

Abstract
The standard temporal method for estimating effective population size (Ne) assumes that generations are discrete, but it is routinely applied to species with overlapping generations. We evaluated bias in the estimates 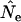 caused by violation of this assumption, using simulated data for three model species: humans (type I survival), sparrow (type II), and barnacle (type III). We verify a previous proposal by Felsenstein that weighting individuals by reproductive value is the correct way to calculate parametric population allele frequencies, in which case the rate of change in age-structured populations conforms to that predicted by discrete-generation models. When the standard temporal method is applied to age-structured species, typical sampling regimes (sampling only newborns or adults; randomly sampling the entire population) do not yield properly weighted allele frequencies and result in biased
caused by violation of this assumption, using simulated data for three model species: humans (type I survival), sparrow (type II), and barnacle (type III). We verify a previous proposal by Felsenstein that weighting individuals by reproductive value is the correct way to calculate parametric population allele frequencies, in which case the rate of change in age-structured populations conforms to that predicted by discrete-generation models. When the standard temporal method is applied to age-structured species, typical sampling regimes (sampling only newborns or adults; randomly sampling the entire population) do not yield properly weighted allele frequencies and result in biased 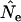 . The direction and magnitude of the bias are shown to depend on the sampling method and the species' life history. Results for populations that grow (or decline) at a constant rate paralleled those for populations of constant size. If sufficient demographic data are available and certain sampling restrictions are met, the Jorde–Ryman modification of the temporal method can be applied to any species with overlapping generations. Alternatively, spacing the temporal samples many generations apart maximizes the drift signal compared to sampling biases associated with age structure.
. The direction and magnitude of the bias are shown to depend on the sampling method and the species' life history. Results for populations that grow (or decline) at a constant rate paralleled those for populations of constant size. If sufficient demographic data are available and certain sampling restrictions are met, the Jorde–Ryman modification of the temporal method can be applied to any species with overlapping generations. Alternatively, spacing the temporal samples many generations apart maximizes the drift signal compared to sampling biases associated with age structure.
BECAUSE effective population size (Ne) is an important parameter in evolutionary biology but is notoriously difficult to measure in natural populations, considerable interest has focused on genetic methods for estimating Ne (reviewed by Beaumont 2003; Leberg 2005; Wang 2005). By far the most widely used genetic approach for estimating contemporary Ne is the temporal method (Krimbas and Tsakas 1971; Nei and Tajima 1981), so called because it depends on estimates of allele frequency taken from a population at two or more points in time. In addition to other simplifying assumptions, the standard temporal method assumes that generations are discrete, whereas many species are age structured and hence have generations that overlap. Two variations of the temporal method can account for effects of age structure, at least in some circumstances. Waples (1990) developed a modified temporal method for species with life histories like Pacific salmon (semelparous with variable age at maturity), but this model is not intended for use with iteroparous species. A more general model for age-structured, iteroparous species was developed by Jorde and Ryman (1995), who showed that the magnitude of allele-frequency change is determined not only by effective size and the sampling interval, but also by age-specific survival and birth rates. They derived an adjustment to the standard model to account for age-structure effects, and subsequent evaluations (e.g., Jorde and Ryman 1996; Palm et al. 2003) documented the biases in estimates of effective size that can result from application of the standard temporal method without accounting for overlapping generations. However, the Jorde–Ryman method requires detailed demographic information and the ability to age individuals or group them into single-cohort samples and perhaps for these reasons has not been widely applied (but see Turner et al. 2002 and Palm et al. 2003 for examples). In spite of its obvious limitations, the discrete-generation temporal method has been and continues to be widely applied to species with overlapping generations (e.g., Scribner et al. 1997; Johnson et al. 2004; Hoffman et al. 2004; Kaeuffer et al. 2004; Poulsen et al. 2006). A priori, we expect bias in the resulting estimate of Ne when an otherwise unbiased method is applied to situations that violate assumptions of the model. With respect to application of the standard temporal method to species with overlapping generations, it is possible that the biases could be small if the elapsed time between samples is long enough that the drift signal strongly dominates sampling considerations (as suggested by Jorde and Ryman 1995 and assumed, for example, by Miller and Kapuscinski 1997 and Hauser et al. 2002). However, under what specific circumstances this might be true cannot be determined without a quantitative analysis, nor can the magnitude and direction of biases associated with samples more closely spaced in time.
In this article we evaluate performance of the standard temporal method when it is applied to iteroparous species with overlapping generations. Central to our evaluations is establishing a point of reference for evaluating the true Ne of such a population. For this we draw on two important contributions of previous authors. First, Hill (1972) showed that discrete-generation models for Ne can be modified to apply to organisms with overlapping generations, provided that the population is of constant size (N) and demographically stable. This means, for example, that over t generations of genetic drift, a population with overlapping generations should experience the same amount of allele-frequency change as a population with discrete generations and the same effective size per generation. But to test this, one must be able to measure (or estimate) the population allele frequency at a given point in time. This is straightforward if generations are discrete, but how can one measure the parametric allele frequency of a population when generations overlap? Felsenstein (1971) proposed that the correct way to calculate a population allele frequency in this case is to weight each individual by its reproductive value. We first show that this method correctly predicts the rate of allele-frequency change in simulated, iteroparous populations of constant size. Next, we evaluate bias of estimates of Ne using the standard temporal method and how they vary as a function of the species' life history and various commonly used sampling strategies.
Populations that change in size are of particular interest to evolutionary biologists and conservation biologists. Changing population size does not present a problem for the standard, discrete-generation temporal model (if population size varies the method estimates the harmonic mean Ne over the time between samples), but most models for Ne in species with overlapping generations depend on the assumption of constant population size. However, Felsenstein (1971) derived an expression for the variance-effective size in species with overlapping generations that grow (or decline) at a constant rate, in which case age structure remains constant. To complete our evaluations, we used Felsenstein's model to establish the benchmark “true” effective size in populations that deterministically change in size and evaluated performance of the standard temporal method under these conditions.
METHODS
Definition of Ne when generations overlap:
Constant N:
The models developed by Felsenstein (1971) and Hill (1972, 1979) both have features that are useful for our analyses. Notation is consistent with that used by Felsenstein (1971) and is summarized in Table 1. The species is a monoecious diploid with the possibility of selfing. Demographic parameters are fixed; exactly N1 individuals are born in each time period, so the population will eventually reach a stable age distribution. Time units, indexed by t, are in years except as noted. The fraction of newborns that survive to age x is lx = Nx/N1, where Nx is the number in age class x. During a single time interval, individuals of age x produce an average of bx offspring that survive to the beginning of age class 1, so the probability that a newborn has a parent of age x is lxbx. The cohort size of newborns is 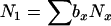 . In this model,
. In this model, 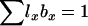 , generation length is given by
, generation length is given by 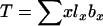 , and reproductive value (Fisher 1958; Felsenstein 1971) can be calculated as
, and reproductive value (Fisher 1958; Felsenstein 1971) can be calculated as 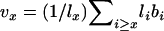 .
.
TABLE 1.
Notation used
| t | Time units (generally in years) |
| N1 | No. of age-1 individuals (newborns) produced in one time interval |
| Nx | No. of individuals of age x in the population |
| ∑Nx | Total no. of individuals in the population |
| j | Age at first maturity |
| q | Age at senescence |
| N | Population size, expressed as the total census size (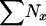 ), no. of reproductive adults (Nadult = ), no. of reproductive adults (Nadult = 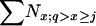 ), or no. of newborns (N1) ), or no. of newborns (N1) |
| T | Generation length (mean age of parents) |
| bx | Mean no. of offspring produced in one time interval by individuals of age x |
| lx | Fraction of a cohort that survives to age x |
| sx | Fraction of individuals of age x that survive to age x + 1 |
| dx | Fraction of individuals of age x that die before reaching age x + 1; dx = 1− sx |
| vx | Reproductive value for individuals of age x |
| αx | Fraction of a cohort that reaches age x and then dies; αx = lxdx |
| λ | Population growth rate per time period |
| k | No. of gametes contributed to the next generation by an individual over its lifetime |
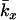 |
Mean lifetime k for individuals that die between ages x and x + 1 |
| σx2 | Variance in lifetime k of individuals that die between ages x and x + 1 |
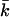 |
Mean lifetime k for all individuals breeding within a generation |
| Vk | Variance in lifetime k among all individuals within a generation |
| Ne | Effective population size for a generation |
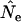 |
An estimate of effective size based on genetic data for two samples taken 1 or more time units apart |
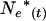 |
Instantaneous effective size that describes the actual rate of change in the population at a point t in time |
| S | No. of individuals per time interval sampled for genetic analysis |
| L | Time interval between samples |
| g | Elapsed no. of generations between samples; g = L/T |
| F | Standardized variance of allele frequencies |
| Px(t) | Allele frequency in age class x at time unit t |
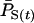 |
Population allele frequency at time t, weighting each individual equally |
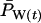 |
Population allele frequency at time t, weighting each individual by its reproductive value |
| C | A constant that depends on life-history parameters of the population, used to estimate Ne in the temporal method of Jorde and Ryman (1995) |
Felsenstein (1971) showed that Ne in species with overlapping generations can be calculated directly from the age-specific survival and fecundity parameters contained in a standard Leslie matrix. In his model, each year within each age class there is random (binomial) variation among individuals in reproductive success, random survival of individuals between age classes, and no correlation between mortality and fecundity. Under these conditions, and assuming constant population size, inbreeding and variance-effective sizes are the same and are given by
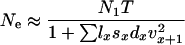 |
(1) |
(Felsenstein 1971, Equation 10), where dx is the probability of death at the end of age x and sx = 1 − dx.
Hill (1972, 1979) considered variance Ne in a model similar to Felsenstein's except that Hill made no particular assumption about variation in reproductive success among individuals. For monoecious diploids with random selfing, Hill showed that effective size is given by
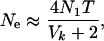 |
(2) |
where N1 and T are as defined above and Vk is the lifetime variance in reproductive success (production of newborns) by the N1 individuals making up a cohort of newborns. With random variation in (lifetime) reproductive success (Vk ≈ 2), Ne = N1T, which is the number of individuals entering the population over a period of one generation. In general, however, age structure leads to Vk > 2 and Ne < N1T.
Equation 2 is more general than Equation 1 but the latter is more convenient to use with data from a typical life table. However, if one assumes that the distribution of reproductive success is Poisson within each age class, Equations 1 and 2 are comparable (Johnson 1977). Under this assumption, a reformulation of Equation 2 (see appendix a) provides a way of implementing Hill's model on the basis of population vital rates,
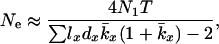 |
(3) |
where 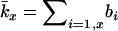 is the average lifetime reproductive success of individuals that die between years x and x + 1. Effective size calculated from Equation 3 is referred to as Ne(H).
is the average lifetime reproductive success of individuals that die between years x and x + 1. Effective size calculated from Equation 3 is referred to as Ne(H).
Changing N:
Felsenstein (1971) also developed an expression for variance Ne in populations that are changing size at a constant rate λ per time period. λ is the dominant eigenvalue of the Leslie matrix and is the unique real solution to the discrete-time version of the Euler–Lotka equation 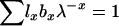 . When population size changes deterministically, analogs to the life-history parameters described above are
. When population size changes deterministically, analogs to the life-history parameters described above are 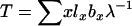 and
and 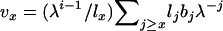 , and effective size is given by
, and effective size is given by
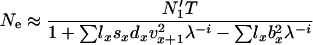 |
(4) |
(Felsenstein 1971, Equation 24), where N1′ is the number of newborns in the next time interval. If λ = 1, N1′ = N1 and Equation 4 is identical to Equation 1 except for the last term in the denominator, which is zero if the number of offspring per parent in one time interval is Poisson (Felsenstein 1971). Effective size calculated from Equation 4 is referred to as Ne(F).
Model species:
We chose three model species (Figure 1; Table 2), each representative of one of the three basic survival schedules. Humans are a classic type I survivorship species, with high survival well into adulthood followed by a period of rapidly increasing mortality. The white-crowned sparrow (Zonotrichia leucophrys nuttalli; Baker et al. 1981) has a modified type II survivorship curve, with a constant survival rate after an episode of high early mortality. The barnacle (Balanus glandula; Connell 1970) exhibits a classical type III survivorship curve with very high early mortality, even after we reduced fecundity and age-1 mortality by an order of magnitude from published values to make the simulations more tractable. We used the human demographic data analyzed by Felsenstein (1971), which are arranged into 5-year age classes. For the other two species, life-history parameters were modified slightly from published values to provide for a constant population size and integer generation lengths in units of years (T = 3 years for the sparrow and 4 years for the barnacle). The published data for all three species are for females only, so for the purposes of this exercise we assumed that the same values apply to the entire population.
Figure 1.—

Survivorship curves for the three model species. See Table 2 for data sources.
TABLE 2.
Life-history parameters for the model species, scaled to constant population size
| Human
|
Sparrow
|
Barnacle
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Age (x) | lx | bx | vx | lx | bx | vx | lx | bx | vx |
| 1 | 1.000 | 0 | 1.000 | 1.000 | 0 | 1.000 | 1.00000 | 0 | 1.0 |
| 2 | 0.979 | 0 | 1.022 | 0.180 | 2.546 | 5.556 | 0.00062 | 359.2 | 1612.9 |
| 3 | 0.978 | 0.017 | 1.023 | 0.095 | 2.754 | 5.679 | 0.00034 | 679.4 | 2279.4 |
| 4 | 0.975 | 0.290 | 1.009 | 0.051 | 2.921 | 5.518 | 0.00020 | 898.1 | 2666.6 |
| 5 | 0.972 | 0.344 | 0.720 | 0.027 | 3.130 | 4.899 | 0.00016 | 991.8 | 2267.2 |
| 6 | 0.968 | 0.215 | 0.378 | 0.014 | 3.339 | 3.339 | 0.00011 | 991.8 | 1771.4 |
| 7 | 0.964 | 0.114 | 0.164 | — | — | — | 0.00007 | 991.8 | 1299.3 |
| 8 | 0.957 | 0.045 | 0.050 | — | — | — | 0.00002 | 991.8 | 991.8 |
| 9 | 0.946 | 0.006 | 0.006 | — | — | — | — | — | — |
| 10 | 0.928 | 0 | 0 | — | — | — | — | — | — |
| Generation (T) | 5.26 | 3.0 | 4.0 | ||||||
Sources: humans, Felsenstein (1971), based on data for United States white females taken from U. S. Department of Health, Education, and Welfare (1969, Vol. II, Part A, Table 5-2); white-crowned sparrow, modified from Baker et al. (1981); barnacle, modified from Connell (1970). Ages are 5-year units in humans and 1 year in the other two species.
Computer simulations:
We used computer simulations to model drift variance in allele frequency in populations with demographic parameters characteristic of the three life-history types. We considered both stable and growing populations.
Constant population size:
For each species we considered a “small” and a “large” population size, indexed by N1, the number of newborns produced each year [“newborns” were enumerated as the number of live births (humans), clutch size (sparrow), and plankton just after hatching (barnacle)]. The small and large N1 values were: human, 102, 103; sparrow, 103, 104; and barnacle, 105, 106 (Table 3). These population sizes translated into Ne values of order 102 for the small population sizes and of order 103 for the large population sizes (Table 3), on the basis of application of Equations 1 and 3 to the life-history data in Table 2. Given a fixed value of N1, the stable age distribution (and hence the number in each age class) is given by the right eigenvector of the Leslie matrix, which we calculated using the power method described by Caswell (2001). For the simulations, we rounded the number of individuals in each age class to the nearest integer, yielding (for each population size in each species) a vector of Nx values representing the (fixed) number of individuals in age class x.
TABLE 3.
Fixed numbers in each age class and effective population size for constant populations of the three model species
| Age | Human | Sparrow | Barnacle | |||
|---|---|---|---|---|---|---|
| 1 | 100 | 1,000 | 1,000 | 10,000 | 100,000 | 1,000,000 |
| 2 | 98 | 979 | 180 | 1,800 | 62 | 620 |
| 3 | 98 | 978 | 95 | 954 | 34 | 341 |
| 4 | 97 | 975 | 51 | 506 | 20 | 205 |
| 5 | 97 | 972 | 27 | 268 | 16 | 160 |
| 6 | 97 | 968 | 14 | 142 | 11 | 115 |
| 7 | 96 | 964 | — | — | 7 | 69 |
| 8 | 96 | 957 | — | — | 2 | 21 |
| 9 | 95 | 946 | — | — | — | — |
| 10 | 93 | 927 | — | — | — | — |
| >10a | 628 | 6,283 | — | — | — | — |
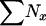 |
1,502 | 15,020 | 1,367 | 13,670 | 105 | 106 |
| Nadult | 676 | 6,760 | 367 | 3,670 | 152 | 1,521 |
| Ne (F) | 511.8 | 5,118.1 | 370.2 | 3,702.2 | 120.0 | 1,199.6 |
| Ne (H) | 511.8 | 5,118.1 | 370.3 | 3,702.8 | 119.9 | 1,199.2 |
| Ne (JR) | 511.8 | 5,118.1 | 370.1 | 3,702.1 | 119.8 | 1,199.1 |
| Ne (F)/∑Nx | 0.34 | 0.34 | 0.27 | 0.27 | 0.0012 | 0.0012 |
| Ne (F)/N1 | 5.12 | 5.12 | 0.37 | 0.37 | 0.0012 | 0.0012 |
| Ne (F)/Nadult | 0.76 | 0.76 | 1.01 | 1.01 | 0.79 | 0.79 |
Nadult = 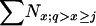 ; Ne (F), Equation 4 from Felsenstein (1971); Ne (H), Equation 3 modified from Hill (1972); Ne (JR), equilibrium value calculated by iterating Equations 2–8 in Jorde and Ryman (1995).
; Ne (F), Equation 4 from Felsenstein (1971); Ne (H), Equation 3 modified from Hill (1972); Ne (JR), equilibrium value calculated by iterating Equations 2–8 in Jorde and Ryman (1995).
Proportion of human females in age classes >10 based on estimate by Felsenstein (1971).
For each fixed demographic trajectory, we modeled the stochastic process of genetic drift by randomly drawing genes to represent birth of newborns and survival from one age class to the next. At each time t, we calculated the frequency of a “gamete pool” of infinite size as the weighted mean of the allele frequencies in each age class of reproductive adults: 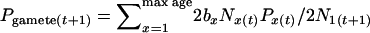 . In generating this gamete pool, all individuals within an age class contribute equally, but the total contribution differs among age classes on the basis of age-specific survival and birth rates. Therefore, the process for reproduction simulated an array of Wright–Fisher subpopulations with the possibility of selfing, stratified by age. [Whether a species is monoecious or dioecious, and whether or not selfing is allowed, has a negligible effect on Ne (Crow and Denniston 1988; Caballero 1994); therefore, this aspect of the model should not have appreciably influenced the results.] Next, we calculated the allele frequency in the N1 newborns in time unit t + 1 (P1(t+1)) by drawing 2N1 genes binomially from this gamete pool. Finally, for each age class x > 1, allele frequency at time t + 1 was calculated by sampling hypergeometrically (without replacement) from the 2Nx−1 genes representing the frequency in age class x − 1 at time t. This entire random process of sampling genes due to births and deaths was then repeated to generate an age-structured vector of allele frequencies for the next time unit. Simulated populations were initialized using the stable age distribution and with the same allele frequency in all age classes at time 0 [Px(0) = 0.5, x = 1, 2, …]. Each replicate simulation was run for 100 time units. Each run of 100 time units was taken to represent the trajectory of one gene locus, and the entire procedure was repeated for each additional locus (5000 loci total).
. In generating this gamete pool, all individuals within an age class contribute equally, but the total contribution differs among age classes on the basis of age-specific survival and birth rates. Therefore, the process for reproduction simulated an array of Wright–Fisher subpopulations with the possibility of selfing, stratified by age. [Whether a species is monoecious or dioecious, and whether or not selfing is allowed, has a negligible effect on Ne (Crow and Denniston 1988; Caballero 1994); therefore, this aspect of the model should not have appreciably influenced the results.] Next, we calculated the allele frequency in the N1 newborns in time unit t + 1 (P1(t+1)) by drawing 2N1 genes binomially from this gamete pool. Finally, for each age class x > 1, allele frequency at time t + 1 was calculated by sampling hypergeometrically (without replacement) from the 2Nx−1 genes representing the frequency in age class x − 1 at time t. This entire random process of sampling genes due to births and deaths was then repeated to generate an age-structured vector of allele frequencies for the next time unit. Simulated populations were initialized using the stable age distribution and with the same allele frequency in all age classes at time 0 [Px(0) = 0.5, x = 1, 2, …]. Each replicate simulation was run for 100 time units. Each run of 100 time units was taken to represent the trajectory of one gene locus, and the entire procedure was repeated for each additional locus (5000 loci total).
At each time unit, population allele frequencies were computed parametrically (by exhaustive population census) and estimated by sampling subsets of individuals. Parametric frequencies were computed two ways: (1) the standard method, which involves counting alleles in all individuals in the population and leads to an unweighted frequency 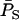 , and (2) the weighted method, in which each individual is weighted by its reproductive value (
, and (2) the weighted method, in which each individual is weighted by its reproductive value (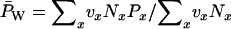 ; Felsenstein 1971).
; Felsenstein 1971).
Sampling of individuals was simulated by random draw of the required number of genes from specified age classes, and sample allele frequencies were computed by counting alleles (hence the sample frequencies were unweighted). We considered three different sample sizes (S1 = S2 = 25, 50, and 100) and three different sampling regimes: (a) all individuals are at equal risk of being sampled regardless of their age, (b) only reproductive adults (those in age classes with bx > 0) are subject to sampling, and (c) only newborns are subject to sampling (so sampling is from a single cohort). In all cases, genes were counted without replacement, so no individual could be sampled more than once in any time period. However, after the sampling (or enumeration) was completed, all genes were returned to the population, so sampling did not affect the future demographic or genetic trajectory of the population.
After initializing all age classes at the same frequency (P0 = 0.5), a period of time is needed before the population reaches a dynamic equilibrium with respect to the amount of random allele-frequency variation among years and among age classes within years. In agreement with results reported by previous authors (Jorde and Ryman 1995; Waples 2002), we found that this dynamic equilibrium was reached quickly (within a few generations) under the conditions we modeled (data not shown). Therefore, we allowed each replicate to “warm up” for 20 time periods (periods 0–19) before collecting data.
Beginning in period 20, parametric or estimated population allele frequencies were compared at several different time intervals (L): 1 time unit and 1, 5, or 10 generations (T, 5T, and 10T time units, respectively). For each time interval, we used the method of Nei and Tajima (1981) to estimate F, the standardized variance of allele frequencies at two points in time. For diallelic loci such as those considered here, this measure is calculated as
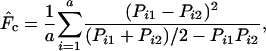 |
(5) |
where a is the number of loci and Pi1 and Pi2 are gene frequencies at the ith locus in the first and second samples, respectively. We also considered a closely related measure (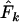 ) proposed by Pollak (1983). As previous authors have shown that the two measures generally lead to comparable results, we focused on
) proposed by Pollak (1983). As previous authors have shown that the two measures generally lead to comparable results, we focused on 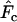 because it has a smaller variance for diallelic loci (Tajima and Nei 1984; Waples 1989). However, we found that in some circumstances
because it has a smaller variance for diallelic loci (Tajima and Nei 1984; Waples 1989). However, we found that in some circumstances 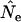 differed substantially depending on which estimator of F was used, and these cases are noted below.
differed substantially depending on which estimator of F was used, and these cases are noted below.
For each time interval, we computed an overall 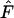 as the mean across the 5000 replicate loci. After advancing the initial time period by one generation (e.g., first sample occurs in time period 20 + T), this process was repeated for each interval of L years, generating a vector of mean
as the mean across the 5000 replicate loci. After advancing the initial time period by one generation (e.g., first sample occurs in time period 20 + T), this process was repeated for each interval of L years, generating a vector of mean 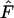 values for a number of intervals of the same duration equally spaced one generation apart across the last 80 years of each replicate. For some analyses, we also averaged across the elements of these vectors to arrive at an overall mean
values for a number of intervals of the same duration equally spaced one generation apart across the last 80 years of each replicate. For some analyses, we also averaged across the elements of these vectors to arrive at an overall mean 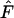 characteristic of a particular sampling interval of L years.
characteristic of a particular sampling interval of L years.
To evaluate accuracy of the demographic formula for Ne, we compared observed rates of allele frequency change in simulated populations with the magnitude of change expected assuming that true Ne was as given in Table 3. The expected variance of parametric population allele frequency at time period t among replicate populations or gene loci is given by
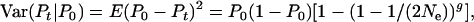 |
(6) |
where P0 = initial allele frequency (0.5) and g = t/T = elapsed time in generations. For the simulated populations, we calculated an observed variance in allele frequency at time period t as the variance in 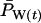 among 5000 replicate gene loci.
among 5000 replicate gene loci.
Estimating Ne with the temporal method:
In the temporal method, effective size is estimated by relating the observed amount of allele frequency change to that expected under pure drift. The standard temporal method (Krimbas and Tsakas 1971; Nei and Tajima 1981; Pollak 1983; Waples 1989) is commonly referred to as the moment method because it relies on an estimate of F. Under a pure drift model, the expected value of 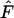 depends on Ne, the number of generations between samples (g), and the sampling regime. Here we consider nonlethal sampling of the population (with replacement, as in collecting a biopsy for DNA analysis). This corresponds to sampling plan I described by Nei and Tajima (1981) and Waples (1989). Under these conditions,
depends on Ne, the number of generations between samples (g), and the sampling regime. Here we consider nonlethal sampling of the population (with replacement, as in collecting a biopsy for DNA analysis). This corresponds to sampling plan I described by Nei and Tajima (1981) and Waples (1989). Under these conditions,
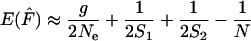 |
and
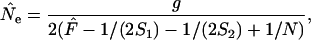 |
where S1 and S2 are the sample sizes at times 1 and 2, N is the number of individuals at risk of being sampled at the time of the first sample, and g = L/T = elapsed time in generations. Because sample sizes S1 and S2 were the same in our analyses of constant population size, the above equation simplifies to
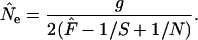 |
(7) |
The term 1/N accounts for the covariance of allele frequencies at times 1 and 2 that arises in plan I sampling because some individuals in the initial sample can also contribute genes to future generations. The appropriate value of N depends on the sampling scheme. If sampling is random with respect to the entire population, the estimator of Ne becomes
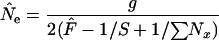 |
(8) |
(sampling from entire population). For an exhaustive population census, S = 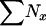 , leading to
, leading to
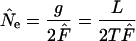 |
(9) |
(complete population census). If only reproductive adults are sampled, then the number subject to sampling is the total in age classes equal to or older than the age at first maturity (j) and younger than the age of senescence, q (at which age bx = 0 again):
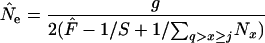 |
(10) |
(sampling mature adults). Finally, if each sample is taken from a single cohort of N1 newborns, the appropriate estimator is
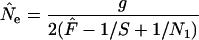 |
(11) |
(sampling newborns). Assuming that survival among age classes is random, Equation 11 also applies to any random sample of a single cohort; it is necessary to substitute for N1 only the total number remaining in the cohort at the time of sampling.
Jorde and Ryman (1995) modified the standard temporal method to account for overlapping generations. Their modified estimator is (in current notation and assuming plan I sampling)
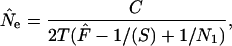 |
(12) |
where C is a constant that depends on life-history features of the population. Jorde and Ryman's model assumes that population size and demographic parameters are constant and is based on a comparison of consecutive cohorts, which can be achieved by sampling single cohorts or sampling mixed cohorts and sorting individuals by age to reconstruct single-cohort samples. We sampled randomly from the N1 newborns in successive years and used Equation 12 to estimate Ne using the Jorde–Ryman method. The recurrent formulas (10–13) and (23) in Jorde and Ryman (1995) were used to calculate C. This iterative process rapidly converged on a constant C value for each life-history scenario we considered.
Changing population size:
We also modeled “human” and “sparrow” populations that grew deterministically, increasing each time period by the multiplicative factor λ. We used the λ values estimated from the original published data (1.037 for humans and 1.063 for the sparrow). In addition, we considered a human population that declined at the rate λ = 0.994 per time period. These populations were initiated and allowed to run for 20 time periods (periods 0–19) at constant size (determined by low N1) and demographic parameters given in Table 2. At time period 20, population growth was generated by scaling the birth rates to provide the desired λ, while the lx values remained unchanged. At time period 20 the age structure was also adjusted to conform to the stable age distribution for a growing (or declining) population. In each successive time period, the vector of Nx values was obtained by applying the birth and death rates (Table 6) to the current population: the newborn cohort was generated according to 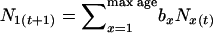 , and random mortality as described above determined which genes survived from one age class to the next. We generated 100 time periods of population sizes using real numbers for the Nx but rounded these to integers in each time period before conducting the simulations. Under the modeled growth rates, the human population (excluding senescent individuals) increased from 826 to >31,000, and the sparrow population size climbed from 1326 to >5 × 105 (Table 6). In the human decline scenario, population size dropped from 5118 to 2807.
, and random mortality as described above determined which genes survived from one age class to the next. We generated 100 time periods of population sizes using real numbers for the Nx but rounded these to integers in each time period before conducting the simulations. Under the modeled growth rates, the human population (excluding senescent individuals) increased from 826 to >31,000, and the sparrow population size climbed from 1326 to >5 × 105 (Table 6). In the human decline scenario, population size dropped from 5118 to 2807.
TABLE 6.
Life-history parameters for the model species in populations that change in size
| Human growth
|
Human decline
|
Sparrow growth
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Age (x) | lx | bx | vx | lx | bx | vx | lx | bx | vx |
| 1 | 1.000 | 0.000 | 1.000 | 1 | 0 | 1 | 1.000 | 0.000 | 1.000 |
| 2 | 0.979 | 0.000 | 1.059 | 0.979 | 0 | 1.016 | 0.180 | 3.050 | 5.905 |
| 3 | 0.978 | 0.021 | 1.100 | 0.978 | 0.017 | 1.010 | 0.095 | 3.300 | 6.090 |
| 4 | 0.975 | 0.352 | 1.123 | 0.975 | 0.281 | 0.990 | 0.051 | 3.500 | 5.987 |
| 5 | 0.972 | 0.416 | 0.816 | 0.972 | 0.333 | 0.705 | 0.027 | 3.750 | 5.404 |
| 6 | 0.968 | 0.260 | 0.432 | 0.968 | 0.208 | 0.370 | 0.014 | 4.000 | 3.763 |
| 7 | 0.964 | 0.138 | 0.189 | 0.964 | 0.110 | 0.160 | |||
| 8 | 0.957 | 0.054 | 0.059 | 0.957 | 0.043 | 0.049 | |||
| 9 | 0.946 | 0.007 | 0.007 | 0.946 | 0.006 | 0.006 | |||
| 10 | 0.928 | 0 | 0.000 | 0.928 | 0 | 0 | |||
| T = 5.2105 | T = 5.2695 | T = 2.919 | |||||||
| λ = 1.037 | λ = 0.994 | λ = 1.063 | |||||||
With changing population size, effective size also changes every time interval. At a given time t, Equation 4 gives a per-generation effective size (Ne(Ft)) that characterizes the expected amount of gene frequency drift that occurs between times t and t + T (Felsenstein 1971). This definition creates an analytical difficulty here. Because there is a different Ne(Ft) for every time period, genetic change in the population is described by an array of Ne(Ft) values that apply to partially overlapping periods of one generation each, and it is not immediately clear how to determine the appropriate Ne value for any given point in time. In the present context, we need to evaluate genetic change over one or more individual time units that are fractions of a generation. Therefore, we need a way to calculate E(Ne(t)), which is the effective size that describes genetic change between times t and t + 1. As shown in appendix b, E(Ne(t)) is just the instantaneous Ne for the point midway between times t and t + 1,
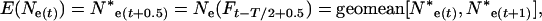 |
(13) |
where Ne*(t) is an instantaneous effective size that describes the actual rate of change in the population at time t. E(Ne(t)) from Equation 13 can be compared with 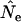 estimated from samples taken in time periods t and t + 1. For longer time intervals, let E(Ne(t,t+x)) represent the effective size that determines the rate of change in the population between time periods t and t + x. Then E(Ne(t,t+x)) can be calculated as the harmonic mean of the E(Ne(t)) values for time periods t through t + x − 1.
estimated from samples taken in time periods t and t + 1. For longer time intervals, let E(Ne(t,t+x)) represent the effective size that determines the rate of change in the population between time periods t and t + x. Then E(Ne(t,t+x)) can be calculated as the harmonic mean of the E(Ne(t)) values for time periods t through t + x − 1.
When effective size changes over time, the expected variance of parametric population allele frequency at time period t among replicate populations or gene loci is
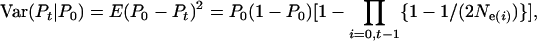 |
where Ne(i) is the effective size for the time period i to i + 1. We used Equation 13 to calculate E(Ne(i)) for each time period.
To estimate Ne, we used the procedure described above to calculate 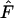 for various time intervals and sampling regimes. With changing population size, however, it is necessary to adjust the terms for N to correspond to the correct time period. Therefore, in Equations 8, 10, and 11 we used the values for
for various time intervals and sampling regimes. With changing population size, however, it is necessary to adjust the terms for N to correspond to the correct time period. Therefore, in Equations 8, 10, and 11 we used the values for 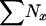 ,
, 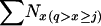 , and N1, respectively, that applied to the time period of the first sample. The sample sizes of 25, 50, and 100 were fixed, but when complete population enumeration was done to calculate parametric population allele frequencies in growing populations, the sample size at time 2 was larger than that at time 1: S2 > S1 =
, and N1, respectively, that applied to the time period of the first sample. The sample sizes of 25, 50, and 100 were fixed, but when complete population enumeration was done to calculate parametric population allele frequencies in growing populations, the sample size at time 2 was larger than that at time 1: S2 > S1 = 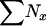 . Therefore, Equation 9 was modified as follows:
. Therefore, Equation 9 was modified as follows:
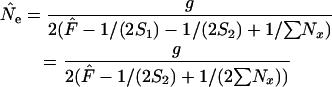 |
(14) |
(complete population census; growing population).
RESULTS
Constant population size:
For the three model species, we compared the demographic calculations of Ne (based on Felsenstein 1971) with values calculated by two other methods (Equation 3, modified from Hill 1972, and Jorde and Ryman 1995). The three methods produced essentially identical values of Ne under both low and high N1 (Table 3). For simplicity, in what follows we use only Equation 4 to calculate Ne from demographic data.
Comparison of expected and observed rates of change in allele frequency:
As shown in Figure 2, the observed variance in weighted population allele frequencies 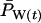 very closely tracked the expected variance in all three model species, indicating that the putative Ne values in Table 3 accurately describe the variance effective size of these populations. For the remainder of this section, therefore, we refer to the values in Table 3 as the “true” Ne.
very closely tracked the expected variance in all three model species, indicating that the putative Ne values in Table 3 accurately describe the variance effective size of these populations. For the remainder of this section, therefore, we refer to the values in Table 3 as the “true” Ne.
Figure 2.—

Comparison of observed and expected Var(Pt) for the three model species under constant population size and low N1. Comparable results were found for high N1 (data not shown).
Genetic estimates of Ne:
Figure 3 compares true Ne values with estimates of Ne based on the standard temporal method, using complete population census to calculate weighted allele frequencies (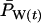 ). For both small and large cohort sizes in all three species, agreement of expected and true Ne was very good. Under every scenario,
). For both small and large cohort sizes in all three species, agreement of expected and true Ne was very good. Under every scenario, 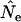 for a given time interval fluctuated randomly around the true value. Figure 3 shows results for low N1 for samples taken 1 year apart; similar results were found for high N1 and longer time between samples (L = 1, 5, or 10T; Table 4). At the extreme, deviations of
for a given time interval fluctuated randomly around the true value. Figure 3 shows results for low N1 for samples taken 1 year apart; similar results were found for high N1 and longer time between samples (L = 1, 5, or 10T; Table 4). At the extreme, deviations of 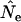 from true Ne were <3% (sampling interval of 10T in humans and barnacle).
from true Ne were <3% (sampling interval of 10T in humans and barnacle).
Figure 3.—

Comparison of estimated Ne (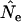 ) and expected Ne [E(Ne)] for the low N1 simulations of the three model species under constant population size. Expected Ne uses Equation 4 and the life-table data in Table 2. Estimated Ne was calculated using the standard temporal method (Equation 9) on the basis of samples taken 1 year apart;
) and expected Ne [E(Ne)] for the low N1 simulations of the three model species under constant population size. Expected Ne uses Equation 4 and the life-table data in Table 2. Estimated Ne was calculated using the standard temporal method (Equation 9) on the basis of samples taken 1 year apart; 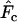 was computed by complete population enumeration, using either unweighted allele frequencies or frequencies weighted by reproductive value.
was computed by complete population enumeration, using either unweighted allele frequencies or frequencies weighted by reproductive value.
TABLE 4.
Effective population size estimated for the three model species using the standard temporal method
| Human
|
Sparrow
|
Barnacle
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Sampling interval | N1: | 100 | 1,000 | 1,000 | 10,000 | 100,000 | 1,000,000 | ||
| Sampling regime | S | Ne (F): | 511.8 | 5,118.1 | 370.2 | 3,702.2 | 120.0 | 1,199.6 | |
| Complete census | |||||||||
| Unweighted P | 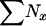 |
1 | 874 | 8,761 | 175 | 1,755 | 67 | 673 | |
| 1T | 893 | 8,868 | 289 | 2,867 | 96 | 955 | |||
| 5T | 568 | 5,708 | 353 | 3,535 | 116 | 1,126 | |||
| 10T | 547 | 5,536 | 365 | 3,631 | 121 | 1,142 | |||
| Weighted P | 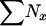 |
1 | 511 | 5,116 | 370 | 3,709 | 120 | 1,203 | |
| T | 510 | 5,094 | 371 | 3,683 | 120 | 1,195 | |||
| 5T | 519 | 5,173 | 370 | 3,718 | 122 | 1,182 | |||
| 10T | 522 | 5,271 | 372 | 3,726 | 123 | 1,174 | |||
| Random sampling | |||||||||
| All age classes | 25 | 1 | Inf | Inf | 1,826 | Inf | 116 | Inf | |
| 1T | Inf | Inf | 572 | Inf | 122 | Inf | |||
| 5T | 626 | Inf | 446 | Inf | 129 | 1,801 | |||
| 10T | 606 | 26,676 | 414 | 8,228 | 131 | 1,435 | |||
| 50 | 1 | Inf | Inf | 226 | Inf | 74 | Inf | ||
| 1T | 1,320 | Inf | 320 | Inf | 102 | 1,631 | |||
| 5T | 645 | 12,865 | 367 | 4,679 | 120 | 1,270 | |||
| 10T | 580 | 5,509 | 377 | 5,004 | 125 | 1,316 | |||
| 100 | 1 | 1,139 | Inf | 184 | 5,838 | 73 | 838 | ||
| 1T | 1,076 | 5,268 | 285 | 2,179 | 100 | 992 | |||
| 5T | 577 | 5,881 | 358 | 3,618 | 118 | 1,159 | |||
| 10T | 563 | 4,940 | 371 | 3,650 | 123 | 1,162 | |||
| Adults | 25 | 1 | Inf | Inf | 222 | Inf | 60 | Inf | |
| 1T | 82,131 | Inf | 377 | Inf | 106 | Inf | |||
| 5T | 711 | Inf | 401 | 76,308 | 122 | 1,706 | |||
| 10T | 607 | Inf | 404 | 9,308 | 127 | 1,415 | |||
| 50 | 1 | 2,579 | Inf | 135 | Inf | 51 | Inf | ||
| 1T | 846 | Inf | 278 | Inf | 100 | 1,620 | |||
| 5T | 552 | 11,641 | 359 | 6,463 | 119 | 1,223 | |||
| 10T | 554 | 8,161 | 367 | 5,222 | 124 | 1,154 | |||
| 100 | 1 | 538 | Inf | 131 | 3,863 | 49 | 566 | ||
| 1T | 893 | 29,797 | 251 | 5,103 | 97 | 896 | |||
| 5T | 563 | 6,041 | 348 | 3,820 | 117 | 1,150 | |||
| 10T | 555 | 5,880 | 365 | 3,847 | 122 | 1,178 | |||
| Newborns | 25 | 1 | 10 | 1,811 | 118 | Inf | 141 | Inf | |
| 1T | 64 | 2,991 | 240 | Inf | 123 | Inf | |||
| 5T | 195 | 5,655 | 352 | 156,798 | 127 | 1,591 | |||
| 10T | 280 | 5,260 | 390 | 9,502 | 131 | 1,519 | |||
| 50 | 1 | 9 | 119 | 82 | Inf | 80 | 3,838 | ||
| 1T | 60 | 922 | 201 | Inf | 101 | 1,437 | |||
| 5T | 184 | 1,754 | 329 | 5,138 | 119 | 1,169 | |||
| 10T | 264 | 2,834 | 351 | 4,591 | 125 | 1,199 | |||
| 100 | 1 | 9 | 93 | 75 | 1,109 | 69 | 892 | ||
| 1T | 59 | 618 | 189 | 3,323 | 96 | 997 | |||
| 5T | 181 | 1,884 | 321 | 3,489 | 118 | 1,137 | |||
| 10T | 265 | 2,790 | 350 | 3,571 | 123 | 1,153 | |||
Allele frequencies were computed on the basis of either a complete population census (using weighted or unweighted allele frequencies) or unweighted allele frequencies in random samples from the population as a whole (all age classes), only adults, or only the cohort of newborns. Samples of size S individuals were taken at the indicated time intervals. Results are based on mean 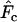 values calculated over 5000 replicate gene loci. Inf, a point estimate of infinity.
values calculated over 5000 replicate gene loci. Inf, a point estimate of infinity.
We also estimated Ne using the temporal method based on a complete population census using unweighted allele frequencies (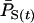 ); this produced estimates of Ne that were substantially biased in all cases: ∼50% too high for humans and ∼50% too low for the other two species (Figure 3; Table 4). That is, complete enumeration of all individuals alive at a given time does not provide a reliable way of calculating parametric population allele frequencies in species with overlapping generations.
); this produced estimates of Ne that were substantially biased in all cases: ∼50% too high for humans and ∼50% too low for the other two species (Figure 3; Table 4). That is, complete enumeration of all individuals alive at a given time does not provide a reliable way of calculating parametric population allele frequencies in species with overlapping generations.
The array of different sampling regimes produced a range of outcomes in the three species (Table 4). Two general patterns can be identified. First, small samples (S = 25) produced estimates of Ne that had large biases under many scenarios, even when 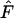 was averaged across 5000 replicate gene loci. Sample sizes of 50 or 100 produced Ne estimates that were less variable and more accurate.
was averaged across 5000 replicate gene loci. Sample sizes of 50 or 100 produced Ne estimates that were less variable and more accurate.
Second, under most sampling regimes in most species, 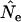 was severely biased when F was estimated over short time intervals but approached true Ne as the sampling interval increased. For example, Figure 4 plots
was severely biased when F was estimated over short time intervals but approached true Ne as the sampling interval increased. For example, Figure 4 plots 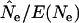 as a function of time between samples for random samples from either newborns or the entire population. In all six scenarios, the longest sampling interval (10T) produced the most accurate estimate of Ne. In five of the six cases the estimates for shorter time intervals were biased more strongly downward; however, sampling from the entire population for humans led to overestimates of Ne, and the bias was high for L = 1 generation (Figure 4). Figure 4 shows results for conditions under which the temporal method has the most power: large sample size (S = 100) and low N1 (hence relatively small Ne). The same basic pattern (more accurate estimates of Ne over longer time periods) is seen, albeit sometimes less clearly, in the scenarios with smaller sample sizes and larger N1 (Table 4). Sampling only from adult age classes produced results qualitatively similar to those for sampling from the entire population under all scenarios (Table 4).
as a function of time between samples for random samples from either newborns or the entire population. In all six scenarios, the longest sampling interval (10T) produced the most accurate estimate of Ne. In five of the six cases the estimates for shorter time intervals were biased more strongly downward; however, sampling from the entire population for humans led to overestimates of Ne, and the bias was high for L = 1 generation (Figure 4). Figure 4 shows results for conditions under which the temporal method has the most power: large sample size (S = 100) and low N1 (hence relatively small Ne). The same basic pattern (more accurate estimates of Ne over longer time periods) is seen, albeit sometimes less clearly, in the scenarios with smaller sample sizes and larger N1 (Table 4). Sampling only from adult age classes produced results qualitatively similar to those for sampling from the entire population under all scenarios (Table 4).
Figure 4.—

The ratio 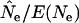 for the three model species under constant population size and low N1. E(Ne) was computed using Equation 4 and the life-table data in Table 2.
for the three model species under constant population size and low N1. E(Ne) was computed using Equation 4 and the life-table data in Table 2. 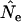 was calculated using
was calculated using 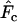 in the standard temporal method, using random samples either from the population as a whole (Equation 8; solid symbols) or only from the cohort of newborns (Equation 11; open symbols). Sample sizes at times 1 and 2 were fixed at S1 = S2 = 100 but the interval between samples varied from L = 1 to 10 generations.
in the standard temporal method, using random samples either from the population as a whole (Equation 8; solid symbols) or only from the cohort of newborns (Equation 11; open symbols). Sample sizes at times 1 and 2 were fixed at S1 = S2 = 100 but the interval between samples varied from L = 1 to 10 generations.
Finally, we evaluated estimates of Ne from samples (rather than complete population census) of individuals weighted by reproductive value. These estimates were consistently biased downward (data not shown), a result that we determined arises because weighting the samples increases the sampling variance of allele frequency beyond that expected for the nominal sample size S. Although it is possible, for a given set of individual weights, to account for this increase in variance to arrive at an effective sample size, in natural populations the information necessary to do so will rarely be available. Consequently, attempting to weight samples by reproductive value is unlikely to be a practical solution and we did not pursue this idea further.
The Jorde–Ryman method was designed for application to data for two successive cohorts (equivalent to sampling newborns in two consecutive years in our model), and under that scenario 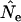 for the Jorde–Ryman method asymptotically approached the true Ne as sample size increased (Table 5). In their published example (which was for a species with type I survivorship), Jorde and Ryman found that with S = 30,
for the Jorde–Ryman method asymptotically approached the true Ne as sample size increased (Table 5). In their published example (which was for a species with type I survivorship), Jorde and Ryman found that with S = 30, 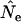 based on
based on 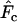 was ∼10% too high. With the human life history, we found comparable results for S = 25 and low N1 (8% upward bias in
was ∼10% too high. With the human life history, we found comparable results for S = 25 and low N1 (8% upward bias in 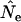 ; Table 5), but this bias was considerably less for larger samples. For the sparrow and barnacle life histories, the Jorde–Ryman method also produced accurate estimates for large sample size but the upward bias was more extreme (up to 100%) for S = 25. A more pronounced upward bias was also found for high N1 simulations, especially for small sample sizes (Table 5). When we used the Jorde–Ryman method with Pollak's measure
; Table 5), but this bias was considerably less for larger samples. For the sparrow and barnacle life histories, the Jorde–Ryman method also produced accurate estimates for large sample size but the upward bias was more extreme (up to 100%) for S = 25. A more pronounced upward bias was also found for high N1 simulations, especially for small sample sizes (Table 5). When we used the Jorde–Ryman method with Pollak's measure 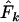 , we found the same general pattern (
, we found the same general pattern (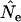 asymptotically accurate for large samples) except that small samples led to underestimates (rather than overestimates) of Ne (data not shown).
asymptotically accurate for large samples) except that small samples led to underestimates (rather than overestimates) of Ne (data not shown).
TABLE 5.
Effective population size estimated for constant populations of the three model species using the method of Jorde and Ryman (1995)
| Human (C = 56.80)
|
Sparrow (C = 4.947)
|
Barnacle (C = 1.793)
|
||||||
|---|---|---|---|---|---|---|---|---|
| N1: | 100 | 1,000 | 1,000 | 10,000 | 100,000 | 1,000,000 | ||
| Sampling regime | S | Ne (F): | 511.8 | 5,118.1 | 370.2 | 3,702.2 | 120.0 | 1,199.6 |
| Newborns | 25 | 553 | 102,835 | 583 | Inf | 251 | Inf | |
| 50 | 523 | 6,778 | 405 | Inf | 143 | 6,848 | ||
| 100 | 515 | 5,286 | 372 | 5,488 | 124 | 1,591 | ||
Estimates are based on unweighted allele frequency differences between samples taken in two consecutive time periods; other conditions are as described in Table 4. Parameter C is calculated by iterating Equations 10–13 in Jorde and Ryman (1995).
Ne/N ratio:
As pointed out by previous authors (e.g., Nunney 1993; Frankham 1995), the Ne/N ratio is sensitive to the choice of which age classes to include in calculating N. Results in Table 3 illustrate this point. For the barnacle, Ne/N is 1.2 × 10−3 if population size is taken to be all individuals alive at a given time (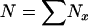 ) but is 0.79 (almost three orders of magnitude higher) if only mature adults are counted (N = Nadult). [Note that to make our simulations more tractable we reduced fecundity and increased age-1 survival of the barnacle by an order of magnitude; the difference between Ne/N ratios would be even more extreme using the published data]. The effects are not as dramatic in the other species, but even for humans the ratio Ne/N is over twice as high if N is taken to be the number of reproductive adults rather than the total population size (0.76 vs. 0.34; Table 3).
) but is 0.79 (almost three orders of magnitude higher) if only mature adults are counted (N = Nadult). [Note that to make our simulations more tractable we reduced fecundity and increased age-1 survival of the barnacle by an order of magnitude; the difference between Ne/N ratios would be even more extreme using the published data]. The effects are not as dramatic in the other species, but even for humans the ratio Ne/N is over twice as high if N is taken to be the number of reproductive adults rather than the total population size (0.76 vs. 0.34; Table 3).
Changing population size:
Comparison of expected and observed rates of change in allele frequency:
For all three scenarios (human growth and decline and sparrow growth; see Table 6), the observed variance in weighted population allele frequencies 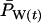 closely tracked the expected variance (data not shown, but results are comparable to those shown in Figure 2), indicating that Equation 13 accurately predicts the effective size as it changes over time.
closely tracked the expected variance (data not shown, but results are comparable to those shown in Figure 2), indicating that Equation 13 accurately predicts the effective size as it changes over time.
Genetic estimates of Ne:
Agreement between 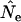 and E(Ne) for populations that changed in size was excellent for all three scenarios considered. Median
and E(Ne) for populations that changed in size was excellent for all three scenarios considered. Median 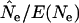 ratios were almost exactly 1.0 for all time periods when weighted population allele frequencies were used to calculate
ratios were almost exactly 1.0 for all time periods when weighted population allele frequencies were used to calculate 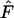 (Table 7). Other sampling regimes produced results that roughly paralleled those for populations of constant size. In humans, a population census with unweighted frequencies led to overestimates of Ne for short time periods, with lower bias over longer time periods. Conversely, sampling newborns led to gross underestimates of Ne for short time periods (Table 7). In the sparrow, a complete population census using unweighted allele frequencies and random sampling from newborns both produced severe underestimates of Ne for short time intervals, but
(Table 7). Other sampling regimes produced results that roughly paralleled those for populations of constant size. In humans, a population census with unweighted frequencies led to overestimates of Ne for short time periods, with lower bias over longer time periods. Conversely, sampling newborns led to gross underestimates of Ne for short time periods (Table 7). In the sparrow, a complete population census using unweighted allele frequencies and random sampling from newborns both produced severe underestimates of Ne for short time intervals, but 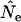 approached E(Ne) for longer time intervals. In contrast, sampling from the entire population or only adults led to substantial overestimates for short time periods. These patterns are all consistent with those found for constant population size (Tables 4 and 7).
approached E(Ne) for longer time intervals. In contrast, sampling from the entire population or only adults led to substantial overestimates for short time periods. These patterns are all consistent with those found for constant population size (Tables 4 and 7).
TABLE 7.
Ratio of estimated (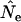 ) to expected [E(Ne)] effective size for the three model species in populations that change in size
) to expected [E(Ne)] effective size for the three model species in populations that change in size
| Sampling regime | L | Human growth | Human decline | Sparrow growth |
|---|---|---|---|---|
| Census | ||||
| Unweighted | 1 | 1.22 | 1.80 | 0.44 |
| T | 1.42 | 1.79 | 0.75 | |
| 5T | 1.03 | 1.12 | 0.94 | |
| 10T | 1.00 | 1.08 | 0.97 | |
| Weighted | 1 | 1.00 | 1.00 | 1.01 |
| T | 1.01 | 1.00 | 1.01 | |
| 5T | 1.01 | 1.01 | 1.01 | |
| 10T | 1.01 | 1.01 | 1.02 | |
| Samplinga | ||||
| All age classes | 1 | Inf | Inf | 3.19 |
| T | 5.28 | 1.83 | 1.28 | |
| 5T | 1.12 | 1.21 | 1.02 | |
| 10T | 1.06 | 1.15 | 1.00 | |
| Adults | 1 | Inf | Inf | Inf |
| T | Inf | Inf | Inf | |
| 5T | 1.17 | 1.27 | 2.56 | |
| 10T | 1.02 | 1.11 | 1.36 | |
| Newborns | 1 | 0.12 | 0.11 | 0.23 |
| T | Inf | Inf | 0.66 | |
| 5T | 1.25 | 1.04 | 0.91 | |
| 10T | 1.06 | 1.07 | 0.97 |
Values shown are median 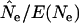 ratios for samples taken L time units apart; other conditions are as in Table 4.
ratios for samples taken L time units apart; other conditions are as in Table 4.
Sample size = 100; unweighted allele frequencies.
DISCUSSION
Felsenstein (1971) proposed, but did not prove, that weighting each individual by its reproductive value is the correct way to calculate parametric population allele frequencies in species with overlapping generations. Our results demonstrate that this is indeed the case, regardless of the species' life history and regardless of whether the population is fixed or changing in size at a constant rate. In contrast, measuring the magnitude of population change using unweighted population allele frequencies led to large departures from expected values in all scenarios considered, even when the entire population was sampled.
These results indicate that caution is needed in interpreting standard-model temporal estimates of effective population size in species with overlapping generations. The resulting estimates will be biased unless the entire population is sampled and individual genotypes are weighted by their reproductive value. Unfortunately, this is not a practical solution to the problem of estimating Ne. Rarely is it feasible to sample an entire population, and, even if this were possible (e.g., as might be the case in small captive populations), the age-specific life history data needed to compute individual reproductive values typically are not available. As discussed above, taking samples and weighting allele frequencies by reproductive value also are generally not feasible. One solution to this problem is to use the Jorde–Ryman method (Jorde and Ryman 1995), which we found generally performed well when it was applied to situations for which it was designed: analysis of samples from consecutive cohorts. This method can be used with any age-structured species, but in practice it has not been widely applied, perhaps because it requires both detailed demographic information to compute the correction factor C and samples from single cohorts in successive years. Most applications of the temporal method for species with overlapping generations continue to use the standard discrete-generation model, so it is important to consider the nature and magnitude of the bias that is likely to result from common sampling schemes. These results are complex, but some patterns are apparent.
First, bias in 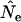 is largest for short time intervals and in many cases largely disappears after 5–10 generations between samples (Figure 4; Table 4). This pattern, which was also seen in analyses of growing populations (Table 7), can be understood by noting that the magnitude of
is largest for short time intervals and in many cases largely disappears after 5–10 generations between samples (Figure 4; Table 4). This pattern, which was also seen in analyses of growing populations (Table 7), can be understood by noting that the magnitude of 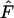 (and hence
(and hence 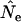 ) is determined by two major components: a component related to drift (the signal) and one related to sampling a finite number of individuals (the noise). The bias arises because the adjustment for sampling derived for the discrete-generation model does not capture all of the complexities associated with sampling from age-structured populations (Jorde and Ryman 1995). The magnitude of this sampling bias is fixed by the sample size and the nature of the sampling in relation to the species' life history; it does not change with the amount of time between samples. In contrast, a longer sampling interval allows more episodes of genetic drift to influence
) is determined by two major components: a component related to drift (the signal) and one related to sampling a finite number of individuals (the noise). The bias arises because the adjustment for sampling derived for the discrete-generation model does not capture all of the complexities associated with sampling from age-structured populations (Jorde and Ryman 1995). The magnitude of this sampling bias is fixed by the sample size and the nature of the sampling in relation to the species' life history; it does not change with the amount of time between samples. In contrast, a longer sampling interval allows more episodes of genetic drift to influence 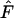 , thus increasing the signal-to-noise ratio in the data.
, thus increasing the signal-to-noise ratio in the data.
The second noteworthy pattern is that the direction of bias in 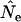 based on
based on 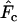 differs markedly between the human and the barnacle: random sampling from the population (or a complete population census with unweighted allele frequencies) leads to an overestimate of Ne in humans but an underestimate in the barnacle. In the barnacle, the vast majority of individuals alive at any time are newborns, so a random sample will primarily reflect frequencies in the newborns, which are derived only from the fraction of the population that reproduced the previous year. The result is a sampling variance greater than would be expected from a random sample from the generation as a whole, with the consequence that
differs markedly between the human and the barnacle: random sampling from the population (or a complete population census with unweighted allele frequencies) leads to an overestimate of Ne in humans but an underestimate in the barnacle. In the barnacle, the vast majority of individuals alive at any time are newborns, so a random sample will primarily reflect frequencies in the newborns, which are derived only from the fraction of the population that reproduced the previous year. The result is a sampling variance greater than would be expected from a random sample from the generation as a whole, with the consequence that 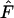 is higher than expected and Ne is underestimated. Humans are much more evenly distributed across age classes, including a substantial contingent of older individuals with low reproductive value that represent a sort of genetic inertia in the population. If their frequencies are weighted equally with younger individuals that have higher reproductive value, the result will be an underestimate of the rate of genetic change and an overestimate of Ne. Sampling only human newborns, however, leads to an underestimate of Ne, as it does in the barnacle, because the progeny are drawn from a relatively small fraction of the population that reproduces in any given time period. Use of
is higher than expected and Ne is underestimated. Humans are much more evenly distributed across age classes, including a substantial contingent of older individuals with low reproductive value that represent a sort of genetic inertia in the population. If their frequencies are weighted equally with younger individuals that have higher reproductive value, the result will be an underestimate of the rate of genetic change and an overestimate of Ne. Sampling only human newborns, however, leads to an underestimate of Ne, as it does in the barnacle, because the progeny are drawn from a relatively small fraction of the population that reproduces in any given time period. Use of 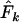 exacerbates the downward bias in
exacerbates the downward bias in 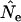 for the barnacle (data not shown), because with diallelic loci
for the barnacle (data not shown), because with diallelic loci 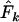 is always larger than
is always larger than 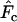 (Waples 1989), resulting in a lower
(Waples 1989), resulting in a lower 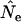 . Use of
. Use of 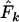 with humans led to quite variable results, with
with humans led to quite variable results, with 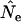 being biased either upward or downward depending on the sampling regime, sample size, and elapsed time between samples (data not shown).
being biased either upward or downward depending on the sampling regime, sample size, and elapsed time between samples (data not shown).
Results for the sparrow are somewhat intermediate but closer to the barnacle than to the human and consistent between the constant size and growth scenarios (Figures 3 and 4; Tables 4 and 7). Presumably this reflects high mortality in the sparrow between ages 1 and 2, which causes a large fraction of the population to have low reproductive value, as in the barnacle. Thus, although the sparrow displays a classical type II survival curve after age 1, the episode of high juvenile mortality has important consequences for estimation of effective population size.
Finally, an interesting result is that, at least for small N1, the magnitude of bias in 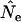 was larger for the human than for the other two species (Table 4, Figure 4). This might mean that species with type I survivorship are particularly prone to bias with use of the standard temporal method. However, the generality of this result requires further evaluation, as other factors such as longer generation time could also be involved.
was larger for the human than for the other two species (Table 4, Figure 4). This might mean that species with type I survivorship are particularly prone to bias with use of the standard temporal method. However, the generality of this result requires further evaluation, as other factors such as longer generation time could also be involved.
Precision:
Our analyses have focused on bias, but precision is also an important consideration for any genetic method of estimating Ne. Although it is beyond the scope of this article to evaluate precision in any comprehensive way, some general observations can be made on the basis of previously published results (Nei and Tajima 1981; Waples 1989; Jorde and Ryman 1995). First, the same proportional increase in sample size, elapsed time between samples, and number of independent alleles used in the estimate of 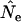 all have approximately the same effect on precision. Our results show that in some cases it is possible to get largely unbiased estimates of Ne from samples taken only a fraction of a generation apart. In general, however, such estimates would have very low precision unless Ne were very small and samples of individuals and loci very large. As the number of alleles used to compute
all have approximately the same effect on precision. Our results show that in some cases it is possible to get largely unbiased estimates of Ne from samples taken only a fraction of a generation apart. In general, however, such estimates would have very low precision unless Ne were very small and samples of individuals and loci very large. As the number of alleles used to compute 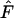 increases, the mean contributions from sampling and drift both stabilize around their expected values and precision increases. With existing technology it is quite feasible to collect data for 10–20 highly polymorphic microsatellite loci in many natural populations, which can produce precise estimates of Ne under certain conditions.
increases, the mean contributions from sampling and drift both stabilize around their expected values and precision increases. With existing technology it is quite feasible to collect data for 10–20 highly polymorphic microsatellite loci in many natural populations, which can produce precise estimates of Ne under certain conditions.
Second, the temporal method can be used most effectively to study populations with small Ne, because the signal from drift is large relative to sampling error. With any genetic method it can be difficult to distinguish a large population from a very large one. For example, under many scenarios with high N1 the point estimates of Ne were infinity, even when 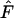 was averaged across 5000 gene loci (Tables 4 and 5). A similar effect was seen in growing populations, which reached a large size before the end of the simulations (Table 7).
was averaged across 5000 gene loci (Tables 4 and 5). A similar effect was seen in growing populations, which reached a large size before the end of the simulations (Table 7).
Finally, results obtained by Jorde and Ryman (1995) suggest that the coefficient of variation of 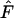 when generations overlap is comparable to that for the discrete-generation model, so parametric confidence intervals for
when generations overlap is comparable to that for the discrete-generation model, so parametric confidence intervals for 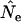 for the standard temporal method (Waples 1989) should also be applicable to iteroparous species.
for the standard temporal method (Waples 1989) should also be applicable to iteroparous species.
Likelihood-based methods for estimating Ne:
In recent years a number of likelihood-based methods (Williamson and Slatkin 1999; Anderson et al. 2000; Wang 2001; Berthier et al. 2002) have been proposed for estimating Ne. In general, results for these methods are roughly comparable to the standard temporal method for moderate allele frequencies but are less biased and more precise when many low-frequency alleles are considered. These methods are all computationally demanding and were not evaluated here. However, all depend on estimates of allele-frequency change in samples taken at two or more points in time. Therefore, they should all be affected in the same general way by the various age-structure biases considered in this article. This conjecture should be tested empirically. The empirical Bayesian method proposed by Tallmon et al. (2004) should be able to deal with overlapping generations by modifying life-history parameters of the modeled species.
Fluctuating population size:
Our results confirmed the basic elements of Felsenstein's (1971) model for populations that change in size at a constant rate, regardless of whether they are increasing or decreasing. However, many populations fluctuate around a “mean” population size. Waples (2005) evaluated performance of the temporal method for semelparous, age-structured species following the Pacific salmon model, but comparable analyses have not been done for iteroparous species. The model recently developed by Engen et al. (2005) for defining Ne in populations with overlapping generations that fluctuate in size could form the basis for such an evaluation in the future.
Conclusions:
Results discussed above lead to the following conclusions regarding application of the temporal method to iteroparous, age-structured species:
The Jorde–Ryman method (Jorde and Ryman 1995) is perhaps the best option if appropriate samples and demographic data are available. Ideally, samples from a number of consecutive cohorts can be analyzed, so that an overall mean
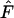 (and associated
(and associated 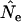 ) can be calculated that accounts for small variations over time in population size and demographic parameters (Jorde and Ryman 1996; Palm et al. 2003). If single-cohort samples cannot be collected directly, it might be possible to reconstruct them by ageing individuals. Jorde and Ryman (1995) suggested that if it is not possible to reconstruct consecutive cohorts, the correction factor C might be modified to reflect a different timing of the samples. Some uncertainty in demographic parameters might not seriously affect the results, depending on the species' life history (Jorde and Ryman 1995). Biases associated with small samples can be minimized by using at least 50 individuals in each cohort.
) can be calculated that accounts for small variations over time in population size and demographic parameters (Jorde and Ryman 1996; Palm et al. 2003). If single-cohort samples cannot be collected directly, it might be possible to reconstruct them by ageing individuals. Jorde and Ryman (1995) suggested that if it is not possible to reconstruct consecutive cohorts, the correction factor C might be modified to reflect a different timing of the samples. Some uncertainty in demographic parameters might not seriously affect the results, depending on the species' life history (Jorde and Ryman 1995). Biases associated with small samples can be minimized by using at least 50 individuals in each cohort.If the available data do not allow use of the Jorde–Ryman model, sampling biases can be minimized (and precision enhanced) by taking samples spaced far apart in time (at least three to five generations, preferably more). This in fact has been the assumption adopted by some authors who have used the temporal method with age-structured species, and our results show that it can be a reasonable one in at least some cases. Recent improvements in the ability to extract DNA from historical material can provide opportunities for retrospective analyses that span large numbers of generations (Hauser et al. 2002; Johnson et al. 2004; Poulsen et al. 2006).
In some cases (e.g., for species with type I survivorship), estimates of Ne using the standard temporal method for closely spaced, random samples might not be severely biased, but precision of such estimates is unlikely to be satisfactory for most applications. Therefore, when generations overlap and samples are closely spaced in time, standard temporal estimates of Ne should be interpreted with extreme caution. This is particularly true because in these cases the choice of which estimator of F to use can have a profound effect on
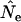 . The minor differences between
. The minor differences between 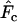 and
and 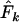 have less impact on
have less impact on 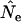 when a longer time elapses between samples and the drift signal is stronger. As a precaution, those employing the temporal method should compute
when a longer time elapses between samples and the drift signal is stronger. As a precaution, those employing the temporal method should compute 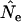 using both
using both 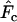 and
and 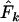 and use considerable caution in interpreting scenarios under which the estimate of effective size depends heavily on the estimator of F.
and use considerable caution in interpreting scenarios under which the estimate of effective size depends heavily on the estimator of F.Large samples of individuals are important, not only for precision but also to help minimize bias arising from various violations of implicit sampling assumptions.
Understanding as much as possible about the species' life history is important to design a sampling strategy to minimize potential biases.
Acknowledgments
We thank Per Erik Jorde, Nils Ryman, and two anonymous reviewers for insightful comments on an earlier draft of the manuscript, and we are grateful to Shuichi Kitada and Toshihide Kitakado for stimulating discussions.
APPENDIX A
Consider a population with constant demographic parameters that produces N1 newborns per time period. The cohort of newborns can be partitioned into x demographic classes of individuals on the basis of age at death. The number of individuals living to age x, reproducing, and then dying before the next time interval is N1lxdx. The average lifetime reproductive success of individuals that die at the end of year x is 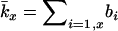 . Population size is constant, so the mean reproductive success of the entire cohort of N1 individuals is
. Population size is constant, so the mean reproductive success of the entire cohort of N1 individuals is 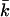 = 2, which is equal to the weighted mean for each age.
= 2, which is equal to the weighted mean for each age.
Now let σx2 be the variance in lifetime reproductive success of individuals that die at the end of age x, and let αx = lxdx be the fraction of the initial cohort that reaches age x and then dies. It can be shown (see online Appendix A in Waples 2006 for details, and see Hill 1972 for a related treatment based on continuous time intervals) that the overall variance in reproductive success for the entire cohort is
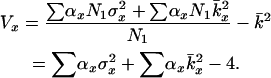 |
(A1) |
If the distribution of reproductive success is Poisson within each age class, so that σx2 = 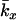 , this simplifies to
, this simplifies to 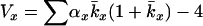 and (from Equation 2)
and (from Equation 2)
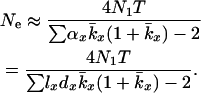 |
(A2) |
APPENDIX B
The objective is to calculate E(Ne(t)), which is the effective size that describes genetic change between times t and t + 1. This can be accomplished by treating Ne as a continuous function of time. Effective size is directly proportional to total reproductive value of the population, which increases at the rate λ per time unit (Felsenstein 1971). Therefore, Ne can be described by the exponential growth model
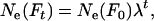 |
(B1) |
where Ne(F0) is the effective size in the generation immediately preceding population growth (506.2 in our human model after adjusting to stable age structure in a growing population). Ne(Ft) describes the “average” rate of change over the generation that starts at time t and ends at time t + T; however, since the population is growing geometrically, the actual effective size must be smaller early in the generation and larger later in the generation. Let us define an instantaneous effective size, denoted by Ne*(t), that describes the actual rate of change in the population at a point t in time. Ne* increases exponentially over the interval t to t + T, and it can be shown that Ne* = Ne(Ft) at the midpoint of the interval. That is, Ne*(t+T/2) = Ne(Ft), which is also equal to the geometric mean of the Ne* values over the period of the generation. It follows that the instantaneous Ne for any time period t can be calculated as
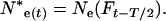 |
(B2) |
That is, instantaneous Ne at time t is just Ne(F) from one-half generation earlier.
We want to find E(Ne(t)), which is analogous to Ne(Ft) except that it describes genetic change over the next time period rather than the next generation. The above logic indicates that E(Ne(t)) is just the instantaneous Ne for the point midway between times t and t + 1:
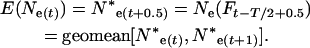 |
(B3) |
References
- Anderson, E. C., E. G. Williamson and E. A. Thompson, 2000. Monte Carlo evaluation of the likelihood for Ne from temporally spaced samples. Genetics 156: 2109–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, M. C., L. R. Mewaldt and R. M. Stewart, 1981. Demography of white-crowned sparrows (Zonotrichia leucophrys nuttalli). Ecology 62: 636–644. [Google Scholar]
- Beaumont, M. A., 2003. Conservation genetics, pp. 751–792 in Handbook of Statistical Genetics, Ed. 2, edited by D. J. Balding, M. Bishop and C. Cannings. Wiley, Chicester, UK.
- Berthier, P., M. A. Beaumont, J.-M. Cornuet and G. Luikart, 2002. Likelihood-based estimation of the effective population size using temporal changes in allele frequencies: a genealogical approach. Genetics 160: 741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero, A., 1994. Developments in the prediction of effective population size. Heredity 73: 657–679. [DOI] [PubMed] [Google Scholar]
- Caswell, H., 2001. Matrix Population Models, Ed. 2. Sinauer, Sunderland, MA.
- Connell, J. H., 1970. A predator-prey system in the marine intertidal region. I. Balanus glandula and several predatory species of Thais. Ecol. Monogr. 40: 49–78. [Google Scholar]
- Crow, J. F., and C. Denniston, 1988. Inbreeding and variance effective population numbers. Evolution 42: 482–495. [DOI] [PubMed] [Google Scholar]
- Engen, S., R. Lande and B.-E. Saether, 2005. Effective size of a fluctuating age-structured population. Genetics 170: 941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 1971. Inbreeding and variance effective numbers in populations with overlapping generations. Genetics 68: 581–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R. A., 1958. The Genetical Theory of Natural Selection, Ed. 2. Dover, New York.
- Frankham, R., 1995. Effective population size/adult population size ratios in wildlife—a review. Genet. Res. 66: 95–107. [DOI] [PubMed] [Google Scholar]
- Hauser, L., G. J. Adcock, P. J. Smith, J. H. B. Ramırez and G. R. Carvalho, 2002. Loss of microsatellite diversity and low effective population size in an overexploited population of New Zealand snapper (Pagrus auratus). Proc. Natl. Acad. Sci. USA 99: 11742–11747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., 1972. Effective size of populations with overlapping generations. Theor. Popul. Biol. 3: 278–289. [DOI] [PubMed] [Google Scholar]
- Hill, W. G., 1979. A note on effective population size with overlapping generations. Genetics 92: 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, E. A., F. W. Schueler and M. S. Blouin, 2004. Effective population sizes and temporal stability of genetic structure in Rana pipiens, the northern leopard frog. Evolution 58: 2536–2545. [DOI] [PubMed] [Google Scholar]
- Johnson, D. L., 1977. Inbreeding in populations with overlapping generations. Genetics 87: 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, J. A., M. R. Bellinger, J. E. Toepfer and P. Dunn, 2004. Temporal changes in allele frequencies and low effective population size in greater prairie-chickens. Mol. Ecol. 13: 2617–2630. [DOI] [PubMed] [Google Scholar]
- Jorde, P. E., and N. Ryman, 1995. Temporal allele frequency change and estimation of effective size in populations with overlapping generations. Genetics 139: 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde, P. E., and N. Ryman, 1996. Demographic genetics of brown trout (Salmo trutta) and estimation of effective population size from temporal change of allele frequency. Genetics 143: 1369–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeuffer, R., D. Pontier, S. Devillard and N. Perrin, 2004. Effective size of two feral domestic cat populations (Felis catus L.): effect of the mating system. Mol. Ecol. 13: 483–490. [DOI] [PubMed] [Google Scholar]
- Krimbas, C. B., and S. Tsakas, 1971. The genetics of Dacus oleae. V. Changes of esterase polymorphism in a natural population following insecticide control—Selection or drift? Evolution 25: 454–460. [DOI] [PubMed] [Google Scholar]
- Leberg, P., 2005. Genetic approaches for estimating the effective size of populations. J. Wildl. Manage. 69: 1385–1399. [Google Scholar]
- Miller, L. M., and A. R. Kapuscinski, 1997. Historical analysis of genetic variation reveals low effective population size in a Northern pike (Esox lucius) population. Genetics 147: 1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., and F. Tajima, 1981. Genetic drift and estimation of effective population size. Genetics 98: 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunney, L., 1993. The influence of mating system and overlapping generations on effective population size. Evolution 47: 1329–1341. [DOI] [PubMed] [Google Scholar]
- Palm, S., L. Laikre, P. E. Jorde and N. Ryman, 2003. Effective population size and temporal genetic change in stream resident brown trout (Salmo trutta, L.). Conserv. Genet. 4: 249–264. [Google Scholar]
- Pollak, E., 1983. A new method for estimating the effective population size from allele frequency changes. Genetics 104: 531–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen, N. A., E. E. Nielsen, M. H. Schierup, V. Loeschcke and P. Grønkjær, 2006. Long-term stability and effective population size in North Sea and Baltic Sea cod (Gadus morhua). Mol. Ecol. 15: 321–331. [DOI] [PubMed] [Google Scholar]
- Scribner, K. T., J. W. Arntzen and T. Burke, 1997. Effective number of breeding adults in Bufo bufo estimated from age-specific variation at minisatellite loci. Mol. Ecol. 6: 701–712. [DOI] [PubMed] [Google Scholar]
- Tajima, F., and M. Nei, 1984. Note on genetic drift and estimation of effective population size. Genetics 106: 569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallmon, D. A., G. Luikart and M. A. Beaumont, 2004. Comparative evaluation of a new effective population size estimator based on approximate Bayesian computation. Genetics 167: 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, T. F., P. Wares and J. R. Gold, 2002. Genetic effective size is three orders of magnitude smaller than adult census size in an abundant, estuarine-dependent marine fish (Sciaenops ocellatus). Genetics 162: 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U. S. Department of Health, Education, and Welfare, 1969. Vital Statistics of the United States, 1967. Volume II: Mortality. U. S. Government Printing Office, Washington, D.C.
- Wang, J., 2001. A pseudo-likelihood method for estimating effective population size from temporally spaced samples. Genet. Res. 78: 243–257. [DOI] [PubMed] [Google Scholar]
- Wang, J., 2005. Estimation of effective population sizes from data on genetic markers. Philos. Trans. R. Soc. Ser. B 360: 1395–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples, R. S., 1989. A generalized approach for estimating effective population size from temporal changes in allele frequency. Genetics 121: 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples, R. S., 1990. Conservation genetics of Pacific salmon. III. Estimating effective population size. J. Hered. 81: 277–289. [Google Scholar]
- Waples, R. S., 2002. Effective size of fluctuating salmon populations. Genetics 161: 782–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples, R. S., 2005. Genetic estimates of contemporary effective population size: To what time periods do the estimates apply? Mol. Ecol. 14: 3335–3352. [DOI] [PubMed] [Google Scholar]
- Waples, R. S., 2006. Seed banks, salmon, and sleeping genes: effective population size in semelparous, age-structured species with fluctuating abundance. Am. Nat. 167: 118–135. [DOI] [PubMed] [Google Scholar]
- Williamson, E. G., and M. Slatkin, 1999. Using maximum likelihood to estimate population size from temporal changes in allele frequencies. Genetics 152: 755–761. [DOI] [PMC free article] [PubMed] [Google Scholar]