantifying the relative frequency of intraindividual length variants in an expansion segment of the 18S rRNA gene of the cladoceran crustacean, Daphnia obtusa, in four apomictically propagated lines. We also used quantitative PCR to estimate rDNA copy number. The apomictic lines were sampled every 5 generations for 90 generations, and we considered each significant change in the frequency distribution of length variants between time intervals to be the result of a recombination event. Using this method, we calculated the recombination rate for this region to be 0.02–0.06 events/generation on the basis of three different estimates of rDNA copy number. In addition, we observed substantial changes in rDNA copy number within and between lines. Estimates of haploid copy number varied from 53 to 233, with a mean of 150. We also measured the relative frequency of length variants in 30 lines at generations 5, 50, and 90. Although length variant frequencies changed significantly within and between lines, the overall average frequency of each length variant did not change significantly between the three generations sampled, suggesting that there is little or no bias in the direction of change due to recombination.
THE ribosomal (r)DNA of metazoan animals is a large multigene family consisting of one or more arrays of tandemly repeated units. Each unit contains one copy of the 18S, 5.8S, and 28S rRNA genes separated by spacers. These arrays make up the nucleolar organizing regions and can be located on one or more chromosomes. Generally, rRNA gene copies retain a high degree of sequence similarity within species. This similarity is caused by a homogenization process, known as concerted evolution, which results from recombination within and between rDNA arrays (Dover 1982; Arnheim 1983; Zimmer et al. 1983). Two specific recombination mechanisms that drive concerted evolution are unequal crossing over and gene conversion, both of which can occur during meiosis and mitosis.
Mitotic recombination occurs in all eukaryotes and is intimately involved in the repair of damaged DNA (Helleday 2003). It can be stimulated in many ways including single-strand DNA breaks, mismatches, transcription, and replication. In addition, the formation of structures in the DNA that inhibit normal transcription and replication, such as replication fork blocks (reviewed in Aguilera et al. 2000) and other types of DNA damage (e.g., methylation or oxidation), can stimulate recombination during mitosis. Double-stranded DNA breaks, which are thought to induce the majority of recombination events in meiosis, can also occur during mitosis and induce recombination (Pâques and Haber 1999; Prado et al. 2003; Aylon and Kupiec 2004). While mitotic recombination is a ubiquitous process, its occurrence may not be uniformly distributed throughout the genome. For example, there is some evidence that chromatin structure mediated by the protein SIR2 may play a role in suppressing recombination in rDNA (reviewed in Aguilera et al. 2000).
Despite our growing understanding of the genes and mechanisms involved in recombination, little is known about its rate in rDNA, which has important implications for the process of concerted evolution in rDNA in natural populations. This gap in our knowledge results from the fact that measuring recombination rates experimentally can be a difficult task, even in model organisms. Nevertheless, many elegant experiments have been done to estimate the number of recombination events per generation in the rDNA of Saccharomyces cerevisiae [e.g., 1 × 10−2/generation (Szostak and Wu 1980), 1.3 × 10−3 (Merker and Klein 2002), and 7.4–7.5 × 10−5 (Kobayashi et al. 2004)] and in the rDNA of murine cells [1.2–1.8 × 10−5 (Nelson et al. 1989)]. Statistical approaches have also been developed to estimate recombination rates indirectly from population genetic data (reviewed in Stumpf and McVean 2003), but these methods need empirical confirmation. In this study, we estimate the recombination rate in the rDNA of Daphnia obtusa by quantifying changes in the frequency of length variants of the 18S rRNA expansion segment 43/e4 through time in apomictically propagated lines that were established from a single wild-caught female.
Daphnia (Crustacea: Anomopoda) are small, freshwater organisms that generally reproduce by cyclic parthenogenesis. When environmental conditions are favorable, females produce diploid eggs via apomictic parthenogenesis (apomixis), which develop directly into females. Environmental cues trigger the production of meiotically produced haploid diapausing eggs that require fertilization by males. An analysis of restriction site polymorphism has shown that organisms that reproduce parthenogenetically have highly homogenized rDNA repeats, demonstrating that the frequency of recombination events during germ-line apomixis is sufficiently high for concerted evolution to occur (Crease and Lynch 1991).
Expansion segments are regions within the rDNA that exhibit high sequence diversity within species and, in some cases, within individuals. Regardless of their length, expansion segments tend to fold into energetically stable hairpin or helical secondary structures in the rRNA, which may or may not contain unpaired nucleotides that form bulges or loops. For a given sequence length and base composition, the energetic stability of helices containing unpaired nucleotides is generally lower than that of helices in which all nucleotides are involved in base pairing.
Previously, McTaggart and Crease (2005) examined the frequency of length variants in expansion segment 43/e4 of the 18S rRNA (see Wuyts et al. 2001 for a diagram showing the location of all expansion segments in the 18S rRNA gene) in six individuals from four North American populations of D. obtusa. They identified two pairs of short (di- or trinucleotide) indel sites that pair with each other when the secondary structure of the sequence is formed. They found that the length variants containing energetically stable structures, i.e., those in which indels do not result in a destabilizing bulge (compensated length variants), were present at a wide range of frequencies, while variants containing indels that do cause a bulge (uncompensated length variants) were present only at low frequencies. These results suggest that uncompensated length variants are selectively disadvantageous, while compensated length variants are selectively neutral with respect to one another. Furthermore, the frequency distribution of the compensated length variants suggests that there is no bias in the frequency changes caused by recombination. However, McTaggart and Crease (2005) were unable to show this definitively due to the small number of individuals examined and the fact that the populations sampled may have been experiencing different selective constraints. If there is no bias in the direction of length variant frequency change caused by recombination, then we predict that the frequency of compensated length variants should change randomly within and between the apomictic lines through time in the absence of selection. Here, we test this prediction in addition to providing an estimate of the rate of recombination in the rDNA of apomictically propagated D. obtusa lines.
MATERIALS AND METHODS
Establishment and maintenance of the apomictic D. obtusa lines:
A single female D. obtusa was isolated in May 2001 from the pond in Trelease Woods near Urbana, Illinois. An apomictic line was established and maintained under standard, uncrowded conditions at 20° and well fed. All animals were kept in beakers of filtered (1 μm) lake water. In October 2001, a single individual was randomly chosen to be the stem mother for all of the experimental apomictic lines. A total of 48 apomictically produced daughters were collected from the stem mother and each was used to initiate an experimental line. The standardized procedure for propagating the experimental lines was as follows: 8–10 days following the start of the previous generation, a single randomly chosen female offspring was transferred to a new beaker of lake water. Maturation takes place after ∼7 days at 20°, which ensured that the transferred individual was a daughter and not a granddaughter. If a line had not produced offspring by the time of transfer, the mother was transferred to a new beaker and the generation number for that line was not increased.
In addition to the focal individual transferred, two of her sisters were transferred into separate beakers to serve as backups. Backups were used for a transfer when the focal individual either died before reproducing or produced only male offspring and/or diapausing eggs over her entire life. Throughout the course of the project, backups were used in ∼10% of the transfers. Use of backups neither showed a trend over time nor was clustered in certain lineages (J. L. Dudycha, unpublished data). Approximately every fifth generation, sisters of the focal individual were collected and frozen at −80° for the molecular analyses described below.
Several steps were taken to minimize the risk of exogenous contamination and cross-contamination among the lines. Beakers were kept covered to prevent splash contamination when they were not in use. Pipettes used to handle the animals were rinsed in nearly boiling water after each transfer to kill any neonates that may have adhered to the pipette. To safeguard against exogenous contamination, all lines were scored for 8–12 microsatellite loci at generation ∼40 and were confirmed to be identical to each other (J. L. Dudycha, unpublished data). In addition, all lines were morphologically inspected and diagnostic allozyme loci were analyzed at generation ∼90. Identifying cross-contamination among the apomictic lines was more difficult. However, at generation ∼100, 16 nuclear genes were sequenced in the lines and only two pairs of lines (4 and 10, 8 and 14) had similar sequence profiles (A. Omilian, unpublished data), suggesting shared mutational events or cross-contamination. These lines were included in the final analysis of the coarse-grained time series (see Data analysis) as their exclusion did not alter the results.
DNA analysis:
For each generation that was sampled within each line, total genomic DNA was extracted from 2–50 pooled, apomictically produced sisters using the CTAB method (Doyle and Doyle 1987). DNA samples were obtained for 30 lines at generations 5, ∼50 (generations 49–59), and ∼90 (generations 80–92). These lines were sampled only three times and are referred to as the coarse-grained time series. Due to occasional sampling difficulties, 10 lines were not sampled at all three times. These include 5 lines that were sampled only at generation 5, 2 lines that were sampled only at generations 5 and ∼50, 2 lines that were sampled only at generations 5 and ∼90, and 1 line that was sampled only at generations ∼50 and ∼90. A total of 35 lines were sampled at least twice. The fine-grained time series consists of four lines (3, 12, 29, and 30) that were sampled approximately every 5 generations, starting at generation 5 and ending at approximately generation 90.
The 18S rRNA expansion segment 43/e4 was amplified from 1 μl (20–200 ng) of each genomic DNA sample using the primers 1522F (5′-HEX-ATTCCGATAACGAACGAG) and 1880R (5′-GAAGACTGCGTGACGGAC) in a 10-μl reaction containing 10 mm Tris–HCl, pH 8.3, 20 mm KCl, 1.5 mm MgCl2, 0.05 mm of each of dNTP, 0.75 μm of each primer, and 1 unit of Taq polymerase. Amplification conditions were 94° for 1 min and 35 cycles of 94° for 20 sec, 55° for 20 sec, 72° for 1 min, followed by 72° for 5 min. Each of the fluorescently labeled PCR products was electrophoresed on a 7% polyacrylamide (19:1) denaturing gel at 35 W for 5 hr. Each gel was scanned with a Hitachi FM BIOII scanner on channel 2. The bands within each lane were marked by hand on the resulting gel image using the FM BIOII software. The six length variants that were observed among all of the samples were each marked in every lane, even if they could not be detected by eye. The FM BIOII analysis tool was used to quantify the relative fluorescent signal of each band as a function of the total intensity within each lane. We amplified and analyzed the expansion segment from each DNA sample three times to evaluate the reproducibility of the band intensity estimates. The average intensity of each band from the three PCR products was calculated for each sample at generations 5 (N = 39), ∼50 (N = 33), and ∼90 (N = 33). The average band intensities were used as a measure of the relative frequency of the length variants. In addition, the impact of PCR cycle number on the relative frequency of length variants was determined by amplifying six of the samples at each of 35, 30, and 25 cycles. All other PCR conditions remained the same as those described above.
To sequence each of the length variants, a PCR product was amplified from one DNA sample (line 12, generation 10) with primers 1413F (5′-TCACCAGGCCCGGACACTGGAAGG) and 2004R (5′-TGGGGATCATTGCAGTCCCCAATC), ligated into the pGEM plasmid (Promega, Madison, WI) and transformed into DH5α cells (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions. Plasmid DNA was isolated from 48 of the resulting colonies using the Millipore (Billerica, MA) Plasmid Miniprep kit. To determine the insert size, 2 μl of the plasmid preparation was used in a 10-μl PCR reaction containing the primers 1522F (HEX) and 1880R as described above. One plasmid containing each length variant was sequenced using the BigDye 3.1 terminator kit (Applied Biosystems, Foster City, CA) on a 3730 Genetic Analyzer (Applied Biosystems).
Quantitative PCR:
Haploid rDNA copy number was estimated for a subset of the fine-grained time series samples using the relative quantification method of quantitative PCR (qPCR), which compares the rate of amplification of rDNA to that of a single-copy reference gene (user bulletin no. 2, ABI 7700 SDS; Applied Biosystems). The entire 18S rRNA gene of Daphnia pulex has been cloned and sequenced (GenBank accession no. AF014011) (Crease and Colbourne 1998) and we used this clone to conduct preliminary experiments. We chose two single-copy nuclear genes that were present in cDNA libraries of D. pulex and Daphnia magna. PCR primers were designed in conserved regions shared by these two species and used to amplify these genes from D. obtusa genomic DNA. These PCR products were cloned into the pCR 4-TOPO vector (Invitrogen) according to the manufacturer's instructions. We used one TOPO plasmid clone of each gene for our preliminary qPCR experiments. BLAST analysis of these two genes indicated that one of them is likely to be a member of the Rab subfamily of small GTPases, while the other is likely to encode a transcription initiation factor. For the purposes of this study, we refer to them as GTP and TIF, respectively.
Primers for qPCR were designed from plasmid clones of D. pulex (18S rRNA gene) and D. obtusa (GTP and TIF genes), using the ABI Primer Express software (version 2.0, Applied Biosystems). All primer pairs produce a 50-nt amplicon. The 18S rRNA gene primers are located just downstream of the 43/e4 expansion segment in a conserved region that is identical in cloned sequences from D. obtusa (this study) and D. pulex (AF014011). The primer sequences are as follows: 18S rRNA forward, 5′-CCGCGTGACAGTGAGCAATA; 18S rRNA reverse, 5′-CCCAGGACATCTAAGGGCATC; GTP forward, 5′-TATTCAGCATGGAGAGACGGC; GTP reverse, 5′-GATGTCGACTGACGCTGGAA; TIF forward, 5′-GACATCATCCTGGTTGGCCT; and TIF reverse, 5′-AACGTCAGCCTTGGCATCTT.
To estimate relative amplification efficiency between the 18S rRNA gene and each of the single-copy genes, we did preliminary experiments using the plasmid clones as templates. We created a composite template containing all three genes in equal copy number (3,000,000) by mixing the three plasmids together. This template was then serially diluted to create four additional templates containing 300,000, 30,000, 3000, and 300 copies. We performed a qPCR experiment in which each of these five concentrations for each gene was measured in duplicate and then generated a plot of log (DNA concentration) against ΔCT for each single-copy gene in comparison with rDNA, where CT is the threshold cycle and ΔCT is the difference between CT for rDNA and the single-copy reference gene. CT is the cycle number at which the intensity of fluorescence from a reporter dye in the sample, in this case SYBR Green, exceeds a fixed threshold that is set above the background baseline. The threshold should be set close to the baseline and well within the exponential phase of the amplification reaction.
The absolute value of the slope of the regression line for each gene was <0.1 in both cases (−0.052 for rDNA relative to GTP and 0.074 for rDNA relative to TIF), indicating that the relative amplification efficiency of these genes is sufficiently similar to allow the use of the relative quantification method (user bulletin no. 2, ABI 7700 SDS; Applied Biosystems). In addition, we performed preliminary experiments with several samples of genomic DNA from the D. obtusa lines and found that using a template quantity of ≥10 ng gave a relative copy number very close to one when the single-copy genes were compared to one another. On the other hand, template quantities beyond 20 ng caused a high baseline and thus interfered with establishment of the threshold for subsequent CT estimation.
All qPCR reactions were 20 μl in volume and contained 1× Power SYBR Green Master Mix (Applied Biosystems), 10 pmol of each primer, and 20 ng of template in the case of genomic DNA samples from the D. obtusa apomictic lines. The 260/280 ratio of these DNA samples is ∼2.0, indicating that they also contain RNA, so the actual amount of DNA in each qPCR reaction was substantially less than the estimated value. However, it is not necessary to know the exact amount of DNA template that is used in relative quantification experiments, as long as it is not so high that it interferes with subsequent CT analysis.
Amplification was performed on an ABI 7000 SDS real-time thermal cycler (Applied Biosystems), using the default parameters for thermal cycling (50° for 10 min, 95° for 1 min, followed by 40 cycles of 95° for 15 sec and 60° for 1 min) and for analysis [automatic threshold cycle (CT) and baseline]. The threshold for CT estimation was set to 0.3 (the default value is 0.2) so that we could use template quantities of 20 ng. Each gene from each sample was amplified in duplicate.
We averaged duplicate CT-values within an experiment, calculated the difference in average CT between rDNA and one of the single-copy genes (ΔCT), and then used the value of 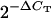 as the estimate of haploid rDNA copy number. On the basis of the range of estimates that we obtained using qPCR, we replicated all relevant analyses using three values of haploid rDNA copy number, 120, 160, and 200.
as the estimate of haploid rDNA copy number. On the basis of the range of estimates that we obtained using qPCR, we replicated all relevant analyses using three values of haploid rDNA copy number, 120, 160, and 200.
Data analysis:
Coarse-grained time series:
To estimate changes in the length variant frequency distribution between lines within a generation (i.e., generation 5), we compared the length variant frequencies of each line to those of one other line (the next line in the numerical sequence) from the same generation with a G-test. The probability values for all G-tests were sequentially Bonferroni corrected for multiple tests (Rice 1989). Results were considered to be significant if the probability values were <0.01. We did not compare the length variant frequency distribution in all pairwise comparisons within a generation due to the loss of statistical power in multiple tests. In addition, we compared the length variant frequency distributions from the same line from two consecutive samples (i.e., from generation 5 to ∼50 or from ∼50 to ∼90) with a G-test.
Fine-grained time series:
Sequentially Bonferroni-corrected (Rice 1989) G-tests were used to determine if differences in the distribution of length variant frequencies between each consecutive time interval (i.e., generations 5–10, generations 10–15, etc.) are significant within each of lines 3, 12, 29, and 30.
Recombination events were counted in two ways. First, we counted the number of time intervals that had a significant G-value. We considered that every time interval with a significant shift in the distribution of length variant frequencies was caused by a single recombination event. The final rate of recombination was calculated as the total number of significant events in each line, divided by the total number of generations elapsed. Second, we calculated the residual values for each length variant from the G-tests within each time interval. We considered any length variant that had a residual value >2 or < −2 to have changed substantially during the time interval. Thus, in this second test, if at least one residual value within a time interval was >2 or < −2, a recombination event was counted, even if the overall G-score was not significant. Both of these methods of counting recombination events are conservative and are likely to underestimate the actual number of events. However, because it is possible for a single recombination event to change the frequency of more than one length variant (for example, if variants are clustered together within the array), we counted only one event for any particular time interval, even if more than one residual value was < −2 or >2. Furthermore, this method is unable to detect equal exchanges between sister chromatids or recombination events that do not change length variant frequencies to the extent that we can detect them on the gels. Finally, if a recombination event occurs between chromatids from homologs, and the four chromatids segregate such that both parental chromatids or both recombinant chromatids end up in the same daughter cell, we will not be able to detect a recombination event because the relative frequencies of length variants will not change from the parental frequencies.
RESULTS
PCR amplification of expansion segment 43/e4 from the D. obtusa experimental lines revealed six length variants (V1–V6), which vary in length from 319 to 333 nt. The length variation is due to six indel sites (1–6) that form three complementary pairs, such that sites 1 and 6, 2 and 5, and 3 and 4 are opposite each other in the secondary structure (Figure 1A). Length variants are designated as compensated if nucleotides are either present [+] or absent [−] at both complementary sites of any pair (Figure 1B). From the sequences of the different length variants, we found that V1, V3, V4, V5, and V6 are compensated, whereas V2 is uncompensated. V2 could be a mixture of four different uncompensated variants of the same length, while V3 could be a mixture of two compensated variants (Figure 2). However, only one representative of each length variant was sequenced. The fact that more than one variant could be present within these two size classes will result in a more conservative estimate of recombination frequency.
Figure 1.—

(A) Secondary structure of a portion of rRNA expansion segment 43/e4 showing the putative nucleotide pairing of six indel sites. Boundaries between adjacent indel sites are indicated with a vertical dashed line. (B) Indel sites are classified as [+] if the nucleotides are present and [−] if the nucleotides are absent. In this example, sites 1–6 and 2–5 are uncompensated, while indel sites 3 and 4 are compensated. Thus, the genotype of this variant would be [−−+ +++].
Figure 2.—

Gel image of the six length variants in rRNA expansion segment 43/e4 from 10 apomictic lines of Daphnia obtusa. The lines have diverged from each other for ∼90 generations. Each lane represents one apomictic line. The lanes are labeled with the line number and the generation at which the line was sampled (e.g., 6-84 denotes line 6 sampled at generation 84). The indel genotype and length of each variant are shown below the gel image (see Figure 1). The genotype that was observed in the sequenced clones is [+−+ +++] for V2 and [+−+ +−+] for V3. The relative mobilities of the six length variants are indicated on the size standards in the first and last lanes.
Expansion segment 43/e4 was amplified three times from a total of 160 samples. The average standard deviations relative to the average band intensities of each length variant (Table 1) show that estimates of relative variant frequency based on band intensity are reproducible. The standard deviation of three amplifications at 35, 30, and 25 cycles was of the same magnitude as that for the three replicates done at 35 cycles (0.002–0.012 across all fragments in the six samples), showing that cycle number of the PCR reaction has no impact on the relative frequency of the length variants.
TABLE 1.
Mean frequency and mean standard deviation of six length variants (V1–V6) of rRNA expansion segment 43/e4 in apomictically propagated lines of Daphnia obtusa
| Length variant
|
||||||
|---|---|---|---|---|---|---|
| V1 | V2 | V3 | V4 | V5 | V6 | |
| Mean frequency | 0.211 | 0.025 | 0.205 | 0.249 | 0.286 | 0.024 |
| Mean standard deviation | 0.022 | 0.014 | 0.027 | 0.027 | 0.028 | 0.013 |
The expansion segment was amplified three times from each of 160 DNA samples. The average frequency and the standard deviation of each length variant were calculated for each sample, and then these values were averaged across all samples.
rDNA copy number:
Twenty-five samples were chosen from the fine-grained time series for copy number analysis, 6 from each of lines 3, 12, and 29, and 7 from line 30. Samples within lines with substantially different length variant frequencies were chosen, as were those whose length variant frequencies were similar to one another (see Fine-grained time series results). Estimates of haploid rDNA copy number range from 53 to 233 (Table 2) and vary substantially between samples within lines even when there are no significant changes in length frequency during the time intervals. The mean copy number of the four lines at generation 5, which is the first generation at which the lines were sampled after establishment from the stem mother, is 160. The mean and median of the 25 samples analyzed are 150 and 159 copies, respectively. Thus, we chose haploid copy numbers of 120, 160, and 200 for the G-tests to determine the effect of the copy number variation on the estimation of length variant frequency change and recombination rate.
TABLE 2.
Quantitative PCR estimates of haploid rDNA copy number in apomictically propagated lines of Daphnia obtusa
| Line | Gena | Plateb | CTc rDNA | CTc GTP | CTc TIF | rDNA copy no. GTPd | rDNA copy no. TIFd | Mean rDNA copy no. | GTP copy no. TIFd | Sig. diff.e |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 5 | B | 16.74 | 24.02 | 23.97 | 156 | 150 | 153 | 0.96 | — |
| 3 | 20 | A | 15.44 | 22.26 | 22.56 | 113 | 139 | 126 | 1.23 | N |
| 3 | 35 | A | 17.47 | 24.38 | 25.10 | 120 | 197 | 159 | 1.64 | Y |
| 3 | 55 | B | 16.49 | 23.62 | 23.92 | 140 | 172 | 156 | 1.23 | Y |
| 3 | 56 | B | 16.41 | 23.85 | 24.26 | 174 | 232 | 203 | 1.33 | N |
| 3 | 65 | B | 15.62 | 22.86 | 23.35 | 151 | 211 | 181 | 1.40 | N |
| 12 | 5 | B | 16.44 | 23.94 | 24.10 | 180 | 201 | 191 | 1.12 | — |
| 12 | 25 | A | 16.05 | 23.61 | 23.70 | 189 | 200 | 195 | 1.06 | Y |
| 12 | 25 | B | 16.23 | 23.84 | 23.89 | 196 | 203 | 200 | 1.03 | Y |
| 12 | 30 | A | 16.03 | 23.87 | 23.98 | 228 | 247 | 237 | 1.08 | N |
| 12 | 30 | B | 16.17 | 24.02 | 24.00 | 231 | 227 | 229 | 0.99 | N |
| 12 | 55 | A | 16.30 | 23.68 | 23.80 | 165 | 181 | 173 | 1.09 | Y |
| 12 | 85 | A | 16.71 | 23.32 | 23.39 | 97 | 102 | 100 | 1.05 | Y |
| 12 | 89 | A | 16.46 | 23.30 | 23.47 | 114 | 129 | 122 | 1.13 | N |
| 12 | 89 | B | 16.80 | 23.62 | 23.50 | 113 | 104 | 109 | 0.92 | N |
| 29 | 5 | B | 16.76 | 24.24 | 24.08 | 178 | 160 | 169 | 0.90 | — |
| 29 | 20 | A | 15.38 | 22.40 | 22.32 | 130 | 123 | 126 | 0.95 | Y |
| 29 | 40 | B | 15.81 | 23.35 | 23.27 | 186 | 177 | 182 | 0.95 | N |
| 29 | 45 | B | 16.38 | 23.95 | 24.06 | 190 | 204 | 197 | 1.08 | N |
| 29 | 60 | A | 16.08 | 23.45 | 23.71 | 166 | 198 | 182 | 1.19 | N |
| 29 | 60 | B | 16.48 | 24.15 | 24.01 | 203 | 186 | 195 | 0.91 | N |
| 29 | 80 | A | 15.56 | 22.90 | 22.94 | 162 | 167 | 165 | 1.03 | N |
| 30 | 5 | A | 16.67 | 23.49 | 23.89 | 113 | 149 | 131 | 1.32 | — |
| 30 | 5 | B | 16.71 | 23.74 | 23.58 | 131 | 117 | 124 | 0.89 | — |
| 30 | 10 | A | 16.53 | 23.82 | 23.66 | 156 | 140 | 148 | 0.90 | N |
| 30 | 25 | A | 16.43 | 23.89 | 24.13 | 176 | 208 | 192 | 1.18 | Y |
| 30 | 60 | A | 16.65 | 22.75 | 22.83 | 69 | 73 | 71 | 1.06 | Y |
| 30 | 65 | B | 17.45 | 23.23 | 23.13 | 55 | 51 | 53 | 0.93 | N |
| 30 | 80 | B | 18.19 | 24.27 | 24.53 | 68 | 81 | 74 | 1.20 | Y |
| 30 | 92 | B | 17.47 | 23.51 | 23.76 | 66 | 78 | 72 | 1.19 | N |
Generation.
The results are based on two experiments. Samples with the same letter were run in the same experiment (A or B). Five samples (12-25, 12-30, 12-89, 29-60, and 30-5) were run in both experiments.
Threshold cycle. Each value is the mean of two replicates.
Haploid copy number of the first gene listed, relative to the second gene (e.g., rDNA relative to GTP in column 7) was calculated as 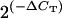 , where ΔCT is the difference in threshold cycle between the two genes.
, where ΔCT is the difference in threshold cycle between the two genes.
A “Y” indicates that there was at least one significant change in length variant frequencies between that generation and the previous one for which rDNA copy number was determined (e.g., between generations 20 and 35 in line 3). An “N” indicates that variant frequencies did not change significantly. Results for all time intervals in each of these four lines are shown in Figure 4.
The relative copy number of the GTP gene relative to TIF is, as expected, very close to 1 in most samples except those from line 3 (Table 2), where the GTP copy number is consistently higher than that of TIF. It is not clear if this represents a duplication of one GTP allele or a mutation in an allele of another member of the GTP gene family that allows the primers to amplify it to some extent or whether it could be a mutation in one allele of the TIF gene that substantially decreases its amplification efficiency. Regardless of the reason for the difference, the estimate of rDNA copy number based on one of the genes is not correct (either too low in the case of GTP or too high in the case of TIF) but it is not clear which one. Thus, we used the mean of the two estimates (Table 2) in the calculation of the mean and median copy number, as we did for the other samples.
Coarse-grained time series:
The length variant frequencies among all lines at generation 5 are similar to one another (Figure 3A), while length variant frequencies vary greatly and without a consistent pattern across all of the lines at generations ∼50 (Figure 3B) and ∼90 (Figure 3C). Twenty-one of the 38 line-to-line differences (55%) in length variant frequency are significant at generation 5, on the basis of an rDNA copy number of 160, while 27 of 32 comparisons (84%) at generation 50 and 28 of 32 (88%) comparisons at generation 90 are significant. The difference between generation 5 and generation 50 or 90 is more evident at an rDNA copy number of 120, where only 29% of the comparisons are significant at generation 5, compared to 84% at generation 50 and 72% at generation 90.
Figure 3.—

Frequency of length variants of rRNA expansion segment 43/e4 in 40 apomictically propagated Daphnia obtusa lines. Each estimate is the average from three PCR reactions. The x-axis of all graphs is the line number (1–40) and the generation at which the line was sampled. (A) Length variant frequencies at generation 5. (B) Length variant frequencies at generation ∼50. (C) Length variant frequencies at generation ∼90.
Twenty-five of 30 (83%) pairwise comparisons between the length variant frequency distributions from the same line at generations 5 and ∼50 and 17 of 30 comparisons between generations ∼50 and 90 (57%) are significantly different on the basis of an rDNA copy number of 160. A similar decrease in the number of significant length frequency distributions was obtained for an rDNA copy number of 120 (73% of the comparisons between generations 5 and ∼50 are significant, compared to 50% of the comparisons between generations ∼50 and ∼90) and an rDNA copy number of 200 (87% of the comparisons between 5 and ∼50 are significant, compared with 63% between generations ∼50 and ∼90). Examination of the residual values from these G-tests shows that the frequency of each length variant changed significantly in at least one line over the entire time interval except for length variants V2 and V6. In addition, the length variant frequency distribution averaged over all the lines is not significantly different between generations 5 and 50 on the basis of an rDNA copy number of 160 (G = 9.46, d.f. = 5, P = 0.09; Table 3). Moreover, the residual values for each length variant fall between −2 and 2. Consistent with this result, there is no significant difference in the average length variant frequency distributions between generations 5 and 50 on the basis of an rDNA copy number of 120 (G = 7, d.f. = 5, P = 0.21) or 200 (G = 12, d.f. = 5, P = 0.04). Similarly, no significant difference was detected between generations 50 and 90 on the basis of an rDNA copy number of 160 (G = 2, d.f. = 5, P = 0.85; Table 3).
TABLE 3.
Frequency of length variants of rRNA expansion segment 43/e4 averaged across all of the apomictically propagated Daphnia obtusa lines at generations 5, ∼50, and ∼90
| Generation
|
|||
|---|---|---|---|
| Variant | 5 | 50 | 90 |
| V1 | 0.257 (0.009) | 0.185 (0.022) | 0.178 (0.023) |
| V2 | 0.032 (0.002) | 0.029 (0.003) | 0.025 (0.002) |
| V3 | 0.210 (0.011) | 0.235 (0.024) | 0.264 (0.032) |
| V4 | 0.296 (0.013) | 0.274 (0.022) | 0.293 (0.031) |
| V5 | 0.179 (0.005) | 0.258 (0.026) | 0.218 (0.029) |
| V6 | 0.026 (0.002) | 0.019 (0.002) | 0.022 (0.002) |
Standard errors of the estimates are given in parentheses. The length variant frequency distributions are significantly different between generations 5 and 50 (see text), although none of the residual values for any of the length variants were < −2 or >2. The number of lines at generation 5 is 39 and the number of lines at generations ∼50 and ∼90 is 33.
Fine-grained time series:
The length variant frequencies within each of the four fine-grained time series appear to be changing randomly through time (Figure 4). For example, V5 increases in line 30, decreases in line 12, and remains fairly constant in lines 3 and 29. Additionally, all length variant frequencies appear to be relatively constant in line 29, whereas in line 30 V5 increases dramatically and remains dominant to the last sample analyzed. This change in the frequency of V5 is also accompanied by a substantial decrease in rDNA copy number, from ∼190 to ∼70, sometime between generations 25 and 60 (Table 2) that is also maintained until the last sample. The only similarity among the four lines in the fine-grained time series is that V2 and V6 are generally found at much lower frequencies (<5%) than the other length variants.
Figure 4.—

Frequency of length variants of rRNA expansion segment 43/e4 in each of four apomictically propagated Daphnia obtusa lines. The lines were sampled approximately every five generations. Asterisks indicate significant G-scores, and arrows indicate length variants with residual scores < −2 or >2 for a particular time interval. These analyses are based on an rDNA copy number of 160 per haploid genome.
As expected, estimates of recombination rate based on the G-tests decrease with decreasing rDNA copy number, such that the average estimates across the four lines for 200, 160, and 120 haploid copies are 0.04, 0.04, and 0.02 recombination events per generation, respectively (Table 4). Estimates based on the residual values are 0.06, 0.06, and 0.04 recombination events per generation for copy numbers of 200, 160, and 120, respectively (Table 4).
TABLE 4.
Number of recombination events per generation in the rDNA of four apomictically propagated Daphnia obtusa lines
| G-test: | Residual: | ||||||
|---|---|---|---|---|---|---|---|
| Haploid rDNA copy no.
|
Haploid rDNA copy no.
|
||||||
| Line | Gen | 120 | 160 | 200 | 120 | 160 | 200 |
| 3 | 92 | 0.01 | 0.03 | 0.04 | 0.04 | 0.05 | 0.09 |
| 12 | 91 | 0.05 | 0.07 | 0.07 | 0.05 | 0.07 | 0.07 |
| 29 | 86 | 0.00 | 0.01 | 0.02 | 0.01 | 0.07 | 0.03 |
| 30 | 92 | 0.03 | 0.03 | 0.03 | 0.03 | 0.04 | 0.04 |
| Mean | 0.02 | 0.04 | 0.04 | 0.04 | 0.06 | 0.06 | |
The recombination rates were calculated using two approaches. In the G-test method, one recombination event was counted if a G-statistic was significant for a given time interval. In the residual method, one recombination event was counted if at least one residual value within a time interval was < −2 or >2. Gen, total number of generations for each line.
DISCUSSION
Length variant frequency distributions changed between one and six times within a line over the ∼90-generation period, on the basis of the G-test and assuming a haploid rDNA copy number of 160. Moreover, we observed changes in individual length variant frequency as large as 33%. The range of rDNA recombination rate estimates available to date spans several orders of magnitude (10−2–10−5 events per generation; see Introduction), and our results are at the high end of this spectrum (2 × 10−2–6 × 10−2 events per generation). The high frequency of recombination is exemplified by the fact that within 5 generations >50% of the coarse-grained lines had changed significantly from the other line to which they were compared. This was unexpected because our technique is likely to yield an underestimate of the true recombination rate, for the reasons outlined in materials and methods.
If our estimated recombination rate is reasonably accurate, then sequence homogenization among rDNA gene copies in D. obtusa could be very rapid. Previous studies have shown that the length variants in expansion segment 43/e4 isolated from individual D. obtusa from widely distributed populations are the same, although not all variants are found in every population (McTaggart and Crease 2005). This suggests that the length variants are old and that ongoing replication slippage events are not a suitable explanation for their persistence. Alternatively, some of the length variants could be generated de novo by recombination. For example, recombination at different locations between the longest (V1) and the shortest (V6) length variant can yield all of the other observed variants. This may also generate length variants that we did not observe, perhaps because they are energetically unstable and therefore do not persist. If the length variants were old, then we would predict that different compensated length variants would go to fixation via genetic drift in D. obtusa populations that are isolated from one another. A preliminary analysis of the frequency of length variants in expansion segment 43/e4 from 24 D. obtusa populations across its North American range (S. J. McTaggart and T. J. Crease, unpublished data) indicates that all individuals surveyed (n = 3–10 per population) within three populations are indeed fixed for one of the compensated length variants (V1 in all cases). This geographic comparison supports the hypothesis that the length variants are old and furthermore shows that the presence of intraindividual length variation in expansion segment 43/e4 is not a ubiquitous characteristic of D. obtusa populations, as was previously thought (McTaggart and Crease 2005).
Although the data from our study cannot identify the recombination mechanisms that are responsible for the changes that we observed, the wide range of length variant frequency changes detected (2–33%) within short time intervals suggests that we are detecting more than one type of event. Recombination events can be divided into two general classes depending on whether or not they change rDNA copy number. Events that do not change rDNA copy number include equal crossover and gene conversion. Previous studies have provided evidence that interchromosomal recombination occurs less frequently than intrachromosomal recombination in rDNA (Schlötterer and Tautz 1994; Liao et al. 1997), and there is some indirect evidence that this is the case in D. pulex rDNA (Crease 1995). Thus, it is likely that the length variant frequency changes we observed are primarily due to unequal sister chromatid or intrachromatid exchange, both of which will result in gene copy number change. Indeed, our qPCR analysis of samples in the fine-grained time series shows that rDNA copy number can indeed change substantially in only a few generations. The largest change occurred in line 30 between generation 25 and 60 when copy number decreased from ∼190 to ∼70 and then remained low until the last sample analyzed. This is consistent with a substantial increase in the frequency of V5 at the expense of all the other length variants in this line. Moreover, the association of substantial changes in variant frequency with copy number changes provides indirect evidence that length variants are clustered along an rDNA array.
Changes in rDNA copy number of the magnitude that we observed have also been observed in replicate lines of Drosophila melanogaster, with copy numbers on the X chromosome varying between 140 and 310 after 400 generations of laboratory culture (Averbeck and Eickbush 2005). Furthermore, the similarity between copy number estimates in D. obtusa (53–233) and D. melanogaster is consistent with the similarity of their genome sizes: 180 Mb for D. melanogaster (Adams et al. 2000) and 200 Mb for D. pulex, a close relative of D. obtusa (draft genome sequence, Joint Genome Institute).
In the coarse-grained time series, each of the compensated length variants is present at a wide range of frequencies, except for V6, which is present only at low frequencies. Even so, the average length variant frequency across all of the lines does not differ significantly among the three generations sampled. These results strongly suggest that there is no bias with respect to the recombination mechanisms that are operating in the rDNA. The significant change in the frequencies of V1 and V5 between the first two time points but not the last two is likely due to sampling error, as it is difficult to imagine a molecular mechanism that could create such a short-lived bias.
The availability of whole-genome information has confirmed that, in humans at least, rates of recombination vary by as much as four orders of magnitude across all nucleotides (McVean et al. 2004). To determine how the rate that we observed in rDNA compares to that in the genome as a whole, it will be necessary to compare our estimate of recombination rate with those obtained from (a) other portions of the rDNA and (b) other genomic regions. There are opposing viewpoints on how rDNA structure will affect recombination rate. For example, the recombination rate in rDNA may be high relative to the genomewide average due to the fact that each primary unit, which is repeated tens to thousands of times, contains a copy of a region containing the replication fork block, which is known to be associated with an increased probability of recombination (Kobayashi 2003). Indeed, selection may favor an elevated rate of recombination in rDNA due to its impact on the homogenization of gene copies along the rDNA array. In contrast, it has been argued that the heterochromatin structure of rDNA inhibits its accessibility to the recombination machinery, which predicts a lower than average recombination rate. Recent work by Kobayashi et al. (2004) has shown that this latter view may not be correct as they did not see a decrease in the overall rate of recombination in mutant SIR2 yeast strains vs. wild-type strains, although the wild-type strains did have a decreased rate of unequal sister chromatid exchange compared to mutant strains.
In conclusion, we have presented a novel procedure for the cost-effective quantification of recombination rate in rDNA, which is typically a difficult and labor-intensive task (Stumpf and McVean 2003). This method was sensitive enough to detect recombination events that occurred during apomixis in D. obtusa lines over five-generation time intervals and is applicable to any multigene family that contains easily detectable sequence variation and to any species that can be propagated in the lab.
Acknowledgments
We thank A. Danielson and C. Puzio for assistance in culturing the experimental lines and M. Cristescu for extracting DNA from some of the Daphnia samples. S.J.M. thanks M. Lynch for the opportunity to visit his lab and discuss this project with him. We also thank T. Eickbush and the anonymous reviewers for helpful comments on an earlier version of the manuscript. This project was funded by a grant from the Natural Sciences and Engineering Research Council of Canada to T.J.C. and a grant from the National Science Foundation to M. Lynch. S.J.M. was supported by an Ontario Graduate Scholarship in Science and Technology, and J.L.D. was supported by a National Institutes of Health Kirchstein Fellowship.
References
- Adams, M. D., S. E. Celnkier, R. A. Holt, C.A. Evans, J. D. Gocayne et al., 2000. The genome sequence of Drosophila melanogaster. Science 287: 2185–2195. [DOI] [PubMed] [Google Scholar]
- Aguilera, A., S. Chávez and F. Malagón, 2000. Mitotic recombination in yeast: elements controlling its incidence. Yeast 16: 731–754. [DOI] [PubMed] [Google Scholar]
- Arnheim, N., 1983. Concerted evolution of multigene families, pp. 38–61 in Evolution of Genes and Proteins, edited by M. Nei and R. K. Koehn. Sinauer Associates, Sunderland, MA.
- Averbeck, K. T., and T. H. Eickbush, 2005. Monitoring the mode and tempo of concerted evolution in the Drosophila melanogaster rDNA locus. Genetics 171: 1837–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon, Y., and M. Kupiec, 2004. New insights into the mechanism of homologous recombination in yeast. Mutat. Res. 566: 231–248. [DOI] [PubMed] [Google Scholar]
- Crease, T., 1995. Ribosomal DNA evolution at the population level: nucleotide variation in intergenic spacer subrepeat arrays of Daphnia pulex. Genetics 141: 1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crease, T., and J. Colbourne, 1998. Unusually long small subunit ribosomal RNA of the crustacean, Daphnia pulex: sequence and predicted secondary structure. J. Mol. Evol. 46: 307–313. [DOI] [PubMed] [Google Scholar]
- Crease, T., and M. Lynch, 1991. Ribosomal DNA variation in Daphnia pulex. Mol. Biol. Evol. 8: 620–640. [Google Scholar]
- Dover, G., 1982. Molecular drive: a cohesive mode of species evolution. Nature 299: 111–117. [DOI] [PubMed] [Google Scholar]
- Doyle, J. J., and J. L. Doyle, 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19: 11–15. [Google Scholar]
- Helleday, T., 2003. Pathways for mitotic homologous recombination in mammalian cells. Mutat. Res. 532: 103–115. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., 2003. The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol. Cell. Biol. 23: 9178–9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, T., T. Horiuchi, P. Tongaonkar, L. Vu and M. Nomura, 2004. SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell 117: 441–453. [DOI] [PubMed] [Google Scholar]
- Liao, D., T. Pavelitx, J. R. Kidd, K. K. Kidd and A. M. Weinder, 1997. Concerted evolution of the tandemly repeated genes encoding human U2 snRNA (the RNU2 locus) involves rapid intrachromosomal homogenization and rare interchromosomal gene conversion. EMBO J. 3: 588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTaggart, S. J., and T. J. Crease, 2005. Selection on the structural stability of a ribosomal RNA expansion segment in Daphnia obtusa. Mol. Biol. Evol. 22: 1309–1319. [DOI] [PubMed] [Google Scholar]
- McVean, G. A. T., S. R. Myers, S. Hunt, P. Deloukas, D. R. Bentley et al., 2004. The fine-scale structure of recombination rate variation in the human genome. Science 304: 581–584. [DOI] [PubMed] [Google Scholar]
- Merker, R. J., and H. L. Klein, 2002. hpr1Δ affects ribosomal DNA recombination and cell life span in Saccharomyces cerevisiae. Mol. Cell. Biol. 22: 421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, F. K., W. Frankel and T. V. Rajan, 1989. Mitotic recombination is responsible for the loss of heterozygosity in cultured murine cell lines. Mol. Cell. Biol. 9: 1284–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pâques, F., and J. E. Haber, 1999. Multiple pathways of recombination induced by double-stranded breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63: 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado, F., F. Cortés-Ledesman, P. Huertas and A. Aguilera, 2003. Mitotic recombination in Saccharomyces cerevisiae. Curr. Genet. 42: 185–198. [DOI] [PubMed] [Google Scholar]
- Rice, W. R., 1989. Analyzing tables of statistical tests. Evolution 43: 223–225. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C., and D. Tautz, 1994. Chromosomal homogeneity of Drosophila ribosomal DNA arrays suggests intrachromosomal exchanges drive concerted evolution. Curr. Biol. 4: 777–783. [DOI] [PubMed] [Google Scholar]
- Stumpf, M. P. H., and G. A. T. McVean, 2003. Estimating recombination rates from population data. Nat. Genet. 4: 959–968. [DOI] [PubMed] [Google Scholar]
- Szostak, J. W., and R. Wu, 1980. Unequal crossing over in the ribosomal DNA of Saccharomyces cerevisiae. Nature 284: 426–430. [DOI] [PubMed] [Google Scholar]
- Wuyts, J., Y. Van de Peer and R. de Wachter, 2001. Distribution of substitution rate and location of insertion sites in the tertiary structure of ribosomal RNA. Nucleic Acids Res. 29: 5017–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer, E. A., S. L. Martin, S. M. Beverley, Y. W. Kan and A. C. Wilson, 1983. Rapid duplication and loss of genes coding for the alpha chains of hemoglobin. Proc. Natl. Acad. Sci. USA 77: 2158–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]