

Abstract
Recent genetic and cell biological studies illustrate the importance of active transforming growth factor-β signaling in preventing the proliferation of estrogen receptor-positive cells in the normal mammary gland, and suggest how the loss of this inhibition may be important in early breast cancer progression.
A recent report by Ewan and colleagues [1] sheds some light on the mechanism by which steroid receptor-positive cells are prevented from proliferating in the normal adult mammary gland by the expression of activated transforming growth factor (TGF)-β. This report is an extension of earlier studies by the same authors that first demonstrated the ability to detect latent versus activated TGF-β expression in situ [2]. Steroid receptor expression is often used as a prognostic indicator and target of endocrine therapy in breast cancer. However, our understanding of the normal distribution and regulation of estrogen receptor (ER)-α and progesterone receptor (PR) expression is still evolving. In normal adult mammary glands from humans, rats, mice, or cows, steroid receptor-positive cells are heterogeneously located in luminal epithelial cells throughout the ducts and rarely co-localize with markers of proliferation [3-6]. Growth factors expressed in steroid receptor-positive cells act on neighboring cells to induce proliferation in a paracrine fashion. An elegant series of experiments by Cathrin Brisken and her colleagues [7,8] have established the ability of estrogen receptor-positive or progesterone receptor-positive cells to rescue ER-null and PR-null cells, respectively, in chimeric fat-pad transplantation experiments. However, a key question in mammary gland development is what prevents ER/PR-positive cells from dividing and why this proliferative block is lost during breast cancer progression. For example, ER/PR-positive cells are often proliferative in mouse models of mammary hyperplasia and in precancerous lesions of the human breast, such as ductal carcinoma in situ, suggesting a switch from a paracrine to an autocrine response to proliferative stimuli [9,10].
TGF-β is a potent inhibitor of epithelial cell proliferation, but little is known about the process involved in activating the latent form secreted by most cells [11]. The relationship between TGF-β activation, ER-α expression, and proliferation were the focus of the recent study by Ewan and colleagues [1]. Using immunostaining to detect the activated form of TGF-β during estrus, they were able to show that the cells positive for active TGF-β also expressed the downstream effector R-SMAD in the nucleus, providing evidence that TGF-β acts in an autocrine manner. Co-immunostaining experiments using double immunofluorescence were performed to show that ER-α co-localized with nuclear R-SMAD staining and activated TGF-β co-localized with both ER-α and PR, supporting their hypothesis that TGF-β activation might inhibit the ability of ER/PR-expressing cells to respond to ovarian hormone-induced proliferation.
Mice heterozygous for the TGF-β1 allele have a 90% reduction in the amount of TGF-β1 protein expressed [12]. Using mammary glands from these mice, the expression of ER-α and Ki67, a marker of proliferation, was assessed. Mammary glands from TGF-β1+/- mice exhibited a 24-fold increase in proliferation, and co-localization of ER-α and Ki67 was increased 16-fold. The same results were found when this analysis was performed on outgrowths after transplantation of TGF-β1+/- mammary epithelial cells, suggesting that epithelial, rather than stromal, TGF-β is responsible for keeping ER/PR-positive cells from proliferating. After ovariectomy, no ER-α/Ki67 double-positive cells were observed in TGF-β1+/- glands in the absence of ovarian hormones. Only when estrogen and progesterone were added back was there a 17-fold increase in double-labeled cells. Although a loss of TGF-β expression resulted in increased proliferation in the presence of ovarian hormones, the expression of constitutively active TGF-β was able to override hormone-induced proliferation. Mammary glands from mouse mammary tumor virus (MMTV)–TGF-β transgenic mice displayed sixfold fewer ER-α/Ki67 double-positive cells than wild-type glands at estrus.
Fewer proliferating ER-α-positive cells are detected in the mammary glands of parous humans or rats than in glands from nulliparous subjects [4,13]. To test whether TGF-β has a function in this phenomenon, parous glands from TGF-β1+/- mice were analyzed for ER-α and Ki67 co-localization. The frequency of double-labeled cells in parous glands from TGF-β1+/- mice was fourfold that in parous wild-type mice, indicating that TGF-β is also involved in inhibiting the proliferation of ER-α-positive cells in parous animals. These data also fit with the findings of Boulanger and colleagues, who demonstrated that TGF-β-positive cells from parous glands did not contribute to repopulating the gland, suggesting that they were incapable of 'expansive cellular proliferation' and stem cell self-renewal [14]. TGF-β expression has also been shown to increase mammary stem cell senescence and to inhibit MMTV-induced mammary carcinogenesis [15]. Finally, the effect of TGF-β1 depletion was analyzed during puberty, a time of rapid proliferation in the terminal end bud structures. Loss of TGF-β1 did not increase the proliferation of ER-α-positive cells in either the terminal end buds or ducts, although proliferation was increased overall. This supports the idea that proliferation of ER/PR-positive cells during puberty is regulated differently from that in the adult gland.
These data advance our understanding of how steroid receptor-positive cells might be prevented from proliferating in the normal mammary gland, and how this process might become deregulated in breast cancer progression. Further evidence for the role of TGF-β in regulating the proliferation of steroid receptor-expressing cells comes from our studies of CCAAT-enhancer-binding protein (C/EBP)β-null mice. Mammary glands from these mice show increased numbers of ER/PR-positive cells and a 10-fold decrease in proliferation [5]. Recently, we discovered that these C/EBPβ-null glands also display a significant increase in activated TGF-β along with increased downstream Smad2 expression and signaling [16]. Another downstream target of TGF-β is the cyclin-dependent kinase inhibitor, p27Kip1, which in turn affects the activity of other cell cycle components such as cyclin E and cyclin-dependent kinase (cdk)2 [17]. All of these molecules were altered in C/EBPβ-null glands, resulting in decreased cyclin E expression, loss of cdk2 kinase activity, increased p27 stability and decreased levels of cdc25A phosphatase activity. These studies with mouse models have led us to speculate that loss of active TGF-β expression in precancerous breast lesions might result in increased expression or stability of cdc25A and increased cyclin E/cdk2 activity in steroid receptor-positive cells, allowing them to proliferate. In support of this hypothesis, cdc25A was recently shown to be induced by genomic and non-genomic actions of estrogen in breast cancer cells [18,19]. The expression of cdc25a is also regulated both transcriptionally and post-transcriptionally by TGF-β, and thus may be a useful downstream indicator of active TGF-β signaling [20]. p27 has also been proposed as a prognostic marker in breast cancer [21]. However, the subcellular localization and phosphorylation state of the protein are critical in regulating its activity, and it would, therefore, be problematic to assess these changes in clinical samples. A hypothetical model summarizing these results with regard to breast cancer progression is shown in Figure 1.
Figure 1.
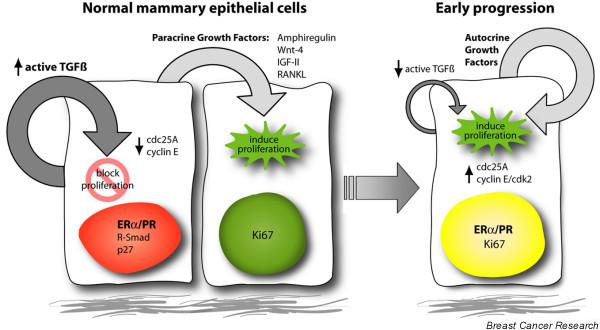
A hypothetical model of paracrine versus autocrine signaling in mammary epithelial cells. Normal mammary epithelial cells expressing estrogen receptor (ER)α and progesterone receptor (PR) are restrained from proliferating, mediated by the growth-inhibitory actions of transforming growth factor (TGF)-β signaling, active in this population. The steroid receptor-positive cells secrete local-acting growth factors to induce neighboring cells to divide in a paracrine fashion. In early breast cancer progression, this normal paracrine mechanism may switch to an autocrine loop, allowing steroid receptor-positive cells to proliferate, possibly through the downregulation of active TGF-β signaling, leading to an upregulation of cell-cycle molecules such as cdc25A, cyclin E and cyclin-dependent kinase (cdk)2. IGF, insulin-like growth factor.
Conclusion
Critical questions that still need to be addressed are what mechanisms are responsible for the activation of TGF-β only in the ER/PR-positive cells, and how the patterning of cells expressing steroid receptors is established both in normal development and breast cancer. With the availability of appropriate immunological reagents, these hypotheses should be tested with patient samples, to determine whether these markers will provide a method for predicting which patients with ductal carcinoma in situ have a higher likelihood of progression to infiltrating ductal carcinoma.
Abbreviations
cdk = cyclin-dependent kinase; C/EBP = CCAAT-enhancer-binding protein; ER = estrogen receptor; MMTV = mouse mammary tumor virus; PR = progesterone receptor; TGF = transforming growth factor.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
This study was supported by NIH grant CA16303.
References
- Ewan KB, Oketch-Rabah HA, Ravani SA, Shyamala G, Moses HL, Barcellos-Hoff MH. Proliferation of estrogen receptor-alpha-positive mammary epithelial cells is restrained by transforming growth factor β1 in adult mice. Am J Pathol. 2005;167:409–417. doi: 10.1016/s0002-9440(10)62985-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewan KB, Shyamala G, Ravani SA, Tang Y, Akhurst R, Wakefield L, Barcellos-Hoff MH. Latent transforming growth factor-beta activation in mammary gland: regulation by ovarian hormones affects ductal and alveolar proliferation. Am J Pathol. 2002;160:2081–2093. doi: 10.1016/s0002-9440(10)61158-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RB, Howell A, Potten CS, Anderson E. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer Res. 1997;57:4987–4991. [PubMed] [Google Scholar]
- Russo J, Ao X, Grill C, Russo IH. Pattern of distribution of cells positive for estrogen receptor alpha and progesterone receptor in relation to proliferating cells in the mammary gland. Breast Cancer Res Treat. 1999;53:217–227. doi: 10.1023/A:1006186719322. [DOI] [PubMed] [Google Scholar]
- Seagroves TN, Lydon JP, Hovey RC, Vonderhaar BK, Rosen JM. C/EBPβ (CCAAT/enhancer binding protein) controls cell fate determination during mammary gland development. Mol Endocrinol. 2000;14:359–368. doi: 10.1210/me.14.3.359. [DOI] [PubMed] [Google Scholar]
- Capuco AV, Ellis S, Wood DL, Akers RM, Garrett W. Postnatal mammary ductal growth: three-dimensional imaging of cell proliferation, effects of estrogen treatment, and expression of steroid receptors in prepubertal calves. Tissue Cell. 2002;34:143–154. doi: 10.1016/S0040-8166(02)00024-1. [DOI] [PubMed] [Google Scholar]
- Brisken C, Park S, Vass T, Lydon JP, O'Malley BW, Weinberg RA. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc Natl Acad Sci USA. 1998;95:5076–5081. doi: 10.1073/pnas.95.9.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallepell S, Krust A, Chambon P, Brisken C. Paracrine signaling through the epithelial estrogen receptor alpha is required for proliferation and morphogenesis in the mammary gland. Proc Natl Acad Sci USA. 2006;103:2196–2201. doi: 10.1073/pnas.0510974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina D, Kittrell FS, Shepard A, Stephens LC, Jiang C, Lu J, Allred DC, McCarthy M, Ullrich RL. Biological and genetic properties of the p53 null preneoplastic mammary epithelium. FASEB J. 2002;16:881–883. doi: 10.1096/fj.01-0885fje. [DOI] [PubMed] [Google Scholar]
- Shoker BS, Jarvis C, Clarke RB, Anderson E, Hewlett J, Davies MP, Sibson DR, Sloane JP. Estrogen receptor-positive proliferating cells in the normal and precancerous breast. Am J Pathol. 1999;155:1811–1815. doi: 10.1016/S0002-9440(10)65498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff MH. Latency and activation in the control of TGF-β. J Mammary Gland Biol Neoplasia. 1996;1:353–363. doi: 10.1007/BF02017391. [DOI] [PubMed] [Google Scholar]
- Tang B, Bottinger EP, Jakowlew SB, Bagnall KM, Mariano J, Anver MR, Letterio JJ, Wakefield LM. Transforming growth factor-beta1 is a new form of tumor suppressor with true haploid insufficiency. Nat Med. 1998;4:802–807. doi: 10.1038/nm0798-802. [DOI] [PubMed] [Google Scholar]
- Medina D, Sivaraman L, Hilsenbeck SG, Conneely O, Ginger M, Rosen J, Omalle BW. Mechanisms of hormonal prevention of breast cancer. Ann NY Acad Sci. 2001;952:23–35. doi: 10.1111/j.1749-6632.2001.tb02725.x. [DOI] [PubMed] [Google Scholar]
- Boulanger CA, Wagner KU, Smith GH. Parity-induced mouse mammary epithelial cells are pluripotent, self-renewing and sensitive to TGF-β1 expression. Oncogene. 2005;24:552–560. doi: 10.1038/sj.onc.1208185. [DOI] [PubMed] [Google Scholar]
- Boulanger CA, Smith GH. Reducing mammary cancer risk through premature stem cell senescence. Oncogene. 2001;20:2264–2272. doi: 10.1038/sj.onc.1204312. [DOI] [PubMed] [Google Scholar]
- Grimm SL, Contreras A, Barcellos-Hoff MH, Rosen JM. Cell cycle defects contribute to a block in hormone-induced mammary gland proliferation in CCAAT/enhancer-binding protein (C/EBPβ)-null mice. J Biol Chem. 2005;280:36301–36309. doi: 10.1074/jbc.M508167200. [DOI] [PubMed] [Google Scholar]
- Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- Foster JS, Henley DC, Bukovsky A, Seth P, Wimalasena J. Multi-faceted regulation of cell cycle progression by estrogen: regulation of Cdk inhibitors and Cdc25A independent of cyclin D1-Cdk4 function. Mol Cell Biol. 2001;21:794–810. doi: 10.1128/MCB.21.3.794-810.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WR, Chen CC, Liu S, Safe S. 17β-estradiol (E2) induces cdc25A gene expression in breast cancer cells by genomic and non-genomic pathways. J Cell Biochem. [DOI] [PubMed]
- Ray D, Terao Y, Nimbalkar D, Chu LH, Donzelli M, Tsutsui T, Zou X, Ghosh AK, Varga J, Draetta GF, et al. Transforming growth factor β facilitates β-TrCP-mediated degradation of Cdc25A in a Smad3-dependent manner. Mol Cell Biol. 2005;25:3338–3347. doi: 10.1128/MCB.25.8.3338-3347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkarain A, Jordan R, Slingerland J. p27 deregulation in breast cancer: prognostic significance and implications for therapy. J Mammary Gland Biol Neoplasia. 2004;9:67–80. doi: 10.1023/B:JOMG.0000023589.00994.5e. [DOI] [PubMed] [Google Scholar]