

Abstract
Our basic observation is that each genome has a characteristic “signature” defined as the ratios between the observed dinucleotide frequencies and the frequencies expected if neighbors were chosen at random (dinucleotide relative abundances). The remarkable fact is that the signature is relatively constant throughout the genome; i.e., the patterns and levels of dinucleotide relative abundances of every 50-kb segment of the genome are about the same. Comparison of the signatures of different genomes provides a measure of similarity which has the advantage that it looks at all the DNA of an organism and does not depend on the ability to align homologous sequences of specific genes. Genome signature comparisons show that plasmids, both specialized and broad-range, and their hosts have substantially compatible (similar) genome signatures. Mammalian mitochondrial (Mt) genomes are very similar, and animal and fungal Mt are generally moderately similar, but they diverge significantly from plant and protist Mt sets. Moreover, Mt genome signature differences between species parallel the corresponding nuclear genome signature differences, despite large differences between Mt and host nuclear signatures. In signature terms, we find that the archaea are not a coherent clade. For example, Sulfolobus and Halobacterium are extremely divergent. There is no consistent pattern of signature differences among thermophiles. More generally, grouping prokaryotes by environmental criteria (e.g., habitat propensities, osmolarity tolerance, chemical conditions) reveals no correlations in genome signature.
Extensive data support the proposal that each living organism possesses a genomic signature consisting of dinucleotide relative abundance values calculated from genomic sequences (1–3). Explicitly, the genomic signature profile consists of the array {ρ*XY = f*XY/f*Xf*Y}, where f*X denotes the frequency of the mononucleotide X and f*XY the frequency of the dinucleotide XY, both computed from the sequence concatenated with its inverted complement. These dinucleotide relative abundance values {ρ*XY} minus 1 (also termed dinucleotide biases) effectively assess differences between the observed dinucleotide frequencies and those expected from random associations of the component mononucleotide frequencies. From data simulations and statistical theory, the estimates ρ*XY ≤ 0.78 or ρ*XY ≥ 1.23 convey significant underrepresentation or overrepresentation, respectively, for 50-kb random DNA contigs (1–3). We present substantial data showing that the genome signatures of bacterial plasmids are pervasively similar to those of their natural hosts. By contrast, the signatures of animal mitochondrial DNA are not close to those of their hosts but are generally concordant with those of other animal mitochondrial (Mt) DNA.
Justifications for Using Genome Signature.
Biochemical experiments in the 1960s and 1970s measuring nearest-neighbor frequencies (4, 5) established that the set of dinucleotide relative abundance values {ρ*XY} is a remarkably stable property of the DNA of an organism. From this perspective, the set of dinucleotide relative abundance values constitutes a genomic signature that is diagnostic and can discriminate sequences from different organisms (3, 6). What causes the uniformity of signature throughout the genome? It pervades both noncoding and coding DNA (7), and hence cannot be explained by preferential codon usage. A reasonable explanation postulates differences in the replication and repair machinery of different species, which either preferentially generate or preferentially select specific dinucleotides in the DNA. These effects might operate through local DNA structures (base step conformational tendencies), context-dependent mutation rates, methylation, and/or other DNA modifications (1–3, 6).
A measure of genomic signature difference between two sequences f and g (from different organisms or from different regions of the same genome) is the average absolute dinucleotide relative abundance difference calculated as
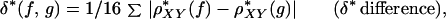 |
where the sum extends over all dinucleotides. Levels of δ* differences of 50-kb contigs for some reference examples are described in the Introduction of the accompanying paper (8).
Genome Signature Comparisons.
Fig. 1 displays the genome signature profiles for all currently available extensive genome sequence sets. These include 22 eubacterial genome ensembles (mostly complete genomes), 5 archaeal chromosomes, the Saccharomyces cerevisiae and Caenorhabditis elegans complete genomes, and extensive nonredundant sequence collections from 5 other eukaryotic species. For each genome, the dinucleotide biases {ρ*XY} are substantially invariant (9).
Figure 1.

Genome signature (dinucleotide relative abundances) of complete genomes and large DNA sequence samples (>500 kb).
The dinucleotide TA is broadly underrepresented or low normal in prokaryotic sequences about the level 0.50 ≤ ρ*TA ≤ 0.82 (exceptions include Rickettsia prowazekii, Clostridium acetobutylicum, and the archaea P. aerophilum, P. horikoshii, and Sulfolobus sp. (full names appear in the legend to Figs. 1 and 2). TA underrepresentation is also pervasive in eukaryotic chromosomes (exception P. falciparum) but not in eukaryotic small viral genomes or in animal mitochondrial genomes (10, 11).
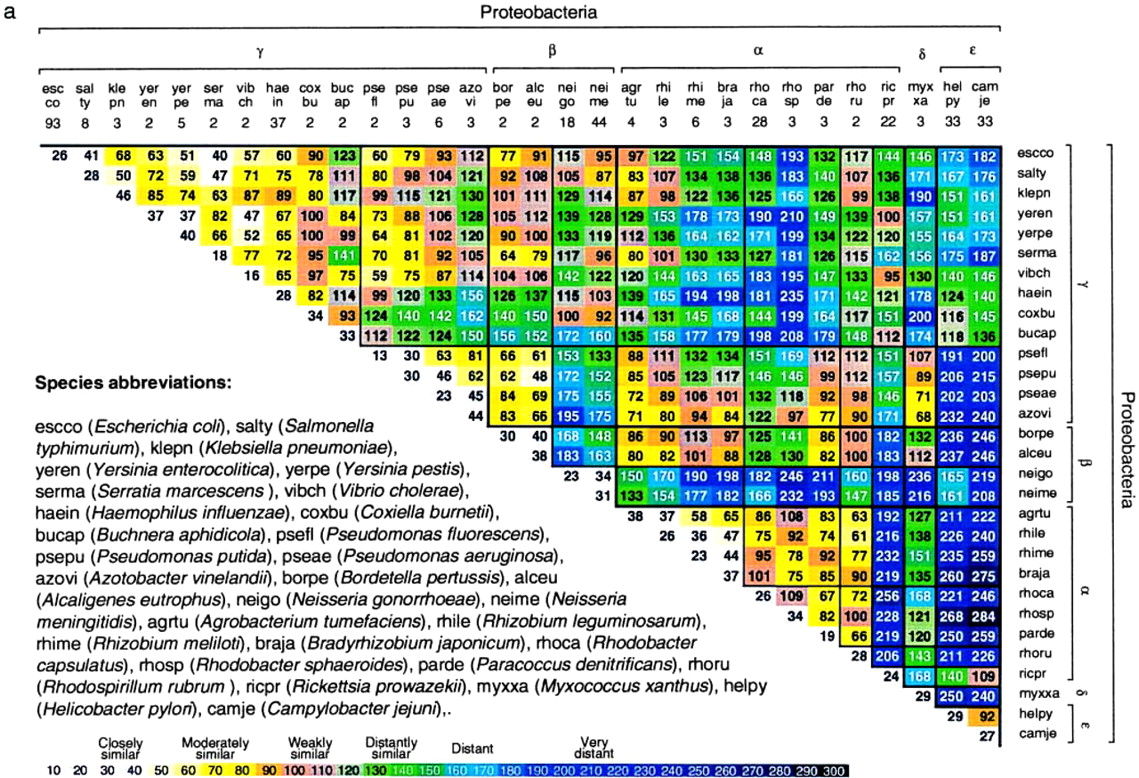
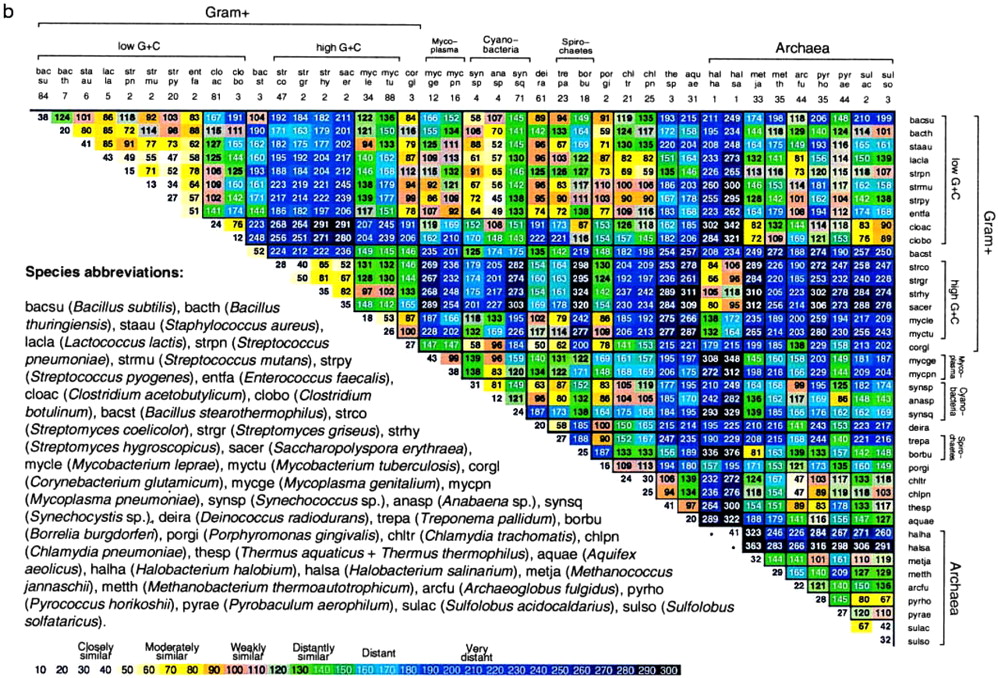
Figure 2.

Average δ* differences within (diagonal entries) and between (nondiagonal entries) prokaryotic DNA sequence samples based on pairwise comparisons of all disjoint nonredundant 50-kb samples available. See also Fig. 5 for 70 prokaryotic species [published as supplemental data on the PNAS web site (www.pnas.org)].
Among prokaryotes, CG is suppressed (underrepresented) in M. genitalium (but not in M. pneumoniae), in R. prowazekii, in B. burgdorferi, in C. jejuni, in the low-G+C Gram-positive sequences of Streptococcus and Clostridium, and in several thermophiles, including M. jannaschii, M. thermoautotrophicum, Sulfolobus spp., but not in P. aerophilum or P. horikoshii. At the other extreme, CG is overrepresented in B. stearothermophilus, in halobacteria, and also in several β- and α-proteobacterial genomes (e.g., Neisseria spp. and Rhizobium spp.). Among eukaryotes, CG shows potent suppression in vertebrates (even in deuterostomes). Overall, ρ*CG values in vertebrates range from 0.23 to 0.40, whereas they are in the normal range for insects, worms, and most fungi. It has been shown (12) that unmethylated CG dinucleotides of normal frequency in most enteroproteobacteria can induce an immune response in mammalian genomes, where CG is very low. Is this a concomitant of genomic signature biases? The reverse dinucleotide, GC, is predominantly overrepresented in many β- and γ-proteobacterial sequences and in several low-G+C Gram-positive bacterial genomes (e.g., B. subtilis and C. acetobutylicum; Fig. 1). In eukaryotes, moderate overrepresentation of GC occurs in Drosophila species but apparently not in other metazoa.
TT/AA is overrepresented in several proteobacteria, in Mycoplasmas, in Synechocystis sp., in Deinococcus radiodurans, in A. aeolicus among prokaryotes, and in insects and worms among eukaryotes. There are no underrepresentations of TT/AA. Overrepresentations of CC/GG include Synechocystis, B. burgdorferi, A. aeolicus, M. jannaschii, M. thermoautotrophicum, and P. horikoshii. There are no underrepresentations of CC/GG.
δ* Differences Among Prokaryotic Sequences.
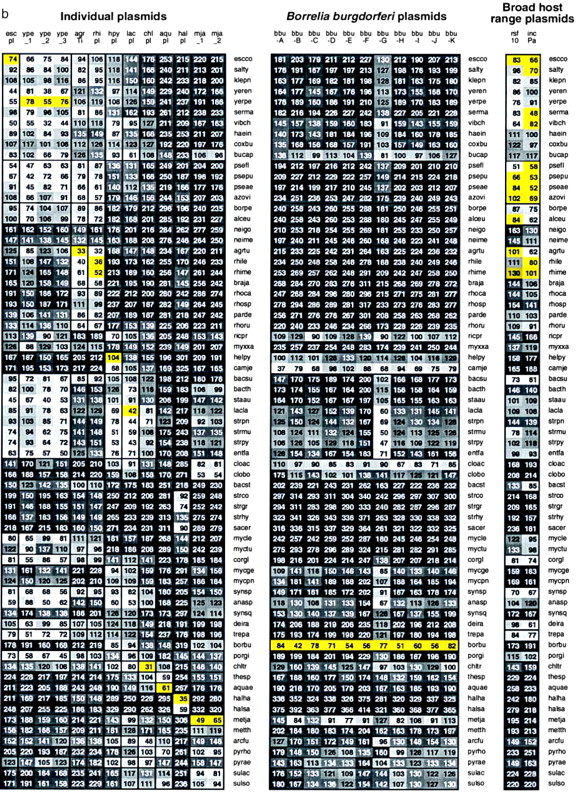
Fig. 2 reports average δ* differences based on multiple disjoint 50-kb contigs among prokaryotic genomic sets, each at least 100 kb in aggregate length and most genome sequence collections exceeding 800 kb. The δ* differences for all pairs of contigs for every pair of genomes are highly stable with limited variation (cf. refs. 1–6).
(i) Within-species δ* differences (diagonal elements of Fig. 2) range from 12 to 52 (all δ* differences are multiplied by 1000), and average between species δ* differences range from 34 to 340.
(ii) The rickettsial sequences are grouped with α-proteobacteria, apparently on the basis of rRNA gene comparisons. Is this consistent? The classical α-proteobacterial types are divided into two major subgroups: 𝒜1, including Rhizobium spp., that function importantly in nitrogen fixation, and 𝒜2, including Rhodobacter spp. and P. denitrificans, found predominantly in soil and marine habitats and doing anoxygenic photosynthesis. A tentative third group, 𝒜3, includes the Rickettsia and Ehrlichia clades (obligate intracellular parasites). Genome signature comparisons indicate drastic discrepancies between the combined groups {𝒜1, 𝒜2} and the group 𝒜3. Moreover, the 𝒜1 and 𝒜2 genomes are pervasively of high G+C content (≥60%), whereas 𝒜3 genomes are of low G+C content (<32%). The δ* differences between Rickettsia and classical α-proteobacterial sequences are generally >200, very distant (see Fig. 2).
(iii) Sulfolobus spp. δ* differences from other prokaryotes. Fig. 2 includes seven archaeal sequence collections consisting of one halobacterial genus (HALSP) (see legends to Figs. 1 and 2 for full names, and we use the Swiss-Prot abbreviations), two methanogens (METJA, METTH), one “derived” methanogen (ARCFU), and three archaeal thermophiles (PYRHO, PYRAE, SULSP). Thermophiles (exception, PYRHO) tend to be relatively closer to vertebrate eukaryotes than to eubacterial sequences, with δ* differences in the range 100–150 (3, 9), whereas HALSP have generally δ* > 200. Thus, the highly diverse δ* differences indicate that a consistent description for the archaeal sequences is problematical. We summarize the genomic signature contrasts for Sulfolobus in the following array.
δ* (Sulfolobus, Clostridium) ≈ 85, moderately similar; δ* (Sulfolobus, Rickettsia, and Buchnera) ≈ 125–130, distantly similar; δ* (Sulfolobus, other thermophilic archaea) ≈ 71–114, weakly to distantly similar; δ* (Sulfolobus, purple proteobacteria and high G+C Gram-positive) ≈ 190–270, very distant; δ* (Sulfolobus, cyanobacteria) ≈ 145–177, distant.
(iv) Halobacterial genome sequences are outliers. Intriguingly, with respect to genome signature comparisons, the closest to Halobacterium spp. are the Streptomyces sequences, weakly similar. The δ* differences of Halobacterium spp. from other prokaryotes mostly exceed the extreme level of 250.
Genome Compatibility Between Plasmids and Host.
Plasmids are genetic mobile elements among bacterial cells, generally laterally transferred by conjugation (on occasion by transduction or transformation). Plasmids carry restriction systems, antibiotic resistance genes, heavy-metal cofactors, Nif (nitrogen fixation) genes, and other contingency functions. Replication of plasmids is largely governed by host machinery. How the genome signature of a plasmid sequence compares with that of its host sequence is assessed for available prokaryotic genomes with completely sequenced plasmids. Fig. 3 reports average δ* differences for each plasmid compared with representatives of 52 prokaryotic genomic collections. Importantly, the δ* differences between each plasmid and its natural host rank among the closest. Thus, the examples of Fig. 3 are consistent with the proposition that plasmids are viable in bacterial hosts only when their genome signatures are sufficiently compatible (moderately similar, 55 ≤ δ* ≤ 85, or at least weakly similar, 90 < δ* < 115) with the host genome signature or they can rapidly be made similar.
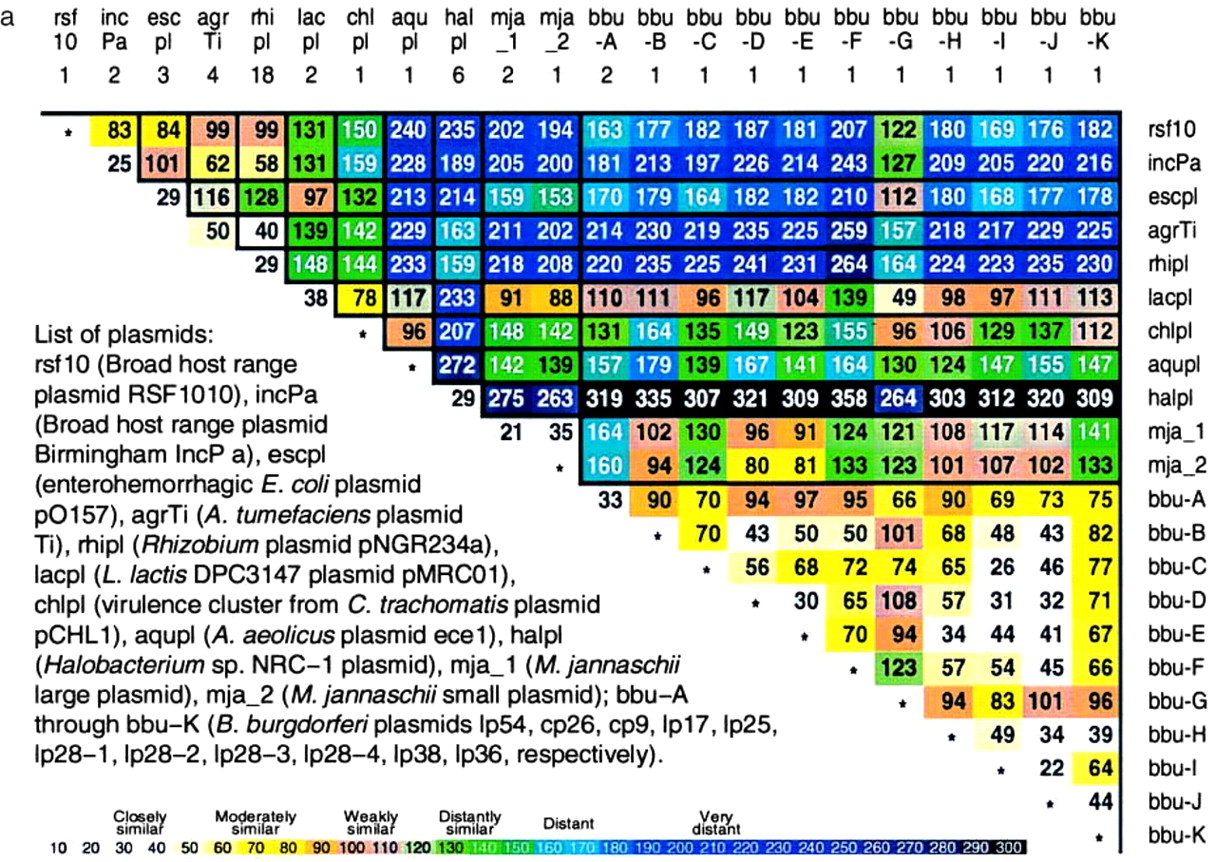
Figure 3.

Plasmids are abbreviated as follows: rsf10 (broad-host-range plasmid RSF1010), incPa (Broad host range plasmid Birmingham IncP α), escpl (enterohemorrhagic E. coli plasmid pO157), ype_1 (Yersinia pestis plasmid pMT-1), ype_2 (Y. pestis plasmid pCD1), ype_3 (Y. pestis plasmid pPCP1), agrTi (A. tumefaciens plasmid Ti), rhipl (Rhizobium plasmid pNGR234a), hpypl (H. pylori plasmid pHPM186); lacpl (L. lactis DPC3147 plasmid pMRC01), chlpl (virulence cluster from C. trachomatis plasmid pCHL1), aqupl (A. aeolicus plasmid ece1), halpl (Halobacterium sp. NRC-1 plasmid), mja_1 (M. jannaschii large plasmid), mja_2 (M. jannaschii small plasmid). See Fig. 2 for prokaryotic species abbreviations. Additional prokaryotes not included in Fig. 2 are salty (Salmonella typhimurium), yeren (Yersinia enterocolitica), yerpe (Yersinia pestis), serma (Serratia marcescens), vibch (Vibrio cholerae), and rhile (Rhizobium leguminosarum). Yellow background indicates δ* differences of a plasmid from its confirmed host. See also Fig. 6, which is published as supplemental data on the PNAS web site (www.pnas.org).
B. burgdorferi. The complete genome extends 0.911 Mb and contains 17 plasmids, of which 11 have been sequenced from 10 kb to 54 kb in size labeled A to K. The average δ* difference within the B. burgdorferi (BORBU) genome is 25 with range 3–66. The δ* differences among all plasmids and compared with a complete set of 50-kb contigs of the host genome range from 42 to 84, clearly moderately similar (data not shown). Mutual plasmid sequence δ* differences show moderate to weak similarity.
Specific plasmids. M. jannaschii, mja. This archaeal genome contains two plasmids of 58 kb (called large) and 16.5 kb (small). Again, we find that the two plasmids are mutually close (δ* = 35) and each moderately similar to the host genome. Rhizobium spp., rhi. We have available about 150 kb of nonredundant aggregate sequences from R. leguminosarum and about 250 kb of sequence from R. meliloti. There is a mammoth plasmid of about 550 kb. Partitioning these sequences into 50-kb contigs yields the average δ* differences given in Fig. 3 of close similarity. Halobacterium spp., hal. An approximately 200-kb plasmid (pNRC100) from H. halobium was sequenced. About 100 kb of aggregate sequences from H. halobium and H. salinarium genomes are also accessible. Fig. 3 reports δ* differences entailing close to moderate similarity. A. tumefaciens, agr-Ti. Compared to nonplasmid sequences, the classic crown gall tumor plasmid Ti (133 kb available) produces an average δ* difference of 33 (closely similar). E. coli, esc. The plasmid O157 of E. coli, of length 93 kb, gives δ* difference of 74 (moderate similarity) compared with E. coli. L. lactis, lac. This bacterium has available about 250 kb of aggregate genomic sequence and a complete plasmid (MRC01) of length about 60 kb. The δ* difference of MRC01 to the average 50-kb genome contig is 42 (closely similar). A. aeolicus, aqu. The complete genome sequence (1.6 Mb) is available. A plasmid contig labeled ece1 has also been sequenced. The δ* difference is 61 (moderately similar). H pylori, hpy. The single plasmid (13 kb) of strain 26695 has δ* difference 104 (weakly similar) to the genome. C. trachomatis, chl. The plasmid (7.5 kb) compared to the complete genome has a close signature difference (δ* = 31).
Broad-host-range plasmids. Two plasmids that stably replicate in many hosts are RSF1010 and RP4 (13, 14); see legend to Fig. 3. We list next those host prokaryotic species with at least 80 kb of available genome sequence which accept stably these plasmids. RSF1010: Escherichia coli, Pseudomonas aeruginosa, Pseudomonas putida, Azotobacter vinelandii, Rhizobium meliloti, Agrobacterium tumefaciens, and Alcaligenes eutrophus. RP4: Serratia marcescens, Azotobacter vinelandii, Pseudomonas aeruginosa, Pseudomonas fluorescens, Escherichia coli, Pseudomonas putida, Rhodospirillum rubrum, Shigella boydii, Salmonella typhimurium, Vibrio cholerae, and Rhizobium meliloti.
The broad-range plasmids are generally moderately or weakly similar to the other plasmids from proteobacterial genomes. The plasmid from L. lactis is distantly similar to the broad-range plasmids. The plasmids from B. burgdorferi, Halobacterium spp., and M. jannaschii are very distant from all proteobacterial plasmids. The δ* differences among B. burgdorferi plasmids are mutually closely or moderately similar, except plasmid G, which is only weakly similar. Plasmid G is close (δ* = 49) to the L. lactis plasmid, perhaps implying that a low G+C Gram-positive bacterium may be the source of plasmid G. The two broad-range plasmids are mutually moderately similar (δ* = 83). This similarity might mean either that relatively close genome signatures promote plasmid establishment or that the plasmids have acquired their hosts’ signatures during long-term residence. Experiments on conjugation can address issues such as specificity vs. wide host range and relevance of size and signature for plasmid compatibility. We interpret the similarities in signature between plasmids and their bacterial hosts as implying that they share much replication and repair machinery, perhaps because the prokaryotic cell is not compartmentalized to the degree that the eukaryotic cell is.
Genomic Signature δ* Differences Among Mitochondrial Mt Genomes.
Fig. 4 details δ* differences among a broad spectrum of Mt genomes. A succinct summary is given in Table 1.
Figure 4.

Average δ* differences among complete Mt genomes based on a single sequence sample for genomes of <60 kb and multiple samples of about 50 kb for larger genomes.
Table 1.
Summary of δ* differences of Mt genomes
| Comparison | δ* | Description |
|---|---|---|
| Among mammalian Mt | <40 | Very close |
| Among all vertebrates including birds, frogs, fish | <67 | Mainly close |
| Vertebrates vs. nonvertebrate deuterostomes | 55–120 | Moderate to weak similarity |
| Deuterostomes vs. protostomes | Mostly 34–152 | Moderate to distant similarity |
| Fungi vs. animals | Mostly 43–128 | Moderate to weak similarity |
| Plants vs. (animals and fungi) | 84–179 | Weak similarity to distant |
| C. reinhardtii vs. animals | Mostly 141–212 | Distant to very distant |
Mammalian Mt genome signatures appear almost as random samples of each other. Why are the genome signatures among vertebrate Mt sequences significantly more similar than those among the corresponding host nuclear genomic sequences (15)? δ* differences among organisms putatively reflect variations in replication and/or repair (1–3, 6). The replication machinery for animal Mt DNA apparently varies less than that for host DNA, perhaps because Mt replication is less affected by changes in external environment or developmental programs than is host DNA replication. δ* differences of Mt from protostomes (e.g., insect, mollusk) versus deuterostomes show δ* differences mostly in the range 90–115, weakly similar. The worm (LUMTE and C. elegans) genomes compared with deuterostome Mt are closely to weakly similar, δ* ≈ 41–101. The fungal Mt sets (excluding S. cerevisiae) compared with deuterostome Mt also yield δ* differences in the range 43–114. The S. cerevisiae mtDNA (≈78 kb) composition is an extreme anomaly attested to by the inordinate δ* differences from all other Mt or nuclear genomes, contributed to by about 100 G+C-rich clusters, each about 50 to 100 bp long, separated by A+T-rich spacers and numerous transposable elements.
The two plant Mt genomes (ARATH, MARPO, see Fig. 4), of about 150-kb length, compared with animal Mt are weakly similar to distant. The green alga CHLRE Mt is very distant from most other Mt sequences (δ* > 180). The other two algal Mt sequences (PROWI, CHOCR) are generally weakly similar to nonprotist Mt sequences. The protist Mt divide into two subgroups, P1 = (RECAM, ACACA) and P2 = (PARAU, TRYBR, LEITA). The RECAM Mt genome signature compared with deuterostome Mt is weakly to distantly similar. ACACA is a bit farther (δ* ≈ 94–144). The protist group P2 are mostly distant to very distant from animal Mt sequences. The plant A. thaliana Mt sequence is distant from mammalian Mt sequences, possibly suggesting a polyphyletic ancestry separating metazoan, plant, and protist Mt.
Animal Mt sequences show significant underrepresentations of CG dinucleotides, ρ*CG ≈ 0.40–0.60 (10), about to the same extent as occurs in vertebrate nuclear genomic sequences. Virtually all animal Mt maintain normal representations of TA dinucleotides, whereas the corresponding nuclear DNAs predominantly have TA in low relative abundance, suggesting that mtDNA may be thermodynamically less stable than nuclear DNA because the dinucleotide TA has the lowest stacking energies compared with all other base steps. The fungal Mt of S. pombe has ρ*CG ≈ 0.54, typical of animal Mt. However, the Podospora anserina fungal Mt genome has ρ*CG in the normal range. The Mt genome of A. thaliana has ρ*CG = 0.73, significantly low. The single persistently high ρ* value occurs for ρ*CC/GG ≥ 1.30 in animal and fungal Mt sequences. δ* differences between species parallel the corresponding nuclear δ* differences, despite large differences between Mt and corresponding host nuclear signatures (15).
Discussion Prokaryotic molecular taxonomy heretofore has been derived predominantly from sequence comparisons among rRNA genes.
There are many uncertainties and controversies regarding divisions among prokaryotes (for recent reviews, see refs. 16 and 17). Protein sequence comparisons and associated phylogenetic tree constructions are even more conflicting relative to evolutionary relationships (18, 19). Conventional methods of phylogenetic reconstruction from sequence information employ only similarity or dissimilarity assessments of aligned homologous genes or regions. Some difficulties intrinsic to this approach compared to the use of genome signature include the following: (i) Alignments of distantly related long sequences (e.g., complete genomes) are generally not feasible for various reasons, including chromosomal rearrangements, whereas signature comparisons do not depend on alignments. (ii) Different phylogenetic reconstructions (trees) may result for the same set of organisms based on analysis of different protein, gene, or noncoding sequences. Attempts to overcome these conflicts by “averaging” over many proteins are problematic because of biases in species sampling, effects of lateral transfer, complications of gene duplications, and inadequacies and artifacts of phylogenetic methods. As the signature has remarkably low variance throughout the genome, a tree based on δ* differences is independent of which genome segments of 50 kb (or longer) is used in its construction. (iii) Chimeric origins and lateral transfer between distantly related organisms complicate alignment-based phylogenies. Signature comparisons are unaffected by these factors. On the other hand, alignment-based comparisons may provide information on individual gene origins that the signature method does not. (iv) Tree construction derived from aligned sequences cannot be applied to organisms for which similar gene sequences are largely unavailable (e.g., for bacteriophages, diverse eukaryotic viruses). (v) That “lateral transfer” is pervasive among prokaryotic genomes is now widely appreciated. Vectors for lateral transfer include exogenous transposons, hitchhiking on plasmids and/or phages, movement via episomes, and cell fusions.
What possible reasons and mechanisms can account for the qualitative parallelism between the evolutionary development of host nuclear genomes and the development of Mt organelle genomes despite the pronounced difference between the Mt and host nuclear genome signatures? The Mt and nuclear genomes for animal and fungal organisms use independent DNA polymerase machinery (e.g., γ vs. α, ɛ, and δ subunits in mammals). Also, the methods of replication and the nature of the replication origins are fundamentally different. Explicitly, the animal and fungal Mt transcription-primed replication machinery is distinctive in that most of the heavy strand is synthesized first and the light strand subsequently, whereas the nuclear genomes are replicated bidirectionally from multiple origins. There appears to be no DNA excision repair mechanism to deal with cyclobutane dimers in the Mt, and no repair of bulky lesions (20). Mt DNAs in animals and fungi show elevated levels of single- and double-strand breaks, mismatches, and generally corrupted base pairings, probably due to a paucity of abasic site correction facilities and mismatch repair capacity in Mt genomes (21). Moreover, repair may be less urgent for Mt activity because each cell has many mitochondria (hundreds or thousands) and a modicum of impaired organelles may not significantly curtail energy production. We, nevertheless, propose that Mt genomes retain signatures close to those of their repair-competent prokaryotic ancestor.
The contrast between plasmids (which track host genomic signatures) and mitochondria (which do not) is sharp. The similar signatures of plasmids and hosts may have two bases: (i) As we have postulated for genome fusions (8), perhaps a plasmid whose signature is too different from that of the host will not be accepted by it. This would prescribe a maximum possible signature deviation. The largest δ* difference observed in our survey is weakly similar (or about the distance from human to sea urchin). (ii) During the plasmid’s residence in its current host, the same pressures that homogenize the signature throughout the chromosome will also drive the plasmid’s signature towards that of the host. Such amelioration has been postulated for the G+C content of laterally transferred DNA (22). We suspect that the signature should ameliorate even more rapidly, for both plasmids and laterally transferred chromosomal segments. Whereas most successful gene transfer between lineages is very likely intraspecific, an appreciable amount of transfer among distantly related bacteria seems to have accumulated over time (22, 23). Despite such transfer, the signatures currently observed are almost invariant throughout the genome. Some means based on codon usage biases for ascertainments of laterally transferred genes in bacterial organisms are set forth in ref. 23.
Supplementary Material
Acknowledgments
We are happy to acknowledge valuable discussions and comments on the manuscript by Drs. B. Edwin Blaisdell, Dale Kaiser, and David Relman. S.K. is supported in part by National Institutes of Health Grants 5R01GM10452-34 and 5R01HG00335-11 and National Science Foundation Grant DMS9704552.
ABBREVIATION
- Mt
mitochondria(l)
References
- 1.Karlin S, Burge C. Trends Genet. 1995;11:283–290. doi: 10.1016/s0168-9525(00)89076-9. [DOI] [PubMed] [Google Scholar]
- 2.Blaisdell B E, Campbell A M, Karlin S. Proc Natl Acad Sci USA. 1996;93:5854–5859. doi: 10.1073/pnas.93.12.5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karlin S, Mrázek J, Campbell A M. J Bacteriol. 1997;179:3899–3913. doi: 10.1128/jb.179.12.3899-3913.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Josse J, Kaiser A D, Kornberg A. J Biol Chem. 1961;263:864–875. [PubMed] [Google Scholar]
- 5.Russell G J, Walker P M, Elton R A, Subak-Sharpe J H. J Mol Biol. 1976;108:1–23. doi: 10.1016/s0022-2836(76)80090-3. [DOI] [PubMed] [Google Scholar]
- 6.Karlin S. Curr Opin Microbiol. 1998;1:598–610. doi: 10.1016/s1369-5274(98)80095-7. [DOI] [PubMed] [Google Scholar]
- 7.Karlin S, Mrázek J. J Mol Biol. 1996;262:459–472. doi: 10.1006/jmbi.1996.0528. [DOI] [PubMed] [Google Scholar]
- 8.Karlin S, Brocchieri L, Mrázek J, Campbell A M, Spormann A M. Proc Natl Acad Sci USA. 1999;96:9190–9195. doi: 10.1073/pnas.96.16.9190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karlin S, Campbell A M, Mrázek J. Annu Rev Genet. 1998;32:185–225. doi: 10.1146/annurev.genet.32.1.185. [DOI] [PubMed] [Google Scholar]
- 10.Cardon L R, Burge C, Clayton D A, Karlin S. Proc Natl Acad Sci USA. 1994;91:3799–3803. doi: 10.1073/pnas.91.9.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karlin S, Doerfler W, Cardon L R. J Virol. 1994;68:2889–2897. doi: 10.1128/jvi.68.5.2889-2897.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krieg A M, Yi A-K, Schorr J, Davis H L. Trends Microbiol. 1998;6:23–27. doi: 10.1016/S0966-842X(97)01145-1. [DOI] [PubMed] [Google Scholar]
- 13.Olsen R H, Shipley P. J Bacteriol. 1973;113:772–780. doi: 10.1128/jb.113.2.772-780.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bagdasarian M, Lurz R, Ruckert B, Franklin F C, Bagdasarian M M, Frey J, Timmis K N. Gene. 1981;16:237–247. doi: 10.1016/0378-1119(81)90080-9. [DOI] [PubMed] [Google Scholar]
- 15.Karlin S, Mrázek J. Proc Natl Acad Sci USA. 1997;94:10227–10232. doi: 10.1073/pnas.94.19.10227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown J R, Doolittle W R. Microbiol Rev. 1997;61:456–502. doi: 10.1128/mmbr.61.4.456-502.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta R S. Microbiol Mol Biol Rev. 1998;62:1435–1491. doi: 10.1128/mmbr.62.4.1435-1491.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta R S, Golding G B. Trends Biochem Sci. 1996;21:166–171. [PubMed] [Google Scholar]
- 19.Budin K, Philippe H. Mol Biol Evol. 1998;15:943–956. doi: 10.1093/oxfordjournals.molbev.a026010. [DOI] [PubMed] [Google Scholar]
- 20.Shadel G S, Clayton D A. Annu Rev Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- 21.Yakes F M, Van Houten B. Proc Natl Acad Sci USA. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawrence J G, Ochman H. J Mol Evol. 1997;44:383–397. doi: 10.1007/pl00006158. [DOI] [PubMed] [Google Scholar]
- 23.Karlin S, Mrázek J, Campbell A M. Mol Microbiol. 1998;29:1341–1355. doi: 10.1046/j.1365-2958.1998.01008.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}