

Abstract
BACKGROUND
Brugada and Long QT type 3 syndromes are linked to sodium channel mutations and clinically cause arrhythmias that lead to sudden death. We have identified a novel threonine to isoleucine missense mutation at position 353 (T353I) adjacent to the pore-lining region of domain I of the cardiac sodium channel (SCN5a) in family with Brugada syndrome. Both male and female carriers are symptomatic at young ages, have typical Brugada-type ECG changes, and have relatively normal corrected QT intervals.
OBJECTIVES
To characterize the properties of the newly identified cardiac sodium channel (SCN5a) mutation at the cellular level.
RESULTS
Using whole cell voltage clamp, we found that heterologous expression of SCN5a containing the T353I mutation resulted in 74% ± 6% less peak macroscopic sodium current when compared to wild-type (WT) channels. A construct of the T353I mutant channel fused with green fluorescent protein failed to traffic properly to the sarcolemma, with a large proportion of channels sequestered intracellularly. Overnight exposure to 0.1 mM mexiletine, a Na+ channel blocking agent, increased T353I channel trafficking to the membrane to near normal levels, but the mutant channels showed a significant late current that was 1.6 ± 0.2% of peak sodium current at 200 ms, a finding seen with long QT mutations.
CONCLUSIONS
The clinical presentation of patients carrying the T353I mutation is that of Brugada syndrome and could be explained by a cardiac Na+ channel trafficking defect. When the defect was ameliorated, the mutated channels had biophysical properties consistent with Long QT syndrome, however. The lack of phenotypic changes associated with the Long QT syndrome could be explained by a T353I-induced trafficking defect reducing the number of mutant channels with persistent currents present at the sarcolemma.
Keywords: Electrophysiology, Patch-clamp, Sodium channel, Arrhythmias, Brugada Syndrome, Long QT
Introduction
Careful regulation of cardiac Na+ channel gating is required to avoid arrhythmias. Disruption of sodium channel function, either by gene mutations or chemical blockade, can lead to fatal arrhythmias.1-12 Brugada and Long QT type 3 syndromes are autosomal dominant inherited sudden death syndromes associated with mutations in the cardiac Na+ channel (SCN5A). Brugada syndrome has been associated with SCN5a mutations that cause decreased sodium current,1-5, 7, 13-21 while mutations that cause sodium current that persists into the plateau region of the action potential are associated with Long QT syndrome.9, 10, 12, 22, 23
Patients with Brugada syndrome are more often male, present in the third decade of life with a syncope or sudden cardiac death, and have electrocardiographic (ECG) findings of right bundle branch block and ST segment elevation in leads V1 to V3.24 Those with Long QT syndrome also present with syncope and/or sudden cardiac death but tend to do so earlier in life and have prolonged repolarization (i.e. QT prolongation) on their ECGs.25
We identified a new cardiac Na+ channel mutation that co-segregates with the clinical manifestations in a moderate-sized family with Brugada syndrome. This newly identified mutation results in a threonine to isoleucine substitution (T353I) in the third extracellular loop of the first domain (Figure 1), approximately 2 amino acids upstream from the N-terminal part of the putative pore lining segment of the sodium channel.26 Here, we characterize the biophysical properties of this channel mutation to determine whether changes are consistent with those expected to cause Brugada syndrome.
Figure 1.

(A) Family pedigree and representative ECG tracings. All affected individuals (darkened squares for males or circles for females) carry the T353I mutation and manifest ECG changes including ST segment elevation in standard precordial leads V1-V3. The arrow identifies the proband. (B) Putative structure of the voltage-gated sodium channel showing the position of the T353I in the first pore lining segment.
Methods
Clinical studies and DNA analysis
The Institutional Review Board at the University of Pittsburgh approved all human studies.
The proband was referred to our institution for clinical evaluation. At-risk family members were asked to participate in this study. For those who agreed, a brief clinical history (including previously diagnosed cardiac conditions, syncope, palpitations, and medications), a physical examination, and a 12-lead ECG were performed and evaluated by the Investigators. Clinical testing, including a 24-hour Holter monitor, an echocardiogram, a cardiac MRI, and drug testing with procainamide, was offered to adult patients. For deceased family members, surviving relatives were questioned and hospital records were examined when available. A single ECG was performed on spouses to exclude inherited cardiac arrhythmopathies. Using criteria from the second consensus report, Brugada Syndrome was diagnosed when a Type 1 ECG repolarization in more than 1 right precordial lead (leads V1 to V3) and one of the following was present: a family history of sudden cardiac death at less than 45 years old, coved-type ECGs in family members or syncope27. Affected family members showed no cardiac anatomical abnormalities on clinical testing that might confound the diagnosis.
Blood samples (∼10 mL) were obtained from participating family members and spouses, and genomic DNA was isolated (Puregene, GENTRA Systems, Inc., Minneapolis, MN). Single stranded conformational polymorphism (SSCP) was used to identify exonic mutations in SCN5A as previously described.28 Mutant bands were subcloned into pBS and sequenced. Direct sequencing of genomic DNA was used to confirm individuals carrying the mutation and to exclude other SCN5A mutations. Additionally, all affected family members were homozygous for H558, a common polymorphism site that can affect sodium channel expression.29
Plasmid constructs
The T353I SCN5A mutation was placed into a wild-type cardiac Na+ channel cDNA (hH1a) cloned into the vector pRC-CMV (Invitrogen, Carlsbad, California) using the QuickChange Kit (Stratagene, La Jolla, CA, USA) and verified by sequencing. This construct encodes for a histidine at amino acid position 558 and for 2015 totals amino acids (i.e., Q1077del). Two types of constructs were made in order to identify cells expressing the hH1A mutation (IRES construct) and also to follow trafficking of the mutated Na+ channel (fusion construct). In the IRES construct, the hH1A gene was subcloned into the pIRES vector (Clontech, Mountain View, CA, USA) containing an internal ribosome entry site (IRES) driving expression of green fluorescent protein (GFP). Therefore, successfully transfected cells showed expression of Na+ channels and GFP simultaneously. In the fusion construct, the cDNA sequence encoding expression of GFP was fused to the C-terminus of hH1A, allowing visual localization of the Na+ channels. In detail, the wild-type human cardiac sodium channel and the channel carrying the missense mutation, T353I, were subcloned from the vector pRC-CMV at the AseI-XbaI sites into the NdeI-XbaI sites of the donor vector pDNR-1r (Clontech, Palo Alto, CA, USA) by compatible cohesive ends. To match the in frame rules, two nucleotides at the upstream codon of SCN5A were deleted by Quick Change II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). The deletion was introduced by the following mutagenic sense and antisense primers: 5′-GAGACCCAAGCTTCCCAGAAGCAGGATG -3′ and 5′-CATCCTGCTTCTGGGAAGCTTGGGTCTC -3′. WT and mutant T353I of SCN5A cDNA were subsequently subcloned into acceptors, either pLP-IRES2-EGFP bicistronic expression vector or the pLPS-AcGFP1-N green fluorescent protein (GFP) fusion report vector using BD creator DNA cloning kit (Clontech, Mountain View, CA) according to the instruction manual. The positive clones were screened by PCR using the primers that the kit suggested. Sequencing of all constructs confirmed correct fragment insertions at the ligation points and the absence of additional mutations.
Expression in a mammalian cell line and patch-clamp analysis.
A human embryonic kidney (HEK) cell line was used for all experiments. A human embryonic kidney (HEK) cell line stably expressing a SCN5A-IRES-GFP construct was maintained by antibiotic selection using Geneticin (G-8168, Sigma-Aldrich) at a concentration of 0.2 mg/mL. SCN5A-fusion constructs were expressed transiently in HEK cells. GFP was used as a marker to identify cells expressing the gene of interest (pIRES vector) or to localize the protein within the cell (fusion vector).
Cells expressing GFP were tested with the whole cell patch-clamp technique in voltage-clamp mode to measure Na+ current levels. For patch-clamping, HEK cells were freshly plated onto plastic coverslips the day of the experiments. Glass pipettes were pulled on a Sutter Model P-97 horizontal puller to a resistance of 1.0 to 2.5 MΩ. The glass pipettes were filled with a solution of (in mM) CsCl 60, Cesium Aspartate 80, EGTA 11, HEPES 10, Na2 ATP 5 and pH 7.2 with CsOH. The bath solution consisted of (in mM) Na 30, NMDG 100, CsCl 5, CaCl2 2, MgCl2 1.2, HEPES 10, Glucose 5 and pH 7.4 with NaOH. Once a seal was established, a small amount of suction was applied to obtain the whole cell configuration. A stepped voltage protocol from -70 to +40 mV from a holding potential of -100 mV was applied to establish the presence of voltage-gated Na+ channels. Currents obtained during steps to -10 mV were used for comparison in determining the relative reduction in sodium current. Cells were tested at 25°C. Data were sampled at 10 kHz and later filtered at 5 kHz for analysis. Currents were recorded and analyzed with an Axopatch 200B amplifier, Axon Digidata 1230A A/D converter and pClamp software (Molecular Devices Corporation, Sunnyvale, CA). For T353I, currents were analyzed when the peak current was greater than 100 pA. Mexiletine was used at a working concentration of 0.1 mM in the culture medium.
Localization of SCN5a protein.
A Nikon (Melville, NY) Diaphot 200 inverted microscope with a fluorescent lamp was used to identify cells expressing GFP. A confocal laser scanning microscope (MRC-1024, Bio-Rad, Hercules, CA) was used to visualize and localize the expression of the SCN5A GFP fusion protein. HEK cells of interest were grown on 22 mm round glass coverslips. The cells were then fixed using 4% paraformaldehyde in PBS, pH7.4. Slides were stored in the dark and immediately viewed for fluorescent imaging. For GFP excitation, the 488 nm emission line of an Argon ion 30 mW water-cooled laser was used. GFP fluorescence was observed with a 507 nm narrow bandpass filter and cells visualized with a 60x oil 1.4 NA objective. Mexiletine (M2727, Sigma-Aldrich) was added as indicated to a final concentration of 0.1 mM to the culture solution.
Computer Modeling
Simulations were conducted using the Luo-Rudy model of the mammalian ventricular action potential 30. The model includes membrane ionic channel currents that are formulated using the Hodgkin-Huxley approach, ionic pumps and exchangers, and processes that regulate intracellular calcium. The transient outward potassium current, Ito, was introduced into the LRd model as previously described 31. To simulate the effects of the T353I mutation, the experimentally observed gating changes were incorporated into the formalism of the mutant sodium current. These include: (1) an 11 mV negative shift in the steady-state inactivation curve, (2) a reduction in the maximum conductance by 75% (to reflect the upper limit of reduced expression), (3) a two-fold decrease in the time constant of fast inactivation (to reflect the faster decay of mutant current), and (4) introduction of a sustained inward non-inactivating component as described earlier 32. To reflect the heterozygous condition present in the patients carrying the mutation, we adjusted the model so that the total Na+ current was a 50/50 mixture of wild-type and mutant channels. Therefore, the total sodium current in the model of affected patients was approximately 62.5% of that in normal patients.
Statistical Analysis
Data are shown as mean ± standard error of the mean (SEM). Significance was determined with the t-test in Sigma Plot or a Fisher’s exact test in SPSS (SPSS, inc., Chicago, IL, USA). Statistical significance was assessed with a p<0.05.
Results
The proband, an asymptomatic 39 year old man of Greek descent with a past medical history significant only for mild hypertension, was identified by an electrocardiogram (ECG) done during a routine physical examination (Figure 1A). A stress echocardiogram showed normal left and right ventricular function without ischemia. An MRI showed no right ventricular abnormalities suggestive of arrhythmogenic right ventricular dysplasia, and an electrophysiology (EP) study was notable for a mildly prolonged HV interval (50 ms), polymorphic ventricular tachycardia with two extrastimuli at the RV apex requiring cardioversion, and a marked increase in both ST elevation and QRS prolongation during procainamide infusion. A prophylactic internal cardiac defibrillator (ICD) was placed, and no arrhythmias have been detected during the subsequent 6.5 years. The proband’s 22-year old niece presented with aborted sudden death (initial rhythm ventricular fibrillation) from which she was resuscitated and underwent ICD placement approximately one year ago. She has since had two appropriate shocks for ventricular arrhythmias. Of note, her ECG is suggestive of Brugada syndrome with up to 1.5 mm saddleback ST elevation in lead V2, and all of her arrhythmic episodes have followed moderate alcohol intake. The proband’s son presented at age 12 with syncope and a Brugada pattern on his ECG following a febrile illness. The proband’s sister has a history of near syncope and an ECG diagnostic of Brugada syndrome, but has refused ICD placement. The proband’s 19-year old daughter has an ECG suggestive of Brugada syndrome, is asymptomatic, and recently underwent prophylactic ICD placement. All affected individuals showed evidence of conduction abnormalities with PR and/or QRS prolongation, while corrected QTc intervals (Bazett’s formula) were near the top of the normal range at resting heart rates. Characteristics of affected members are listed in table 1.
Table I.
Characteristics of the Affected Family Members
| ID | AGE | GENDER | SYMPTOMS | PR* | QRS | QTc | STE (type; mm) | ECGs (Affected) | MEAN QTc (24-hour Holter) | IMAGING STUDIES |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 39 | M | None | 160 | 124 | 443 | C ; 3.5 | 4 / 4 (100%) | 467 ± 17 | MRI nl; Exercise echo nl |
| 2 | 45 | F | Near-Syncope | 176 | 120 | 443 | C ; 2.0 | 2 / 2 (100%) | 477 ± 26 | MRI nl; Cath mild CAD |
| 3 | 12 | M | Syncope | 176 | 118 | 440 | C ; 2.5 | 1 / 3 (33%) | ND | Echo nl |
| 4 | 19 | F | None | 206 | 120 | 443 | SB ; 1.0 | 1 / 2 (50%) | 448 ± 15 | Echo mild LAE |
| 5 | 22 | F | Aborted SCD | 218 | 134 | 420 | SB ; 1.5 | 1 / 2 (50%) | ND | Cath nl; Echo low nl EF |
C, Coved; CAD, coronary artery disease; Cath, cardiac catheterization; Echo, echocardiogram; EF, ejection fraction; F, female; LAE, left atrial enlargement; M, male; MRI, magnetic resonance imaging; ND, Not Done; nl, normal; SB, saddle back; SCD, sudden cardiac death; STE, ST elevation
PR, QRS, and QTc in ms; ECG parameters are from earliest affected ECG.
We screened SCN5A by single stranded conformational polymorphism (SSCP), and an abnormal band in exon 9 was identified. Subcloning of the band identified a T353I (cytosine to thymine substitution at position 1058) mutation, which is predicted to be in the pore-forming loop of domain I (Figure 1B). The mutation was confirmed and other mutations in SCN5A were excluded by direct sequencing of the SCN5A coding region. All members of the family with symptoms and/or abnormal ECGs carried the mutation (Individuals 1-5, Figure 1A).
Using the pIRES mammalian expression vector, the WT and mutant SCN5A sodium channels were transfected into HEK cells (Figure 2). GFP expression indicated a high proportion of successful transfection. Rapidly inactivating sodium currents were seen in HEK cells expressing wild-type SCN5A (Figure 3A). Of the cells successfully transfected with T353I, 65% had no measurable current defined as < 100 pA of current typical for the Na+ channel (Figure 3A, upper traces), and 35% had decreased current (Figure 3A, lower traces, p<0.01 compared to WT). Current could be increased by incubating cells overnight in 0.1 mM mexiletine (T353I+mex, Figure 3, panels A and B). After rescue with mexiletine, T353I channels showed persistent late sodium current. Measured at -10 mV, T353I channels continued to show 1.6 ± 0.2% of the peak current at 200 ms as compared to 0.03 ± 0.02% in the WT (p<0.01; panel C). Comparison of current-voltage curves for cells manifesting T353I current showed a reduced peak current at all voltages for mutant versus WT channels without a shift in the reversal potential (Figure 3D). Detailed investigation of the gating kinetics of the mutant channel revealed that the activation curve for T353I current was similar to WT (p>0.05; Figure 3E). On the other hand, the inactivation curve for T353I was shifted to more negative potentials compared to WT (p<0.01; Figure 3E). After rescue with mexiletine, the T353I channels displayed similar inactivation kinetics as the WT channel (p>0.05, Figure 3E). The cause of this shift is unknown, however. Further, fast inactivation for T353I was more rapid than WT (τ=1.3 ± 0.01 ms versus 0.78 ± 0.02 ms, WT vs. T353I at -40 mV, p<0.005).
Figure 2.

Representative phase contrast and fluorescent images of HEK cells transfected with the SCN5a and GFP on a bicistronic vector. Control HEK cells showed no fluorescence. HEK cells transfected with a vector containing the coding sequences of either wild-type (WT) or mutant (T353I) Na+ channels and green fluorescent protein (GFP) showed a high percentage of fluorescent cells. Cells transfected with the mutant T353I channel tended to be larger than native cells. For these pictures, cells were plated at similar density 48 hr prior to imaging.
Figure 3.
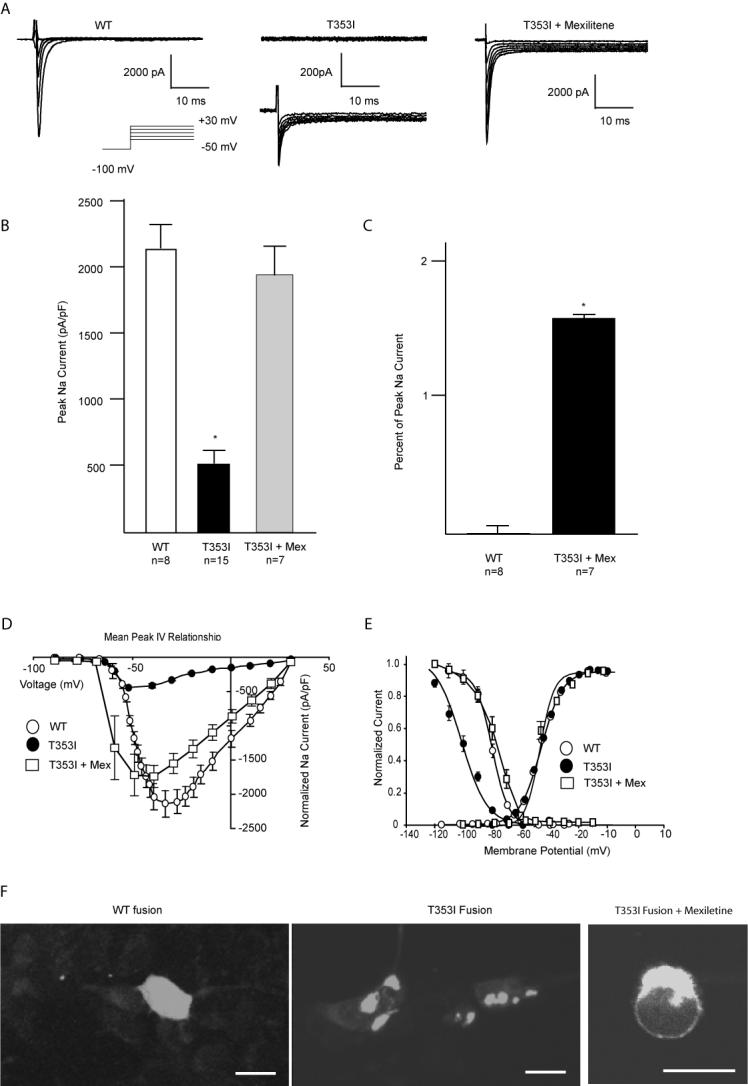
A summary of the effects of the T353I mutation. Panel A summarizes the results of patch-clamp current recordings of wild-type (WT) and T353I channels expressed in HEK cells. Cells expressing T353I demonstrated little (lower traces) or no current (upper traces). Cells expressing T353I and incubated overnight with mexiletine express significantly more current (T353I + Mexiletine). Panel B compares peak whole cell Na+ current normalized to cell capacitance. Peak current was significantly reduced for T353I (50 ± 12 pA/pF) compared to WT (212 ± 23 pA/pF; p<0.01) but was largely restored when T353I expressing cells were treated with mexiletine. Panel C compares the late current of cells expressing WT and T353I channels after exposure to mexiletine (T353I + Mex). Cells expressing T353I and treated with mexiletine showed a persistent late current at 200 ms (1.6 ± 0.2%) compared to WT (0.03 ± 0.02%; p<0.01). Panel D compares current-voltage relationships for WT and T353I with or without preceding mexiletine treatment showing a decreased peak current for T353I without changes in the reversal potential. Mexiletine resulted in nearly full recover of current. Panel E compares steady state activation and inactivation curves. Steady state activation parameters for WT, T353I and T353I+mex were V½ =-50 ± 0.4, k=4.6 ± 0.4 (n=15) and V½ =-49 ± 0.8, k=4.7 ± 0.3 (n=8), V½ =-54 ± 1.3, k= 1.5 ± 0.5 (n=7), respectively. Steady state inactivation parameters for WT, T353I, T353I+mex were V½ =-98 mV ± 2.2 mV, k=8.7 ± 1.9 (n=15) and V½ =-87 ± 0.3 mV, k=8.1 ± 0.2 (n=8), V½ =-84 ± 0.3 mV, k=7.1 ± 0.2 (n=6), respectively. Panel F demonstrates that T353I results in a trafficking defect that can be ameliorated by mexiletine. The WT SCN5a-GFP fusion construct was evenly distributed on the membrane surface. In contrast, the T353I-GFP channels appear sequestered within the cell. Overnight incubation with mexiletine resulted in mutant channel localization to the cell membrane. In each micrograph, the calibration bar represents 10 micrometers.
Because most cells successfully transfected with T353I showed no current, we created fusion constructs of the WT and mutant SCN5A channels with GFP to assess the distribution of channels. Confocal microscopy of cells transfected with the WT fusion protein showed uniform surface membrane channel expression, whereas the T353I protein was largely sequestered with the cell (Figure 3F). Incubating cells overnight in 0.1 mM mexiletine, a clinically useful Na+ channel pore blocking agent, resulted in increased cell surface expression of the T353I-GFP fusion channel when compared to untreated cells. This effect is consistent with the rescue of other mutated Na+ channels showing trafficking defects by Na+ channel pore blocking antiarrhythmic drugs.21, 33
We used action potential (AP) modeling to better understand the implications of reduced overall current with a persistent late component (figure 4). The action potential (AP) simulations mimicked a heterozygous 1:1 mixture of WT and mutant channels. Because of the decreased inward current from mutant channels, the AP upstroke was reduced from 371 V/s to 232 V/s. The sustained inward current of mutant channels resulted in an only slightly prolonged AP duration. These results are consistent with the limited changes in QT duration seen in affected family members, and these results suggest that the reduced Na+ current associated with Brugada syndrome effectively masked any expected QT prolongation associated with a persistent Na+ current. Increasing the mutant current to that of normal resulted in a markedly prolonged AP duration and generation of early after depolarizations, however.
Figure 4.
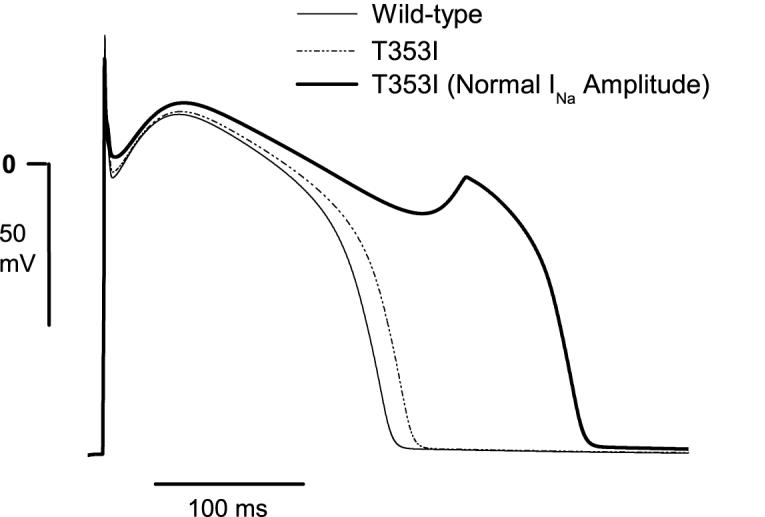
Computer modeling of action potentials (APs) of cells expressing only wild-type Na+ channels (solid line), a 1:1 (i.e., heterozygous) mixture of mutant (MT) and wild-type (WT) channels (dashed line), or a mixture of MT and WT channels after simulated rescue with mexiletine (bold line). The decreased inward current from MT channels reduced the AP upstroke velocity from 371 V/s to 232 V/s and the sustained inward current slightly prolonged AP duration. Upon simulated rescue of all MT channels (normal INa amplitude) and without continued drug present, the AP was significantly prolonged and an early afterdepolarization was generated.
Discussion
We describe a new sodium channel mutation (T353I) associated with Brugada syndrome and show that the mutation causes a significant reduction in Na+ current, suggesting its involvement in the pathogenesis of the arrhythmic phenotype seen in this family. The clinical presentation of Brugada syndrome, with the typical ECG pattern and the absence of torsades de pointes on ICD telemetry, was consistent with a loss of function of the cardiac sodium channel as the predominant manifestation the T353I mutation, despite a demonstrable persistent current in this mutation. This loss of function was the result of a trafficking defect that provided the reduced current substrate for Brugada syndrome and seemed to mask the effects of the persistent Na+ current usually associated with Long QT syndrome.
The T353I mutation is predicted to be in the pore-lining segment of the first channel domain. Several other arrhythmia-causing mutations have been described in this region. All except one are associated with Brugada syndrome. A cluster of Brugada mutations (currently fourteen as listed in Gene Connection for the Heart, http://pc4.fsm.it:81/cardmoc/) is located in the first pore forming domain. Several mutations have not been characterized at the cellular level including G292S,34 V294M,35 G319S,35 R367C,36 and M369K. 36 One mutation, L325R was shown to have temperature dependent effects in addition to having significant current reduction.18 The mutations R282H,17 G351V, and R376H20, 37 were shown to have significant current reduction5 while the D356N,38 and R367H22 had no current expression. The T353I mutation produced significantly decreased sodium current in those cells in which it could be measured, consistent with the expected Brugada cellular phenotype.
Unlike other mutations in this segment, the T353I mutation, after mexiletine rescue, caused a significant late inactivation current, a finding usually associated with Long QT syndrome. This is consistent with other evidence that the pore region can be involved in gating.1, 17, 39-44 Furthermore, a cysteine mutagenesis study of a pore lining amino acid in the third domain shows this amino acid to be part of a structural rearrangement of the pore that leads to changes in gating.45 In addition to the evidence that the domain I S5-S6 segment forms the outer vestibule of the pore,26, 46 it has also been shown that this segment plays a role in inactivation gating.39
The biophysical characteristics of the T353I channel are consistent with the potential for a mixed syndrome. Interestingly, several other amino acid positions in SCN5A, when mutated, also result in more than one pathophysiological syndrome. These sites are mostly located in the intracellular C-terminus of the channel, however, an area thought to be involved in channel regulation.47, 48 For example, residue Y1795, when mutated to a cysteine, causes LQT-3 syndrome and, when mutated to an histidine, causes Brugada Syndrome.19 Another mutation identified in the C-terminus, an insertion of an aspartic acid at position 1795, causes both Brugada and Long QT Syndrome15. A recently identified LQT3 mutation L1825P, also has properties of decreased peak current, but its main effect is a persistent late inward current, that is augmented by channel rescue with cisapride.33 Two mutations in domain I have been associated with more than one clinical phenotype, but neither has the Long QT phenotype. The E161K missense mutation in segment 2 of domain I produced significantly reduced sodium current when expressed in a mammalian embryonic kidney cell line and is associated with Brugada syndrome, sick sinus syndrome, and conduction disease.49 A second mutation in domain I, located in the pore lining segment, is a missense mutation that results in no detectable current and is associated with Brugada syndrome and atrial standstill.50 Our mutation is the first identified in the first pore lining segment to have both Brugada and LQT syndrome channel features.
Polymorphisms and splice variants of SCN5A can have an important effect on sodium current density in vitro.21, 29, 51-53 The H588R polymorphism when compared to WT channels has reduced sodium current density when expressed in HEK cells29 but also can rescue a Brugada Syndrome mutation.51 Nevertheless, affected family members in our study did not have this polymorphism. Humans simultaneously express two WT splice variants at position 1077, a glutamine (Q1077) and a deletion (Q1077del).53 The Q1077 decreases sodium current density when co-expressed with certain polymorphisms whereas the Q1077del does not significantly alter sodium current density.53 Our mutation was expressed using a clone encoding for the Q1077del splice variant. Therefore, the current reductions that we observed in vitro are likely to represent an upper limit on the in vivo current.
Our GFP-SCN5A fusion construct identified a trafficking problem with the T353I channel that is likely to underlie the reduced current seen with this mutation. Assessing sodium channel trafficking within a cell with a GFP fusion construct was previously reported.54 Makielski et al. published results on a trafficking defective SCN5A mutation (G1743R) that was rescued with mexiletine.21 Liu et al. also identified a trafficking defective mutant (L1825P), which was shown to localize to the endoplasmic reticulum and was rescued with cisapride.33 We show that the T353I channels get sequestered in intracellular vesicles. Intracellular sequestration of protein may be one reason that certain Brugada syndrome patients are seen to have altered cardiomyocyte structure.37
In summary, the ECG changes and arrhythmic symptoms in our family are consistent with a mutation in the pore region of the cardiac Na+ channel that reduces channel trafficking to the sarcolemma. This trafficking defect could be ameliorated with mexiletine, a Na+ channel blocking drug. After rescue by mexiletine, channels showed a sustained current, thought to contribute to Long QT syndrome. Since our family had no evidence of QT prolongation, the likely explanation is that the trafficking defect masked any electrocardiographic changes that might have arisen from a lack of full channel inactivation.
Acknowledgments
This study was supported by National Institutes of Health (NIH) grants HL64828 and HL73753 (SCD), HL62300 (BL), by a Department of Veterans Affairs merit grant (SCD), an American Heart Association Established Investigator Award (SCD), and research fellowship from the American Heart Association (AEP).
Footnotes
Sources of financial support and potential conflicts of interest:
Samuel C. Dudley, National Institutes of Health (NIH) grants HL64828 and HL73753, a Department of Veterans Affairs merit grant, an American Heart Association Established Investigator Award
Barry London, National Institutes of Health (NIH) grant HL62300
Arnold E. Pfahnl, AHA Research Fellowship Award
References
- 1.Amin AS, Verkerk AO, Bhuiyan ZA, Wilde AA, Tan HL. Novel Brugada syndrome-causing mutation in ion-conducting pore of cardiac Na+ channel does not affect ion selectivity properties. Acta Physiol Scand. 2005;185:291–301. doi: 10.1111/j.1365-201X.2005.01496.x. [DOI] [PubMed] [Google Scholar]
- 2.Baroudi G, Napolitano C, Priori SG, Del BA, Chahine M. Loss of function associated with novel mutations of the SCN5A gene in patients with Brugada syndrome. Can J Cardiol. 2004;20:425–30. [PubMed] [Google Scholar]
- 3.Baroudi G, Acharfi S, Larouche C, Chahine M. Expression and intracellular localization of an SCN5A double mutant R1232W/T1620M implicated in Brugada syndrome. Circ Res. 2002;90:E11–E16. [PubMed] [Google Scholar]
- 4.Baroudi G, Pouliot V, Denjoy I, Guicheney P, Shrier A, Chahine M. Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G) Circ Res. 2001;88:E78–E83. doi: 10.1161/hh1201.093270. [DOI] [PubMed] [Google Scholar]
- 5.Vatta M, Dumaine R, Antzelevitch C, Brugada R, Li H, Bowles NE, Nademanee K, Brugada J, Brugada P, Towbin JA. Novel mutations in domain I of SCN5A cause Brugada syndrome. Mol Genet and Metab. 2002;75:317–24. doi: 10.1016/S1096-7192(02)00006-9. [DOI] [PubMed] [Google Scholar]
- 6.Wang DW, Makita N, Kitabatake A, Balser JR, George AL., Jr. Enhanced Na+ channel intermediate inactivation in Brugada syndrome. Circ Res. 2000;87:E37–E43. doi: 10.1161/01.res.87.8.e37. [DOI] [PubMed] [Google Scholar]
- 7.Yokoi H, Makita N, Sasaki K, Takagi Y, Okumura Y, Nishino T, Makiyama T, Kitabatake A, Horie M, Watanabe I, Tsutsui H. Double SCN5A mutation underlying asymptomatic Brugada syndrome. Heart Rhythm. 2005;2:285–92. doi: 10.1016/j.hrthm.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 8.Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. N Engl J Med. 1989;321:406–12. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- 9.Abriel H, Cabo C, Wehrens XHT, Rivolta I, Motoike HK, Memmi M, Napolitano C, Priori SG, Kass RS. Novel arrhythmogenic mechanism revealed by a long-QT syndrome mutation in the cardiac Na+ channel. Circ Res. 2001;88:740–5. doi: 10.1161/hh0701.089668. [DOI] [PubMed] [Google Scholar]
- 10.Dumaine R, Wang Q, Keating MT, Hartmann HA, Schwartz PJ, Brown AM, Kirsch GE. Multiple mechanisms of Na+ channel-linked long-QT syndrome. Circ Res. 1996;78:916–24. doi: 10.1161/01.res.78.5.916. [DOI] [PubMed] [Google Scholar]
- 11.Makita N, Horie M, Nakamura T, Ai T, Sasaki K, Yokoi H, Sakurai M, Sakuma I, Otani H, Sawa H, Kitabatake A. Drug-induced long-QT syndrome associated with a subclinical SCN5A mutation. Circulation. 2002;106:1269–74. doi: 10.1161/01.cir.0000027139.42087.b6. [DOI] [PubMed] [Google Scholar]
- 12.Wehrens XHT, Abriel H, Cabo C, Benhorin J, Kass RS. Arrhythmogenic mechanism of an LQT-3 mutation of the human heart Na+ channel α-subunit : A computational analysis. Circulation. 2000;102:584–90. doi: 10.1161/01.cir.102.5.584. [DOI] [PubMed] [Google Scholar]
- 13.Baroudi G, Carbonneau E, Pouliot V, Chahine M. SCN5A mutation (T1620M) causing Brugada syndrome exhibits different phenotypes when expressed in Xenopus oocytes and mammalian cells. FEBS Lett. 2000;467:12–6. doi: 10.1016/s0014-5793(00)01099-1. [DOI] [PubMed] [Google Scholar]
- 14.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O’Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–6. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 15.Clancy CE, Rudy Y. Na+ channel mutation that causes both Brugada and long-QT syndrome phenotypes: A simulation study of mechanism. Circulation. 2002;105:1208–13. doi: 10.1161/hc1002.105183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong K, Guerchicoff A, Pollevick GD, Oliva A, Dumaine R, de ZM, Burashnikov E, Wu YS, Brugada J, Brugada P, Brugada R. Cryptic 5’ splice site activation in SCN5A associated with Brugada syndrome. J Mol Cell Cardiol. 2005;38:555–60. doi: 10.1016/j.yjmcc.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 17.Itoh H, Shimizu M, Mabuchi H, Imoto K. Clinical and electrophysiological characteristics of Brugada syndrome caused by a missense mutation in the S5-pore site of SCN5A. J Cardiovasc Electrophysiol. 2005;16:378–83. doi: 10.1046/j.1540-8167.2005.40606.x. [DOI] [PubMed] [Google Scholar]
- 18.Keller DI, Rougier JS, Kucera JP, Benammar N, Fressart V, Guicheney P, Madle A, Fromer M, Schlapfer J, Abriel H. Brugada syndrome and fever: Genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc Res. 2005;67:510–9. doi: 10.1016/j.cardiores.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 19.Rivolta I, Abriel H, Tateyama M, Liu H, Memmi M, Vardas P, Napolitano C, Priori SG, Kass RS. Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. J Biol Chem. 2001;276:30623–30. doi: 10.1074/jbc.M104471200. [DOI] [PubMed] [Google Scholar]
- 20.Rossenbacker T, Carroll SJ, Liu H, Kuiperi C, de Ravel TJL, Devriendt K, Carmeliet P, Kass RS, Heidbuchel H. Novel pore mutation in SCN5A manifests as a spectrum of phenotypes ranging from atrial flutter, conduction disease, and Brugada syndrome to sudden cardiac death. Heart Rhythm. 2004;1:610–5. doi: 10.1016/j.hrthm.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Valdivia CR, Tester DJ, Rok BA, Porter CB, Munger TM, Jahangir A, Makielski JC, Ackerman MJ. A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc Res. 2004;62:53–62. doi: 10.1016/j.cardiores.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 22.Hong K, Berruezo-Sanchez A, Poungvarin N, Oliva A, Vatta M, Brugada J, Brugada P, Towbin J, Dumaine R, Pinero-Galvez C, Antzelevitch C, Brugada R. Phenotypic characterization of a large European family with Brugada syndrome displaying a sudden unexpected death syndrome mutation in SCN5A:. Female predominance in the signs and symptoms of the disease. J Cardiovasc Electrophysiol. 2004;15:64–9. doi: 10.1046/j.1540-8167.2004.03341.x. [DOI] [PubMed] [Google Scholar]
- 23.Chandra R, Starmer CF, Grant AO. Multiple effects of KPQ deletion mutation on gating of human cardiac Na+ channels expressed in mammalian cells. Am J Physiol Heart Circ Physiol. 1998;274:H1643–H1654. doi: 10.1152/ajpheart.1998.274.5.H1643. [DOI] [PubMed] [Google Scholar]
- 24.Brugada J, Brugada R, Antzelevitch C, Towbin J, Nademanee K, Brugada P. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation. 2002;105:73–8. doi: 10.1161/hc0102.101354. [DOI] [PubMed] [Google Scholar]
- 25.Shimizu W. The long QT syndrome: Therapeutic implications of a genetic diagnosis. Cardiovasc Res. 2005;67:347–56. doi: 10.1016/j.cardiores.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 26.Choudhary G, Yotsu-Yamashita M, Shang L, Yasumoto T, Dudley SC., Jr. Interactions of the C-11 hydroxyl of tetrodotoxin with the sodium channel outer vestibule. Biophys J. 2003;84:287–94. doi: 10.1016/S0006-3495(03)74849-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, Wilde A. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–70. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 28.Wang Q, Li Z, Shen J, Keating MT. Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics. 1996;34:9–16. doi: 10.1006/geno.1996.0236. [DOI] [PubMed] [Google Scholar]
- 29.Makielski JC, Ye B, Valdivia CR, Pagel MD, Pu J, Tester DJ, Ackerman MJ. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ Res. 2003;93:821–8. doi: 10.1161/01.RES.0000096652.14509.96. [DOI] [PubMed] [Google Scholar]
- 30.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res. 1994;74:1071–96. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- 31.Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, Brugada J, Brugada R, Antzelevitch C. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–9. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- 32.Viswanathan PC, Rudy Y. Pause induced early afterdepolarizations in the long QT syndrome: a simulation study. Cardiovasc Res. 1999;42:530–42. doi: 10.1016/s0008-6363(99)00035-8. [DOI] [PubMed] [Google Scholar]
- 33.Liu K, Yang T, Viswanathan PC, Roden DM. New mechanism contributing to drug-induced arrhythmia: rescue of a misprocessed LQT3 mutant. Circulation. 2005;112:3239–46. doi: 10.1161/CIRCULATIONAHA.105.564008. [DOI] [PubMed] [Google Scholar]
- 34.iimura H, Matsunaga A, Kumagai K, Ohwaki K, Ogawa M, Noguchi H, Yonemura K, Saku K. Genetic analysis of Brugada syndrome in Western Japan: two novel mutations. Circ J. 2004;68:740–6. doi: 10.1253/circj.68.740. [DOI] [PubMed] [Google Scholar]
- 35.Priori SG, Napolitano C, Gasparini M, Pappone C, la Bella P, Brignole M, Giordano U, Giovannini T, Menozzi C, Bloise R, Crotti L, Terreni L, Schwartz PJ. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome : A prospective evaluation of 52 families. Circulation. 2000;102:2509–15. doi: 10.1161/01.cir.102.20.2509. [DOI] [PubMed] [Google Scholar]
- 36.Smits JP, Eckardt L, Probst V, Bezzina CR, Schott JJ, Remme CA, Haverkamp W, Breithardt G, Escande D, Schulze-Bahr E, LeMarec H, Wilde AA. Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J Am Coll Cardiol. 2002;40:350–6. doi: 10.1016/s0735-1097(02)01962-9. [DOI] [PubMed] [Google Scholar]
- 37.Frustaci A, Priori SG, Pieroni M, Chimenti C, Napolitano C, Rivolta I, Sanna T, Bellocci F, Russo MA. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation. 2005;112:3680–7. doi: 10.1161/CIRCULATIONAHA.105.520999. [DOI] [PubMed] [Google Scholar]
- 38.Makiyama T, Akao M, Tsuji K, Doi T, Ohno S, Takenaka K, Kobori A, Ninomiya T, Yoshida H, Takano M. High risk for bradyarrhythmic complications in patients with Brugada Syndrome caused by SCN5A gene mutations. J Am Coll Cardiol. 2005;46:2100–6. doi: 10.1016/j.jacc.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 39.Balser JR, Nuss HB, Chiamvimonvat N, Perez-Garcia MT, Marban E, Tomaselli GF. External pore residue mediates slow inactivation in mu 1 rat skeletal muscle sodium channels. J Physiol (Lond) 1996;494:431–42. doi: 10.1113/jphysiol.1996.sp021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kambouris NG, Hastings LA, Stepanovic S, Marban E, Tomaselli GF, Balser JR. Mechanistic link between lidocaine block and inactivation probed by outer pore mutations in the rat μ1 skeletal muscle sodium channel. J Physiol (Lond) 1998;512:693–705. doi: 10.1111/j.1469-7793.1998.693bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hilber K, Sandtner W, Kudlacek O, Schreiner B, Glaaser I, Schutz W, Fozzard HA, Dudley SC, Todt H. Interaction between fast and ultra-slow inactivation in the voltage-gated sodium channel. Does the inactivation gate stabilize the channel structure? J Biol Chem. 2002;277:37105–15. doi: 10.1074/jbc.M205661200. [DOI] [PubMed] [Google Scholar]
- 42.Hilber K, Sandtner W, Kudlacek O, Glaaser IW, Weisz E, Kyle JW, French RJ, Fozzard HA, Dudley SC, Todt H. The selectivity filter of the voltage-gated sodium channel is involved in channel activation. J Biol Chem. 2001;276:27831–9. doi: 10.1074/jbc.M101933200. [DOI] [PubMed] [Google Scholar]
- 43.Sandtner W, Szendroedi J, Zarrabi T, Zebedin E, Hilber K, Glaaser I, Fozzard HA, Dudley SC, Todt H. Lidocaine: A foot in the door of the inner vestibule prevents ultra-slow inactivation of a voltage-gated sodium channel. Mol Pharmacol. 2004;66:648–57. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 44.Todt H, Dudley S, Kyle JW, French RJ, Fozzard HA. Ultra-slow inactivation in μ1 Na+ channels is produced by a structural rearrangement of the outer vestibule. Biophys J. 1999;76:1335–45. doi: 10.1016/S0006-3495(99)77296-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ong BH, Tomaselli GF, Balser JR. A structural rearrangement in the sodium channel pore linked to slow inactivation and use dependence. J Gen Physiol. 2000;116:653–62. doi: 10.1085/jgp.116.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marban E, Yamagishi T, Tomaselli GF. Structure and function of voltage-gated sodium channels. J Physiol. 1998;508(Pt 3):647–57. doi: 10.1111/j.1469-7793.1998.647bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wingo TL, Shah VN, Anderson ME, Lybrand TP, Chazin WJ, Balser JR. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat Struct Mol Biol. 2004;11:219–25. doi: 10.1038/nsmb737. [DOI] [PubMed] [Google Scholar]
- 48.Young KA, Caldwell JH. Modulation of skeletal and cardiac voltage-gated sodium channels by calmodulin. J Physiol. 2005;565:349–70. doi: 10.1113/jphysiol.2004.081422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smits JPP, Koopmann TT, Wilders R, Veldkamp MW, Opthof T, Bhuiyan ZA, Mannens MMAM, Balser JR, Tan HL, Bezzina CR, Wilde AAM. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J Mol Cell Cardiol. 2005;38:969–81. doi: 10.1016/j.yjmcc.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 50.Takehara N, Makita N, Kawabe J, Sato N, Kawamura Y, Kitabatake A, Kikuchi K. A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J Intern Med. 2004;255:137–42. doi: 10.1046/j.0954-6820.2003.01247.x. [DOI] [PubMed] [Google Scholar]
- 51.Poelzing S, Forleo C, Samodell M, Dudash L, Sorrentino S, Anaclerio M, Troccoli R, Iacoviello M, Romito R, Guida P, Chahine M, Pitzalis M, Deschenes I. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation. 2006;114:368–76. doi: 10.1161/CIRCULATIONAHA.105.601294. [DOI] [PubMed] [Google Scholar]
- 52.Tan BH, Valdivia CR, Song C, Makielski JC. Partial expression defect for the SCN5A missense mutation G1406R depends upon splice variant background Q1077 and rescue by mexiletine. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.00101.2006. [DOI] [PubMed] [Google Scholar]
- 53.Tan BH, Valdivia CR, Rok BA, Ye B, Ruwaldt KM, Tester DJ, Ackerman MJ, Makielski JC. Common human SCN5A polymorphisms have altered electrophysiology when expressed in Q1077 splice variants. Heart Rhythm. 2005;2:741–7. doi: 10.1016/j.hrthm.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 54.Zimmer T, Biskup C, Dugarmaa S, Vogel F, Steinbis M, Bohle T, Wu YS, Dumaine R, Benndorf K. Functional expression of GFP-linked human heart sodium channel (hH1) and subcellular localization of the a subunit in HEK293 cells and dog cardiac myocytes. J Membr Biol. 2002;186:1–12. doi: 10.1007/s00232-001-0130-1. [DOI] [PubMed] [Google Scholar]