

Abstract
We provide data and analysis to support the hypothesis that the ancestor of animal mitochondria (Mt) and many primitive amitochondrial (a-Mt) eukaryotes was a fusion microbe composed of a Clostridium-like eubacterium and a Sulfolobus-like archaebacterium. The analysis is based on several observations: (i) The genome signatures (dinucleotide relative abundance values) of Clostridium and Sulfolobus are compatible (sufficiently similar) and each has significantly more similarity in genome signatures with animal Mt sequences than do all other available prokaryotes. That stable fusions may require compatibility in genome signatures is suggested by the compatibility of plasmids and hosts. (ii) The expanded energy metabolism of the fusion organism was strongly selective for cementing such a fusion. (iii) The molecular apparatus of endospore formation in Clostridium serves as raw material for the development of the nucleus and cytoplasm of the eukaryotic cell.
Many perspectives have been proffered on domains of life, the origin and early evolution of eukaryotes, and the genesis of organelles. However, 16S rRNA genes and protein sequence comparisons give mixed and conflicting results (1–3) attributed in part to artifacts and inadequacies of phylogenetic methods, sensitivity to unequal evolutionary rates, biases in species sampling, unrecognized paralogy, and widespread reductions and expansions of genome content. That genomes of many prokaryotes and primitive eukaryotes are “heterogeneous unions” in which lateral transfer and/or close associations have been at work is increasingly accepted (3, 4). It is surmised that primitive prokaryotic cells did not possess rigid walls and therefore could easily fuse and exchange and modify genetic material. Several archaeal–bacterial partnerships about the time of the origin of eukaryotes have been proposed. These include the following: fusion of an archaeon and an unspecified bacterium (5), a union of an eocyte and a Gram-negative bacterium (6), the Hydrogen Hypothesis based on symbiosis of a “methanogen” and an “α-proteobacterium” (7), and the Syntrophic hypothesis, proffering an archaeon and δ-proteobacterial consortium plus a secondary symbiont of an α-proteobacterium (4). It has frequently been advocated that mitochondria (Mt) are descended from a bacterial endosymbiont that colonized a primitive amitochondrial (a-Mt) eukaryote cell. Various α-proteobacteria have been favored candidates. However, there are several current arguments that mitochondria have an anaerobic origin (3, 4, 7).
Genome Signature.
Every living organism has its characteristic “genome signature” related to the frequencies with which two particular types of nucleotides neighbor each other in the DNA chain (8–11). Explicitly, the genome signatures are not the raw frequencies themselves but rather dinucleotide relative abundances defined as the ratios between the observed frequencies and the frequencies expected if neighbors were chosen at random. This is the array {ρ*XY = f*XY/f*Xf*Y} extended over all dinucleotides, where f*X is the frequency of X and f*XY is the dinucleotide frequency of XY, both computed from the sequence concatenated with its inverted complementary sequence. The genome signatures {ρ*XY} of DNA contigs ≥50 kb from different regions of the same species are substantially congruent (8–11). Moreover, closely related species have more similar genome signatures than distantly related species. We reiterate the hypothesis (8–11) that species-specific properties of DNA stacking energies, DNA methylation and other modifications, and the replication and repair machinery that processes the whole genome contribute decisively to maintaining the genome signature of an organism and that differences in this machinery create meaningful genomic differences between organisms. For example, among phages, the dinucleotide relative abundance values strongly correlate with the extent to which host machinery is used and with the nature of the host. Analysis of phage genomes supports a picture in which temperate phages dependent on host replication machinery converge toward the DNA signatures of the host, whereas autologously replicating phages (T4 and T7) maintain their own signatures (10).
A measure of genomic signature difference between two sequences 𝒮 and 𝒯 (from different organisms or from different regions of the same genome) is the average absolute dinucleotide relative abundance difference calculated as δ*(𝒮, 𝒯) = 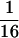 ∑|ρ*XY(𝒮) − ρ*XY(𝒯)| where the sum extends over all dinucleotides (11). We describe levels of δ* differences for 50-kb contig samples (all values multiplied by 1,000): closely similar (δ* < 50), human vs. cow, Escherichia coli vs. Salmonella typhimurium; moderately similar (55 < δ* < 85), human vs. chicken, E. coli vs. Haemophilus influenzae; weakly similar (90 < δ* < 115), human vs. sea urchin, Sulfolobus sp. vs. Methanococcus jannaschii; distantly similar (120 < δ* < 145), human vs. Saccharomyces cerevisiae, human vs. Sulfolobus; distant (150 < δ* < 185), human vs. Drosophila, E. coli vs. Helicobacter pylori; and very distant (δ* > 190), human vs. E. coli, Halobacterium sp. vs. Sulfolobus sp.
∑|ρ*XY(𝒮) − ρ*XY(𝒯)| where the sum extends over all dinucleotides (11). We describe levels of δ* differences for 50-kb contig samples (all values multiplied by 1,000): closely similar (δ* < 50), human vs. cow, Escherichia coli vs. Salmonella typhimurium; moderately similar (55 < δ* < 85), human vs. chicken, E. coli vs. Haemophilus influenzae; weakly similar (90 < δ* < 115), human vs. sea urchin, Sulfolobus sp. vs. Methanococcus jannaschii; distantly similar (120 < δ* < 145), human vs. Saccharomyces cerevisiae, human vs. Sulfolobus; distant (150 < δ* < 185), human vs. Drosophila, E. coli vs. Helicobacter pylori; and very distant (δ* > 190), human vs. E. coli, Halobacterium sp. vs. Sulfolobus sp.
Examination of plasmids in prokaryotic hosts reveals generally close or moderate similarity of the plasmid genome signature to the host genome signature, suggesting that an organism may accept a plasmid of similar genome signature or may rapidly convert a weakly similar plasmid genome to a host-compatible signature and render the union stable. We have also verified that broad-host-range plasmids almost always have a genome signature moderately similar to the genome signature of a potential host (11). The foregoing observations lead us to the following postulate: Genome signature similarity is essential for compatibility and coexistence between two genomes.
Primitive organisms probably engaged in much reduction, acquisition, and lateral transfer of DNA, producing chimeric genomes. Current studies of molecular evolution emphasize lateral gene transfer as a major evolutionary mechanism (4, 12). For some genomes, it is estimated that 10–20% of genes have been laterally transferred within the last 100 million years (13). Established vehicles for lateral transfer include transposition, conjugative plasmids, phage hitchhiking, and transformation. One would expect that genes advantageous to the recipient organism bear potential for successful interspecies gene flow. However, successful incorporations are rare. The Hydrogen Hypothesis of Martin and Müller (7) “requires the genetic systems of the eubacterium and archaebacterium to be sufficiently compatible as to allow expression of the transferred gene(s).” We interpret this compatibility to require at least moderate similarity for the genomic signatures of the organisms, in agreement with the comparative analysis of the genome signatures of plasmids and their hosts (11).
The Clostridium–Sulfolobus Nexus.
We propose a fusion ancestor, derived from a Clostridium-like organism and a Sulfolobus-like organism, that was ancestral to all eukaryotes and whose DNA features are preserved in animal Mt and several a-Mt eukaryotes (see Fig. 1). The following outlines the arguments for a Clostridium-like and Sulfolobus-like prokaryotic fusion: (i) Just as with plasmid–host compatibility (11), genome DNA signature comparisons show that Sulfolobus spp. and Clostridium spp. sequences are moderately similar and, therefore, mutually compatible. (ii) The Sulfolobus and Clostridium signatures are also moderately similar to deuterostome (and most protostome) Mt sequences, whereas the genome signatures of other available prokaryotes, including classical α-proteobacteria, are generally very distant from animal Mt. (iii) Expanded energy metabolism of an ancient Clostridium-like organism with an ancient Sulfolobus-like organism can be realized. A Clostridium-like organism contributes to a fusion organism metabolic properties for degradation of organic matter to acetate and H2. Clostridium spp. lack a pathway for acetate oxidation and a respiratory chain for energy conservation. To an organismic fusion, a Sulfolobus-like organism contributes hydrogen utilization and a respiratory chain, including quinones and cytochrome cofactors (14). (iv) Clostridium spp. can form endospores, which, in the context of a Clostridium–Sulfolobus fusion, provide a means to establish nuclear and Mt compartments. (v) Clostridium spp. possess enzymes typical of the hydrogenosome (hydrogen-producing organelles) present in some anaerobic eukaryotes. (vi) Sulfolobus and most archaea express two kinds of genes: eubacteria-like and eukaryote-like. (vii) Sulfolobus has no cell wall and possesses some sort of cytoskeleton (15). Its flexible cell membrane is shaped as irregular lobes, which are composed of protein–lipid layers that morphologically resemble those of the animal inner mitochondrial membrane, and it contains or adsorbs eukaryote-like steroids, which enhance membrane flexibility (16). The fusion cell provided an environment where gene transfer between both genomes was frequent. Furthermore, these transfers were stable and complex cellular functions were preserved, because the transferred genes could act in trans in these compartments. A remnant of the Sulfolobus-like genome presumably evolved to the animal Mt genome and the Clostridium-like genome evolved to a nucleus compartment.
Figure 1.

Model for formation of primitive eukaryotic cells by a fusion between a Clostridium-like and Sulfolobus-like organism. The fusion cell of a Clostridium-like eubacterium (blue genome) and a Sulfolobus-like archaeon (yellow genome) (A), or a close association of these (B), provided an environment for extensive gene transfer between both genomes (red arrow, and color change of genomes). These transfers were successful and stable because of their compatible moderately similar genome signatures. Complex cellular functions were preserved because the transferred genes could act in trans. (1) Septation of the dikaryotic fusion cell. (2) One of the separated cells (Clostridium-like) engulfs the other cell in a mechanism resembling the process of endospore formation (29). (C) The engulfment process is complete. The Sulfolobus-like genome is enclosed by two membranes and develops into the proto-Mt. (4) The nuclear membrane evolves. (D) Prototype of a primitive, mitochondriated eukaryotic cell. Different selective pressure resulted in cells where primitive Mt degenerated into hydrogenosomes (E) (e.g., Trichomonads), into cells containing many Mt (F) (e.g., animal cells), and into a-Mt cells (G). Subsequent events (not shown) in Mt cells may have included endosymbiosis and/or parasitism.
In the fusion process, many genes are lost and others are relocated to the nucleus, and some genes are also acquired from other organisms. Transfer of genes and fragments of genes from plastids to Mt, from plastids to the nucleus, and from Mt to the nucleus has been documented for numerous organisms (e.g., see refs. 17 and 18)). These transfers may include transient breaks in organellar membranes during budding, degradation of organelles combined with release of nucleic acids to the cytoplasm, use of protein import machinery (chaperonins), or fusion between heterotypic membranes. We further contemplate that the intermediate Clostridium-like/Sulfolobus-like fusion organism was modified by an assortment of transient endosymbiont events and/or parasitic invasions. Thus, it is conceivable that an obligate intracellular parasite or an endosymbiont (e.g., an α-proteobacterium and/or other eubacteria) contributed genes and other DNA sequences to the Clostridium-like/Sulfolobus-like fusion or to some descendants of this partnership.
Variation Among Mt Organelles and Among a-Mt Eukaryotes.
There is great diversity among mitochondria of animal, plant, fungal, and protist lineages, including size variation, contrasting patterns of genome organization, and gene content and expression (17). Animal Mt genomes are compact, about 13 to 19 kb, are intronless, generally possess an altered DNA code, have nonstandard tRNA structures, and contain about 5–13 genes. Plant Mt extend from about 150 kb to 2,000 kb and contain “foreign” DNA, partly derived from the chloroplast organelle (17, 18). Fungal Mt are of mixed character. Protist mitochondria are diverse, as exemplified by the kinetoplasts of Trypanosoma brucei and the Mt (68-kb size) of the freshwater zooflagellate Reclinomonas americana, which contains at least 44 protein coding genes, retains the universal genetic code, and shows several strictly eubacterial characteristics (19).
Absence of Mt and plastid organelles is observed in some primitive eukaryotes. The a-Mt Diplomonads (e.g., Giardia lamblia, GIALA) (using the Swiss-Prot abbreviation, the first three letters of the genus name joined with the first two letters of the species name), Trichomonads (e.g., Trichomonas vaginalis, TRIVA), Microsporidia (e.g., Vairimorpha necatrix, VAINE), Entamoebidae (e.g., Entamoeba histolytica, ENTHI) each have small genome size (generally 6 to 20 Mb), are anaerobic, and are generally obligate intracellular parasites of mammals (20). TRIVA possesses a hydrogenosome organelle that contains the key enzyme, pyruvate:ferredoxin oxidoreductase, which uses pyruvate as substrate and oxidizes it to CO2 and acetyl-CoA that is used to generate acetate + ATP (20). In this process, hydrogenase specific to the hydrogenosome releases the reducing equivalents recovered from pyruvate oxidation as molecular hydrogen. This energy system is also characteristic of prokaryotic Clostridium species (21).
Genomic Signature Comparisons of Prokaryote and Mitochondrial Genomes.
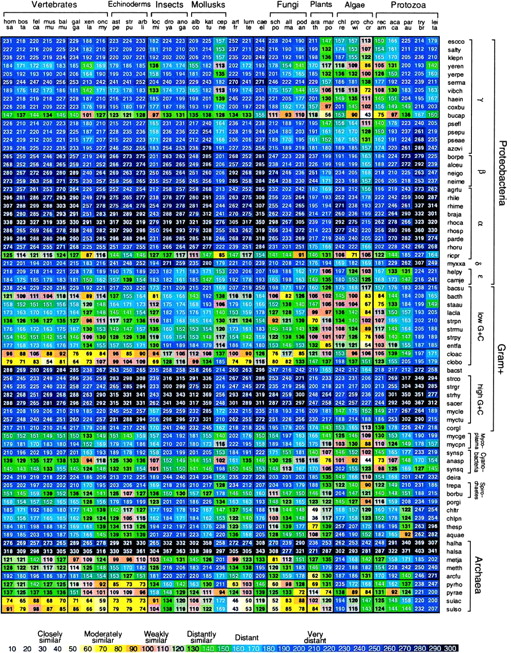
Fig. 2 displays δ* differences of representative Mt vs. prokaryotic sequences. Among the prokaryotic sequences the most similar (moderately to weakly similar) to animal and fungal Mt genomes are the Sulfolobus and Clostridium genome sequences. δ* differences comparing Sulfolobus to animal Mt genomes generally register moderate to weak similarity to all deuterostomes, protostomes, and Caenorhabditis elegans, but are very distant from Chlamydomonas reinhardtii (δ* ≈ 193) and protists (δ* > 150 to Trypanosoma brucei). Clostridium comparisons with Mt parallel those of Sulfolobus. δ* differences between the Rickettsia genome and animal Mt genomes are generally 50% greater than the corresponding δ* differences between Sulfolobus sp. and animal Mt genomes. Anabaena sp. shows δ* values most similar to the Mt of plants and green algae and remarkably close to the red alga Chondrus crispus, δ* = 44. Most comparisons of the Mt-genome signatures relative to α-, β-, and γ-proteobacteria, and to high-G+C Gram-positive eubacteria entail δ* > 200. The R. americana protist mitochondrion is moderately or weakly similar to several bacterial sequences. The halobacterial sequences are very distant from all Mt sequences.
Figure 2.

Average δ* differences between prokaryotic and Mt DNA sequence samples based on comparisons of 50-kb samples. The complete genome was used for Mt sequences of less than 50 kb length. A representative collection of complete Mt genomes is compared to a representative set of prokaryotes, each with at least 100 kb of nonredundant DNA. See also Fig. 3 for 70 prokaryotes [published as supplemental data on the PNAS web site (www.pnas.org)].
The Animal Mt–Sulfolobus/Clostridium Connection.
The classical α-proteobacterial types 𝒜1 include Rhizobium spp., Rhodobacter spp., and Paracoccus denitrificans. A tentative group, 𝒜2, includes the Rickettsia and Ehrlichia clades (obligate intracellular parasites). Genome signature comparisons reveal drastic disparities between 𝒜1 and 𝒜2. Recently, a Rickettsia prowazekii (RICPR) genome has been completely sequenced and advocated as a possible forebear of Mt organelles (22). However, there are genomic reasons for a Sulfolobus-like/Clostridium-like ancestor fusion leading to animal Mt. (i) Virtually all animal Mt are significantly CG underrepresented and among prokaryotes, Sulfolobus and Clostridium sequences have CG underrepresented (9, 11). Other thermophilic archaea, including Methanobacterium thermoautotrophicum and Methanococcus jannaschii species, also possess significantly low CG relative abundances, but not Pyrobaculum aerophilum or Pyrococcus horikoshii. RICPR is marginally CG suppressed (11). By contrast, most Gram-negative and Gram-positive bacteria show normal CG relative abundances. (ii) Animal Mt carry high CC/GG relative abundances, and the same for Sulfolobus and Clostridium, but RICPR is normal for ρ*CC/GG. The proteobacteria tend to be normal in CC/GG representations. (iii) The dinucleotide TA is broadly underrepresented in most prokaryotic and eukaryotic sequences and drastically underrepresented in classical α- and β-proteobacteria. In contrast, TA representations are normal in animal Mt genomes and also in Sulfolobus spp., Clostridium spp., and RICPR (11). (iv) Relative abundance values for the dinucleotide GC are high in β- and γ-proteobacteria, normal in classical α-proteobacteria, drastically high in RICPR (ρ*GC = 1.53), but normal in animal Mt and in Sulfolobus and Clostridium sequences. (v) The disparity in genomic C+G content between the classical α-proteobacteria (generally ≥60%) versus animal Mt (21–46%) is outstanding. The Sulfolobus, Clostridium, and RICPR sequences are C+G poor to the same extent as animal Mt. (vi) The signature comparisons of Fig. 2 place animal Mt farther from each classical α-proteobacterium, by a factor exceeding 3, than from Sulfolobus and Clostridium, which show moderate to weak similarity. (vii) Several subdivisions of proteobacteria, including the γ- and classical α-proteobacteria, high C+G Gram-positive bacteria, and Halobacterium spp. show very distant differences from all animal Mt genomes.
The pervasive similarities of the dinucleotide relative abundance extremes of animal Mt to those of Sulfolobus and Clostridium, coupled with the manifest deviations of these relative abundance extremes with respect to classical α-proteobacteria and partially for RICPR, argue against the hypothesis of an α-proteobacterium ancestry of animal Mt but are consistent with a Sulfolobus-like/Clostridium-like combination. The Mt of Trypanosoma/Leishmania are very distant (δ* > 200) from almost all prokaryotic sequences, suggesting that the prokaryotic forebears of these protist Mt are remote from the collection of prokaryotic genomes reviewed in Fig. 2. Unusual structural attributes in these Mt protist genomes include the kinetoplast “cryptogenes,” insertional RNA editing, and trans-splicing mechanisms.
Possible Expansions of Energy Metabolism in a Clostridium–Sulfolobus Fusion.
Metabolic properties of Clostridia and Sulfolobales. Clostridia are strictly anaerobic, endospore-forming, organotrophic bacteria that ferment carbohydrates, amino acids, or nucleic acids to products such as H2, CO2, and low molecular weight fatty acids and alcohols (21). Pyruvate, which is a key intermediate in many of those fermentations, is generally oxidized in clostridia by the enzyme pyruvate:ferredoxin oxidoreductase to acetyl-CoA + CO2 with the concomitant reduction of ferredoxin (23). Acetyl-CoA is converted to acetate, and reduced ferredoxin is oxidized by a hydrogenase that reduces H+ to molecular hydrogen. The order Sulfolobales includes the genus Sulfolobus spp., Acidianus spp., and Metallosphera spp. (14). All members are thermophilic and sulfur-metabolizing. Their energy metabolism includes either the oxidation of sulfur with molecular oxygen or Fe3+ as electron acceptors, or the reduction of sulfur to H2S (14). Energy is conserved during these processes by respiration via an electron transport chain. The respiratory chain in Sulfolobus acidocaldarius includes a unique quinone (caldariellaquinone), NADH dehydrogenase, and a cytochrome aa3 oxidase that oxidizes reduced caldariellaquinone (24, 25). Most members of this order are chemolithoautotrophic, although Sulfolobus and Metallosphera can also grow organotrophically. Enzymes for the complete oxidation of acetate via the citric acid cycle are present in Sulfolobus. It is conceivable that an ancestor to the Sulfolobales was an anaerobic chemolithoautotrophic organism capable of growth by oxidation of H2 or H2S with sulfur or Fe3+, respectively, as electron acceptor.
Expanded energy metabolism.
A Clostridium-like/Sulfolobus-like fusion organism would have the metabolic capability to completely mineralize organic matter with sulfur or Fe3+ as terminal electron acceptor. The fusion organism would have contained enzymes derived from clostridia for conversion of organic matter to acetate via pyruvate as intermediate, and a pyruvate:ferredoxin oxidoreductase, as well as Sulfolobales-derived enzymes for acetate oxidation to CO2, a respiratory chain to use ferric iron as terminal electron acceptor, and a hydrogenase to funnel hydrogen into the respiratory chain. Dissimilatory Fe3+ reduction has been proposed to be an early form of microbial respiration (26), and several hyperthermophilic Fe3+-reducing prokaryotes have been identified recently (27). Electron acceptors such as oxygen, nitrate, and sulfate are considered to have been limited in primitive times, whereas Fe(III), derived from photochemical oxidation or anaerobic phototrophic oxidation (28) of Fe(II), is thought to have been abundant on early Earth.
Evolution of the Eukaryotic Cell.
Compartmentalization in the early fusion cell. After the fusion of a Clostridium-like and a Sulfolobus-like organism, the chimeric microbe contained two genomes with compatible genome signatures. This compatibility also permitted frequent exchange of genetic material between these genomes. Under special conditions clostridia differentiate into endospores by a process that includes septation of a diploid cell and subsequent engulfment of the prespore by the mother cell (29). The genetic determinants of this process could have been present in the Clostridium-like ancestor of the fusion organism. Therefore, a similar process occurring in the fusion microbe could have resulted in engulfment of one genome and its subsequent compartmentalization to a protomitochondrion being ultimately enclosed by two membranes (Fig. 1). Also in that organism, gene transfer between the protomitochondrion (Sulfolobus-derived) and Clostridium-like genome were frequent because of compatibility in genome signatures. During subsequent evolution of the compartmentalized cell and after the appearance of molecular oxygen, some lateral gene transfer from external sources would have allowed establishment of an aerobic electron transport chain in the mitochondrion.
Evolution of the a-Mt cell.
From the primitive compartmentalized fusion organism, the protomitochondrion could have degenerated in the sense that with the exception of the genes encoding pyruvate:ferredoxin oxidoreductase (POR), all genes for respiration were lost from the compartment as well as the entire protomitochondrial genome, thus forming the hydrogenosome. That the hydrogenosome of a-Mt protists may be of clostridial origin is consistent with molecular data. For example, in the a-Mt protist Trichomonas vaginalis and in Clostridium acetobutylicum, the molecular properties of the POR enzyme are highly similar; both enzymes are homodimers with a molecular mass of the subunit of 123 kDa (23).
Lateral Transfer and Heat Shock Proteins.
HSP60 (GroEL) and HSP70 (DnaK) are abundant essential proteins in all E. coli life stages (30, 31) and in most bacteria. Several chaperonin and degradation proteins that function in Mt organelles of eukaryotes have been advanced as good marker sequences for tracing the evolution of Mt genomes (1, 2, 12, 32). Chaperonins play pivotal roles in protein folding, proteolysis, secretion, and translocation across membranes, and also help to stabilize organelle activity. In particular, HSP60 and HSP70 may facilitate bidirectional traffic between the Mt and the nucleus. Specialized complex structures in cells often need their own “dedicated” chaperonins (e.g., see refs. 33 and 34).
There are plausible scenarios indicating that the nuclear encoded HSP60 and HSP70 sequences functioning in Mt are a result of lateral transfer and are probably derived from an α-proteobacterial progenitor. This hypothesis relates to the plethora of duplicated HSP60 sequences among the classical α-proteobacteria, contrasted to no duplications of HSP60 sequences in all clades of other proteobacterial genomes. Thus, Rhizobium meliloti (RHIME) possesses at least three HSP60 sequences of 75–95% pairwise identity and Bradyrhizobium japonicum (BRAJA) contains at least five very similar HSP60 sequences. Rhodobacter sphaeroides (RHOSH) contains at least two HSP60 sequences of 74% identity, and similarities to other HSP60 α-sequences are also high. Multiple HSP60 sequences also exist in cyanobacteria, in Streptomyces spp., in Mycobacterium spp., and in Chlamydia spp., but a single HSP60 in RICPR. The a-Mt eukaryote TRIVA contains two HSP60 sequences, but GIALA apparently possesses a single representative, and no HSP60 sequences have been detected in ENTHI. The two TRIVA HSP60 copies are about 73% identical; one of these functions in the hydrogenosome, the other is localized to the cytoplasm. There is HSP60 that binds ribulose-bisphosphate carboxylase/oxidase (Rubisco) in plastids, generally with multiple copies. The existence of multiple copies of a gene sequence might be attributed to at least four causes. (i) Gene duplication conceivably can increase expression of the encoded protein at various times and places and under special conditions. (ii) The duplicated copies can functionally diverge or participate in heterooligomer complexes. Also, duplicated genes freed from functional constraints can evolve faster and adapt to new needs. This allows an increase in complexity, intrinsic in eukaryotes, and appears to be the nature of some HSP60 structures, for example, the HSP60 Rubisco binding protein in plastids. (iii) Duplications may provide insurance against extreme fluctuations in expression. (iv) The genome simply may be large enough to tolerate duplicated benign genes.
Most eukaryotes have at least three isoforms of HSP70 with cellular location in the endoplasmic reticulum (ER) lumen, in the cytoplasm (CYT), and in organelles. The eukaryote CYT-HSP70 sequences predominantly carry multiple copies, generally with ≥70% identity. Eubacteria that contain multiple copies of HSP70 include E. coli, H. influenzae, Borrelia burgdorferi, and several cyanobacteria. A single HSP70 homolog was detected in ENTHI that is most similar to eukaryote HSP70 cytoplasmic sequences (74–77%). TRIVA encodes three HSP70 sequences, one Mt-like and the other two most similar to eukaryotic CYT and ER versions, respectively. GIALA possesses two strictly eukaryotic HSP70 sequences, an ER isoform and a CYT isoform. These sequences align relatively poorly with all prokaryotic and organelle sequences. In summary, our analysis envisions events of lateral transfer especially for HSP60 and HSP70 sequences (35).
Some Protein Sequence Comparisons.
It is useful to summarize results on pairwise similarity for various classes of protein sequences, emphasizing comparisons among classical α-proteobacteria, RICPR, and Mt sequences. A paramount conclusion emerging from the data on protein sequence comparisons is the lack of a prokaryotic group that is consistently most similar to animal Mt.
Proteins encoded in animal Mt genomes.
For cytochrome oxidase I (CoxI), CoxIII, ATPase F1, cytochrome c, NADH 2 and NADH 4, the Mt versions match better with classical α-types over RICPR. NADH 5, NADH 11, and cytochrome b sequence similarities of α-proteobacteria vs. Mt are about the same as for RICPR vs. Mt. Only CoxII and NADH 7 adhere to the inequality RICPR vs. Mt > α vs. Mt.
Mt aminoacyl-tRNA synthetases.
Arginyl: yeast Mt vs. γ-proteobacterial sequences have 19–22% identity, threefold better than yeast Mt vs. RICPR of only 7% identity. Aspartyl: yeast Mt vs. BORBU 31 significantly dominates yeast Mt vs. RICPR 22. Threonyl: fungal Mt sequences from Schizosaccharomyces pombe, Saccharomyces cerevisiae, and Candida albicans compared with γ-types and BACSU entail 30–36% identity, but in alignment to RICPR 27–29% identity. Tyrosyl: BACSU matches with yeast Mt at 38% identity, whereas RICPR vs. yeast Mt attains 28% identity. Glutamyl: yeast Mt vs. BACSU 31% compared with RICPR 22%.
Chaperones and proteases that function in the Mt.
Lon: (BACSU vs. Mt) 38–40% > (α vs. Mt) 34–38% and RICPR vs. Mt 33–35% identity. FtsH: Mt sequences tentatively match best with Streptococcus pneumoniae with substantially diminished identity to RICPR. ClpP: RICPR vs. γ, 70–75%, RICPR vs. α, 62–66%, indicating that RICPR is more similar to γ-types than to α-types. Comparisons of the single HSP60 of RICPR and HSP60 of classical α to Mt HSP60 sequences show 50–65% identity. The other “Rickettsial” HSP60 ORITS (Orientia tsutsugamushi) aligns to Mt sequences and to corresponding α-sequences 10–20% points lower. The HSP60 sequences of Ehrlichia, considered a sister group to Rickettsia, align weakly with almost all bacterial sequences, including RICPR.
General proteins.
For the proteins DnaA, elongation factor EF-Tu, superoxide dismutase (SOD), and RNA polymerase III β′, RICPR matches γ-proteobacterial sequences significantly better than its matches to classical α-types. For example, with respect to SOD, RICPR vs. γ-types align at 50–56% identity but RICPR vs. RHOCA produces only 40% identity. Rosenthal et al. (36) examined eight fermentation enzymes and proteins and assessed protein similarities among the a-Mt protists ENTHI, GIALA, and TRIVA together with a broad span of bacterial sequences. No consistent picture emerged among a-Mt species, indicating that fermentation enzymes apparently derive from diverse bacterial sources and are liable to lateral transfer events, although in each case it is not clear which bacterial classes are the forebears.
Discussion.
We first state implicit assumptions and summarize and interpret key observations relevant to the analysis and hypothesis discussed in the preceding text; see also ref. 11.
Assumptions.
(i) Genome signatures have frequently remained almost unchanged over long time intervals for Sulfolobus, Clostridium, and animal Mt, whereas eukaryotic nuclear sequences have undergone much more divergence in genome signature. (ii) Evolutionarily successful cell fusion requires that the signatures of the two partners be generally moderately similar or at least weakly similar (11). (iii) Lateral gene transfer into Mt genomes from nuclei, plastids, and invading bacteria has been frequent throughout evolution, especially in the early stages of eukaryotic cell development. Laterally transferred genes evolve rather rapidly toward the signature of their new host. (iv) The eukaryotic cell encompassing the complexities of mitosis, meiosis, the cytoskeletal matrix, differentiation with multiple compartments, etc. argues that eukaryotes probably arose successfully once in evolution, but endosymbiosis (primary, secondary, nucleomorphs) and phagocytosis have occurred multiple times.
Genome signature comparisons.
Current dogma claims that some α-proteobacterium is the natural forebear of the Mt endosymbiont. However, genome signature assessments (δ* differences) show that Clostridium spp. and Sulfolobus spp. genomic sequences of mutual moderate similarity are significantly closer to animal Mt genomes than these Mt genomes are to other available prokaryotic sequences (Fig. 2). Also, genomic signatures of classical α-proteobacteria are very distant from all Mt DNA, and Rickettsia sequences are distantly similar to animal Mt. For plant Mt, the best agreements in genomic signature occur with Anabaena sp. and Buchnera aphidicola sequences. This similarity may stem from influences of plastid organelles through intracellular lateral transfer (18). Excepting Reclinomonas americana, comparisons with protist Mt genomes reveal no recognizable prokaryotic genomic sequences of even weak similarity.
Accumulating evidence indicates that α-proteobacteria replicate bidirectionally from a unique origin (demonstrated for Caulobacter crescentus (37) and suggested for RHIME and RHOCA). The replication asymmetry of the two animal Mt strands (heavy and light) would entail drastic alterations from the replication processes of an α-proteobacterial genome. The replication asymmetry, however, can rather easily be accounted for if the prokaryotic ancestor contained multiple replication origins, as archaeal genomes apparently do, with reduction to a single different origin for each strand.
Animal Mt genomes tend to be at least mutually weakly similar and mammalian Mt are very close, almost replicas of each other, much closer than the corresponding nuclear signatures are to each other. Why this difference? We have hypothesized that signature differences among organisms primarily reflect variations in replication and/or repair mechanisms, context-dependent DNA mutations, and modifications (8–10). The replication machinery for animal Mt DNA apparently varies less than that for host DNA, perhaps because Mt replication is less affected by changes in external environment or developmental programs than is host DNA replication. By contrast, the nuclear signature of Drosophila is distant from vertebrate signatures. This might be attributed to at least three mechanisms underscoring differences in replication processes, repair systems, and DNA modification operations between vertebrate and invertebrate species (e.g., lack of methylation in invertebrates). Drosophila (and apparently all protostomes), unlike mice, lack embryonic transcription-coupled-repair capacity (38). Moreover, Drosophila DNA replicates frenetically immediately after fertilization, with replication bubbles distributed about every 10 kb. At about 12 hr, effective origins are spread to about 40 kb apart. In mice the rate of replication seems to be uniform throughout developmental and adult stages (39). The observed narrow limits to intragenome signature heterogeneity putatively correlate with conserved features of DNA structure.
Supplementary Material
Acknowledgments
We are happy to acknowledge valuable discussions and comments on the manuscript by Profs. Dale Kaiser and David Relman. S.K. is supported in part by National Institutes of Health Grants 5R01GM10452-34 and 5R01HG00335-11 and by National Science Foundation Grant DMS9704552.
ABBREVIATIONS
- Mt
mitochondria(l)
- a-Mt
amitochondrial
References
- 1.Budin K, Philippe H. Mol Biol Evol. 1998;15:943–956. doi: 10.1093/oxfordjournals.molbev.a026010. [DOI] [PubMed] [Google Scholar]
- 2.Gupta R S. Microbiol Mol Biol Rev. 1998;62:1435–1491. doi: 10.1128/mmbr.62.4.1435-1491.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moreira D, Lopez-Garcia P. J Mol Evol. 1998;47:517–530. doi: 10.1007/pl00006408. [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Garcia P, Moreira D. Trends Biochem Sci. 1999;24:88–93. doi: 10.1016/s0968-0004(98)01342-5. [DOI] [PubMed] [Google Scholar]
- 5.Zillig W, Klenk H P, Palm P, Leffers H, Puhler G, Gropp F, Garrett R A. Cell Res. 1989;6:1–25. doi: 10.1139/m89-011. [DOI] [PubMed] [Google Scholar]
- 6.Gupta R S, Golding G B. Trends Biochem Sci. 1996;21:166–171. [PubMed] [Google Scholar]
- 7.Martin W, Müller M. Nature (London) 1998;392:37–41. doi: 10.1038/32096. [DOI] [PubMed] [Google Scholar]
- 8.Karlin S, Burge C. Trends Genet. 1995;11:283–290. doi: 10.1016/s0168-9525(00)89076-9. [DOI] [PubMed] [Google Scholar]
- 9.Karlin S. Curr Opin Microbiol. 1998;1:598–610. doi: 10.1016/s1369-5274(98)80095-7. [DOI] [PubMed] [Google Scholar]
- 10.Blaisdell B E, Campbell A M, Karlin S. Proc Natl Acad Sci USA. 1996;93:5854–5859. doi: 10.1073/pnas.93.12.5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell A, Mrázek J, Karlin S. Proc Natl Acad Sci USA. 1999;96:9184–9189. doi: 10.1073/pnas.96.16.9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doolittle W F. Trends Genet. 1998;14:307–311. doi: 10.1016/s0168-9525(98)01494-2. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence J G, Ochman H. Proc Natl Acad Sci USA. 1998;95:9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segerer A H, Stetter K O. In: The Prokaryotes. Balows A, Trüper H G, Dworkin M, Harder W, Schleifer U H, editors. New York: Springer; 1992. pp. 684–701. [Google Scholar]
- 15.Searcy D G, Hixon W G. Biosystems. 1991;25:1–11. doi: 10.1016/0303-2647(91)90008-9. [DOI] [PubMed] [Google Scholar]
- 16.Kates M, Kushner D J, Matheson A T, editors. The Biochemistry of Archaea. Amsterdam: Elsevier; 1993. [Google Scholar]
- 17.Wolstenholme D R, Jeon K W, editors. Mitochondrial Genomes, International Review of Cytology. Vol. 141. San Diego: Academic; 1992. [Google Scholar]
- 18.Douglas S E. Curr Opin Genet Dev. 1998;8:655–661. doi: 10.1016/s0959-437x(98)80033-6. [DOI] [PubMed] [Google Scholar]
- 19.Lang B F, Burger G, O’Kelly C J, Cedergren R, Golding G B, Lemieux C, Sankoff D, Turmel M, Gray M W. Nature (London) 1997;387:493–497. doi: 10.1038/387493a0. [DOI] [PubMed] [Google Scholar]
- 20.Fenchel T, Finlay B J. Ecology and Evolution in Anoxic Worlds. Oxford: Oxford Univ. Press; 1995. [Google Scholar]
- 21.Hippe H, Andreesen J R, Gottschalk G. In: The Prokaryotes. Balows A, Trüper H G, Dworkin M, Harder W, Schleifer U H, editors. New York: Springer; 1992. pp. 1800–1866. [Google Scholar]
- 22.Andersson S G, Zomorodipour A, Andersson J O, Sicheritz-Ponten T, Alsmark U C, Podowski R M, Naslund A K, Eriksson A S, Winkler H H, Kurland C G, et al. Nature (London) 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- 23.Adams M W W, Kletzin A. Adv Protein Chem. 1996;48:101–180. doi: 10.1016/s0065-3233(08)60362-9. [DOI] [PubMed] [Google Scholar]
- 24.Wakao H, Wagaki T, Oshima T. J Biochem (Tokyo) 1987;102:255–262. doi: 10.1093/oxfordjournals.jbchem.a122049. [DOI] [PubMed] [Google Scholar]
- 25.Anemüller S, Schäfer G. Eur J Biochem. 1989;191:297–305. doi: 10.1111/j.1432-1033.1990.tb19123.x. [DOI] [PubMed] [Google Scholar]
- 26.Lovley D R. Annu Rev Microbiol. 1993;47:263–290. doi: 10.1146/annurev.mi.47.100193.001403. [DOI] [PubMed] [Google Scholar]
- 27.Vargas M, Kashefi K, Blunt-Harris E L, Lovley D R. Nature (London) 1998;395:65–67. doi: 10.1038/25720. [DOI] [PubMed] [Google Scholar]
- 28.Ehrenreich A, Widdel F. Appl Environ Microbiol. 1994;60:4517–4526. doi: 10.1128/aem.60.12.4517-4526.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Young I E, Fitzjames P. J Biophys Biochem Cytol. 1959;6:467–505. doi: 10.1083/jcb.6.3.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karlin S, Mrázek J, Campbell A M. Mol Microbiol. 1998;29:1341–1355. doi: 10.1046/j.1365-2958.1998.01008.x. [DOI] [PubMed] [Google Scholar]
- 31.Matin A, Baetens M, Pandza S, Park C H, Waggoner S. In: Microbial Ecology and Infectious Disease. Rosenberg E, editor. Washington DC: Am. Soc. Microbiol.; 1999. pp. 30–48. [Google Scholar]
- 32.Syvanen M. Annu Rev Genet. 1994;28:237–261. doi: 10.1146/annurev.ge.28.120194.001321. [DOI] [PubMed] [Google Scholar]
- 33.Ogawa J, Long S R. Genes Dev. 1995;9:714–729. doi: 10.1101/gad.9.6.714. [DOI] [PubMed] [Google Scholar]
- 34.Kuehn M J, Ogg D J, Kihlberg J, Slonim L N, Flemmer K, Bergfors T, Hultgren S J. Science. 1993;262:1234–1241. doi: 10.1126/science.7901913. [DOI] [PubMed] [Google Scholar]
- 35.Karlin S, Brocchieri L. J Mol Evol. 1998;47:565–577. doi: 10.1007/pl00006413. [DOI] [PubMed] [Google Scholar]
- 36.Rosenthal B, Mai Z, Caplivski D, Ghosh S, De la Vega H, Graf T, Samuelson J. J Bacteriol. 1997;179:3736–3745. doi: 10.1128/jb.179.11.3736-3745.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marczynski G T, Lentine K, Shapiro L. Genes Dev. 1995;9:1543–1559. doi: 10.1101/gad.9.12.1543. [DOI] [PubMed] [Google Scholar]
- 38.de Cock J G, Klink E C, Ferro W, Lohman P H, Eeken J C. Mutat Res. 1992;293:11–20. doi: 10.1016/0921-8777(92)90003-l. [DOI] [PubMed] [Google Scholar]
- 39.Blumenthal A B, Kriegstein H J, Hogness D S. Cold Spring Harbor Symp Quant Biol. 1974;38:205–223. doi: 10.1101/sqb.1974.038.01.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}