

Abstract
The aim of this study was to test the hypothesis that, within a specific cortical unit, fractional changes in cerebral blood flow (CBF) and cerebral metabolic rate of oxygen consumption (CMRO2) are coupled through an invariant relationship during physiological stimulation. This aim was achieved by simultaneously measuring relative changes in these quantities in human primary visual cortex (V1) during graded stimulation with patterns designed to selectively activate different populations of V1 neurons. Primary visual cortex was delineated individually in each subject by using phase-encoded retinotopic mapping. Flow-sensitive alternating inversion recovery MRI, in conjunction with blood oxygenation-sensitive MRI and hypercapnic calibration, was used to monitor CBF and CMRO2. The stimuli used included (i) diffuse isoluminant chromatic displays; (ii) high spatial-frequency achromatic luminance gratings; and (iii) radial checkerboard patterns containing both color and luminance contrast modulated at different temporal rates. Perfusion responses to each pattern were graded by varying luminance and/or color modulation amplitudes. For all stimulus types, fractional changes in blood flow and oxygen uptake were found to be linearly coupled in a consistent ratio of approximately 2:1. The most potent stimulus produced CBF and CMRO2 increases of 48 ± 5% and 25 ± 4%, respectively, with no evidence of a plateau for oxygen consumption. Estimation of aerobic ATP yields from the observed CMRO2 increases and comparison with the maximum possible anaerobic ATP contribution indicate that elevated energy demands during brain activation are met largely through oxidative metabolism.
The increasing importance in neuroscience of noninvasive functional imaging techniques has spurred intense interest in the metabolic physiology of human brain (1, 2); in particular, lack of a detailed understanding of the relationship between cerebral blood flow (CBF) and cerebral metabolic rate of oxygen consumption (CMRO2) in activated brain has impeded rigorous physiological interpretation of images produced by using methods such as blood oxygenation level-dependent (BOLD) functional MRI (fMRI). Moreover, an improved understanding of cerebral metabolic requirements may aid in management of disorders ranging from stroke to Alzheimer’s disease.
In most individual studies of brain activation physiology conducted previously, responses in specific neuronal systems have been examined under a small number of activation conditions (3–11). The stimuli used have generally been chosen to produce intense activation, thereby ensuring robust imaging signals in what are often low signal-to-noise-ratio experiments. Under such conditions, fractional changes in oxygen consumption have been consistently found to be significantly less than those of blood flow. Fox et al., who additionally measured disproportionately large changes in glucose uptake, suggested that this disparity arose because the activity of aerobic metabolic enzymes reached maximal capacity near baseline levels and the resultant energy shortfalls were therefore met through anaerobic glycolysis (5). Observations of elevated tissue lactate during brain activation were felt to support this hypothesis (12–14), although the duration and magnitude of lactate increase reported, as well as the implied glycolytic rates, have varied (possibly reflecting the different experimental stimuli used). Magistretti and Pellerin have suggested that these large increases in anaerobic glycolysis support conversion of glutamate to glutamine in astrocytes, a step required for recycling of the latter neurotransmitter (15).
An alternate explanation for the observed imbalances between aerobic metabolism and flow, proposed initially by Gjedde et al. (16), is that the diffusibility of oxygen from blood to brain is the predominant factor limiting the rate at which it can used at a given perfusion level. Recently, several quantitative models of oxygen delivery have been introduced (17–20), all predicting that disproportionately large flow increases are required to produce a given CMRO2 increase by increasing blood–brain O2 gradients (although the exact flow–metabolism relationships predicted differ; this is discussed below).
Although disparity between fractional changes in CBF and CMRO2 at a single level of activation is consistent with both of the hypotheses described above (limited enzymatic capacity and diffusion-limited delivery of O2), the two theories predict different relationships between these quantities during graded activation. If aerobic metabolic capacity is limited to near-baseline levels, then CMRO2 should reach an upper limit during moderate stimulation that is not exceeded with increasingly intense input even though perfusion may accelerate further. In this case, the upper limit on CMRO2 observed may depend on the enzymatic properties of the stimulated tissue. On the other hand, if the diffusion-limitation hypothesis is correct, then CMRO2 should rise monotonically with increasing blood flow during normal physiological stimulation.
In addition to the divergent predictions of the enzyme-limitation and diffusion-impedance theories, the different quantitative diffusion models themselves predict distinct relationships between percent changes (Δ%) in CBF and CMRO2. The models of Buxton and Gjedde (17, 18) generally forecast a markedly sublinear dependence of Δ%CMRO2 on Δ%CBF, leading to relatively low CMRO2 increases with a plateau at high perfusion levels. The model of Hyder et al. (19), which incorporates increased capillary O2 permeability during elevated perfusion, is consistent with larger CMRO2 increases and a linear relationship between Δ%CMRO2 and Δ%CBF (although the model does not predict a specific slope). The model of Hudetz (20), which accounts for O2 diffusion gradients along and around capillaries, predicts a linear relationship with 1:1 proportionality between these quantities for CBF increases of up to ≈50% with an abrupt transition to sublinearity at higher perfusion levels.
Selection of the appropriate model from the alternatives described above requires detailed information on the relationship between perfusion and oxygen consumption during graded neuronal activation in well-characterized cortical tissue samples. Because of the technical difficulty of measuring multiple physiological quantities during many activation conditions, however, few individual studies have tracked CBF and CMRO2 across graded activation states under precisely controlled conditions in a single group of subjects. The existing human literature in this area can be divided into three categories: (i) studies that measured both CBF and CMRO2 but only at a single level of stimulation (4, 5, 8–11, 21–30); (ii) studies that measured either CBF or CMRO2 (but not both) during graded stimulation (31–36); and (iii) studies that measured both CBF and CMRO2 during graded stimulation (37). There is currently no published data in the last category for awake humans during physiological stimulation. The reference given describes experiments during graded anesthesia, but the agent administered (cyclopropane) was found to be a potent cerebral vasodilator at high concentration levels (23).
Because of the wide range of methods used and experimental conditions present in the above studies, the cross-study retrospective analysis of response amplitudes is confounded by the virtual impossibility of distinguishing between stimulus-dependent response modulation and random measurement error or method-related bias. The goal of the present study was therefore to determine the steady-state relationship between CBF and CMRO2 in human primary visual cortex (V1) over a broad continuum of flow rates under carefully controlled conditions. These measurements were performed by using a standardized functional MRI protocol, permitting detailed and repeated study of a single group of subjects.
Three of the previous studies cited above used fMRI to estimate CBF and CMRO2 increases at a single level of stimulation. Kim et al., who computed stimulus-induced CMRO2 increases from BOLD and CBF changes by solving an expression derived entirely from estimated literature values for NMR-relevant biophysical parameters, concluded that oxygen consumption did not increase during visual stimulation in spite of a significant (43%) increase in perfusion (8). Davis et al. used a reformulation of the basic BOLD signal model that aggregated all of the influential model parameters into a single constant that could be determined, for specific experimental conditions, in a hypercapnic calibration procedure (10). With this approach, a significant increase in CMRO2 (16%) was detected during visual stimulation, in conjunction with a 45% increase in perfusion. In a more recent study, Schwarzbauer and Heinke measured relatively small CMRO2 changes (4.4%) during visual stimulation, also using a hypercapnic calibration approach. Their BOLD signal model (Eq. 1 in ref. 28) erroneously attributes transverse relaxation entirely to deoxyhemoglobin, however, rendering subsequent transverse relaxation time constant (T2*) ratio calculations questionable. The approach used in the present study is an extension of the method of Davis et al.
METHODS
In this section, we first describe general procedures used in this study, followed by specific methodological information regarding MRI data acquisition, region-of-interest definition, visual stimulation, hypercapnic modulation of CBF, and CMRO2 calculation.
A standardized experimental protocol (Fig. 1), in which BOLD and CBF responses were simultaneously measured and the different stimulation conditions interleaved in random order, was used for all experiments. Data were averaged over multiple sessions (in 12 subjects except where noted) with different randomization orders, and baseline signal levels were sampled before and after each stimulation interval. The response to a given condition, for both CBF and BOLD signals, was defined as the mean value during a stimulation interval minus the average level during the adjacent baseline periods, expressed as a percentage of baseline. To ensure that only physiological steady-state responses were included in percent change calculations, data acquired less than 1 min after changes in stimulation state were excluded [a period based on analysis of response waveforms (38)]. All relative signal changes for all conditions were referenced to the same baseline state, and spatial averages were restricted to V1 according to retinotopic maps (Fig. 2) acquired in preliminary scanning sessions. Subjects gave informed consent, and the experimental protocol was approved by the Research Ethics Committee at the Montreal Neurological Institute.
Figure 1.

Outline of approach used for simultaneous measurement of BOLD and perfusion signals during graded visual stimulation and hypercapnia. (a) Timeline of experimental session, comprising eight scanning runs. Graded stimulus levels, in random order, for different runs are indicated by the variable-height square pulses. (b) Timeline of a single scanning run within a session. A single cycle of stimulation was performed in each run, denoted by the square pulse. Each run resulted in the acquisition of 30 BOLD/perfusion image pairs (rectangular blocks). Image pairs shaded in grey were excluded from calculations of steady-state percent change. (c) Interleaved MRI pulse sequence used to acquire BOLD/perfusion image pairs. Scans 1 and 3 (white blocks) constitute a FAIR acquisition, whereas scans 2 and 4 (grey blocks) are T2*two-weighted (long echo time) acquisitions that are added to produce a single BOLD image overlapping in time with the perfusion image. (d) Examples of perfusion and BOLD images for a single subject acquired simultaneously by using the interleaved pulse sequence.
Figure 2.

Example of resampled retinotopic mapping data, used for restriction of spatial averages, from a single subject. (a) Normalized Fast Fourier Transform modulus map showing retinotopic responses at the fundamental frequency at which an annular visual stimulus was periodically swept in the radial direction during BOLD fMRI scanning (three periods in 6 min). (b) Map of visual field eccentricity in retinotopically organized areas, interpolated onto BOLD image acquired with interleaved sequence in a subsequent session. Only the region receiving input from 5 to 10° (≈green → yellow, inclusive, in eccentricity map) in each subject was included in percent change calculations. (c) Polar angle map for the same subject overlaid on BOLD image. Retinotopic representation of the contralateral visual hemifield within the left and right calcarine sulci was a criterion for V1 identification (image left = subject left).
The following approach was used to detect changes in CMRO2 at different levels of activation: In one set of measurements, we determined the average BOLD MRI signal in peripheral V1 at four different perfusion rates while oxygen consumption was held at a fixed baseline level. This was achieved by using inhalation of different CO2/air mixtures to induce graded hypercapnia in human subjects, resulting in direct vasodilatory stimulation with negligible effect on tissue metabolic rates (39, 40). In a second set of measurements, randomly interleaved with the first, we presented a visual stimulus (a four-cycle/degree black and white drifting squarewave grating) at contrast levels adjusted (based on pilot studies) to match the perfusion increases caused by hypercapnia while measuring the resultant BOLD signals.
The objective of this phase of the study was to provide a direct demonstration that acceleration of aerobic metabolism during brain activation produces a systematic attenuating effect on the BOLD fMRI signal [because of increases in tissue deoxyhemoglobin levels (41–43)]. By comparing BOLD signals at matched perfusion levels, we were able to isolate this effect. Quantification of the corresponding CMRO2 increases was performed by using a biophysical model (10, 44), described below, and plots of relative CMRO2 vs. CBF were generated.
To test the hypothesis that the CMRO2-related attenuating effect should be quantitatively reproducible in experiments by using identical MRI pulse sequences and consistent criteria for region-of-interest identification, we performed four successive replications of the graded visual stimulation procedure described above. Three of these replications were averaged over the same 12 subjects used in the original experiment (i.e., a total of 36 additional experimental sessions), whereas the fourth was performed by repeating the experiment six more times in one of the subjects and averaging the results (extensive averaging was necessitated by the low signal-to-noise ratio of the perfusion measurements). For each replication study, CMRO2 vs. CBF relationships were computed by using the calibration data derived from the original hypercapnia experiment. Assuming that repeated application of an identical graded stimulation protocol to a specific group of subjects should produce reproducible CBF-CMRO2 coupling relationships, statistical agreement of the calculated CMRO2 vs. CBF slopes would support the use of calibration data across sessions.
In addition to the high spatial-frequency grating described above, we induced graded V1 activation (in successive experiments in the same 12 subjects) by using other visual stimuli, including radial checkerboards modulated at different temporal frequencies and diffuse chromatic stimuli. In some cases, the patterns were chosen to produce preferential activation in V1 cell populations with different levels of the aerobic metabolic enzyme cytochrome oxidase (45, 46); in others, stimulus parameters were adjusted to increase blood flow well above the range seen in hypercapnia. Each of these additional stimuli was presented at graded contrast levels (or, in one case, different temporal modulation frequencies), and most were randomly interleaved into a grating replication session (taking the place of the hypercapnia runs in the original experiment). The grating, which was directly calibrated against hypercapnia, thus served as a standard reference to ensure applicability of the hypercapnically derived calibration factor used to compute CMRO2 in these sessions.
MRI Data Acquisition.
The BOLD signal and relative CBF were simultaneously recorded by using an interleaved MRI pulse sequence consisting of a standard flow-sensitive alternating inversion recovery (FAIR) acquisition (47) with a T*2-weighted (BOLD) echo-planar acquisition added after each of the two inversion-recovery acquisitions used in the basic FAIR technique (see Fig. 1c). The inversion time used in the FAIR acquisitions was 900 ms, with an echo time of 20 ms. A longer echo time of 50 ms was used in the T*2-weighted BOLD acquisitions. Single-slice images were acquired on a 64 × 64 matrix with 5 × 5 mm2 in-plane voxel dimensions and 7 mm slice thickness, along an oblique axial plane parallel to the calcarine sulcus. Thickness of the selective inversion slab in the FAIR acquisition was 15 mm, chosen to null difference signals in tissue-equivalent static phantoms (48). Both FAIR and BOLD images used identical echo-planar imaging readouts, resulting in exact spatial correspondence between voxels in the two modalities. Excitation pulses were separated by a 3-s repetition time, and audible gradient activity associated with the inversion prepulses of the FAIR acquisition was duplicated before the BOLD acquisitions, making the different phases of the sequence indistinguishable to the subjects. BOLD contamination of FAIR data and inflow effects in BOLD signals, assessed by examination of nonselective inversion recovery images and comparison of BOLD images after selective vs. nonselective acquisitions, were found to be negligible (38).
Head immobilization was achieved by using an assembly incorporating a bite bar, rigidly mounted ear cups that could be tightly clamped against the head, and a small saddle-shaped fixture pressed firmly into the subject’s nose bridge. Subject motion was negligible by using this apparatus. A receive-only circularly polarized surface coil was built into the head immobilization apparatus, providing high signal-to-noise ratio MRI signals from the occipital lobe. Excitation and Inversion were performed using the system body coil. All experiments were performed on a 1.5 T Siemens (Iselin, NJ) Magnetom Vision MRI system.
Region-of-Interest Definition.
To ensure a homogeneous and uniformly responding tissue sample, all measurements in this study included only tissue in primary visual cortex from 5–10° eccentricity. Maps of eccentricity and polar angle of retinal projection for voxels in topographically organized visual cortex were generated for each subject in separate preliminary fMRI sessions, by using phase-encoded retinotopic mapping (49, 50). These were then interpolated onto the voxel grids used in subsequent experiments according to the computed transformation required to align high-resolution anatomic images acquired at the beginning of each session [by using the package mni_autoreg, available via public file transfer protocol (51)]. Primary visual cortex was defined as being the retinotopically organized region within the left or right calcarine sulcus containing a mirror-image representation of the contralateral visual hemifield. The 5–10° eccentricity range was chosen because it lay within the region stimulated by the test patterns used in our experiments while avoiding sagittal sinus interference and confluence of multiple visual areas that can occur near the fovea. Because all stimuli used in the present study encompassed this portion of the visual field, every voxel within the retinotopically defined region-of-interest was guaranteed to contain activated neurons during stimulation. Also, by using a restricted patch within a larger region of activated cortex, we sought to minimize the possibility that any differences in FAIR or BOLD sensitivity to global vs. focal changes might invalidate our comparison of responses during hypercapnia and visual stimulation. Examples of images produced by using this procedure are shown in Fig. 2. The MRI pulse sequence used for retinotopic mapping was identical to the BOLD phase of the interleaved sequence described above, except that 16 slices with an isotropic resolution of 4 × 4 × 4 mm3 were acquired.
Visual Stimulation.
Visual stimuli were generated in real time by using a Silicon Graphics (Mountain View, CA) O2 computer with OpenGL-based software (glstim, available via public file transfer protocol). The red-green-blue output was used to drive a NEC MT820 LCD projector operating in 640 × 480 mode at 60 Hz. Subjects viewed stimuli projected onto a screen mounted above their heads via a mirror while they lay prone in the scanner. Stimulus presentation was automatically synchronized to data acquisition, and alertness and fixation were continually verified and logged in all subjects by requiring them to report, at 3-s intervals throughout all experiments, the orientation of a small low-contrast triangular fixation marker that was present at the center of the display in a left–right orientation (◂ or ▸) that was varied at random intervals. Feedback was given via an MRI-compatible two-button mouse.
The four-cycle-per-degree black and white squarewave grating stimulus used to match hypercapnia-induced perfusion increases drifted across the visual field at one degree per second at systematically varied orientations. This stimulus has been found, in previous autoradiographic studies (45, 46), to selectively activate interblob regions in primate V1 (particularly in cortex projecting to peripheral visual field, where the spatial frequency of this stimulus relative to receptive field sizes is higher). A red uniform field, changed to isoluminant grey and back at 3 Hz, was used in subsequent experiments to bias activation toward V1 blob domains, which contain higher levels of cytochrome oxidase. Radial checkerboard patterns were used as nonspecific stimuli. These contained both color (yellow/blue) and luminance contrast, with 30 spokes and 6.5 rings (counting from 0.5–10° eccentricity) of equal radial thickness, modulated in a temporal squarewave at various frequencies. The same baseline condition, consisting of a uniform grey field at the mean luminance of the stimulus patterns, was presented at the beginning and end of all scanning runs in this study, as indicated in Fig. 1b. Potency of the various stimuli was varied by changing their luminance contrast and, where applicable, color saturation (by dilution with variable amounts of white). All stimuli converged in appearance to the uniform grey field as contrast/saturation approached zero.
Hypercapnic Modulation of CBF.
We induced graded hypercapnia by administering different concentrations of a CO2/air mixture through a nonrebreathing face mask (Hudson RCI Model 1069, Temecula, CA) worn by subjects. The baseline condition was always inhalation of atmospheric composition medical air ([CO2] < 300 ppm) delivered at 16 L/min while attending to the standard baseline visual display (uniform grey field with attention/fixation task). Hypercapnic episodes were initiated during scanning runs by switching the breathing gas to a mixture of 5:21:74% CO2/O2/N2 (BOC Canada) and medical air. Different levels of hypercapnia (inhaled CO2 concentrations of 1.25%, 2.5%, 3.75%, and 5%) were achieved by combining the premixed CO2/air preparation with medical air in a Y-connector and adjusting respective flow rates to achieve the desired proportions while maintaining a total flow rate of 16 L/min. End-tidal CO2, which was measured via a nasal cannula with monitoring aspirator [Normocap 200, Datex (Oakville, Ontario, Canada)], increased by 5 ± 1 mmHg (1 mmHg = 133 Pa) on average during inhalation of the highest concentration CO2 mixture. Subjects were instructed to breathe at a constant rate, which was easily maintained to within ± one breath per minute. Pulse rate and arterial oxygen saturation were also monitored [Oxygen/Pulse Monitor, Nonin Medical, Plymouth, ME], and these remained constant throughout hypercapnia experiments.
CMRO2 Calculation.
The method of Davis et al. (10) was extended to incorporate our graded hypercapnia measurements and a formalism for interpreting simultaneously acquired BOLD and CBF data in terms of iso-CMRO2 contours in the BOLD–CBF plane. All raw BOLD and perfusion measurements were spatially averaged within peripheral V1 and then pooled across subjects before subsequent display and processing, although averaging of multiple sessions performed on a single subject suggested that individual responses resemble group averages. BOLD vs. perfusion data measured during graded hypercapnia (Fig. 4a) were fit with the function
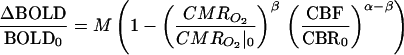 |
1 |
by setting CMRO2/CMRO2|0 to one and adjusting the parameter M (zero subscripts denote baseline values). The value of M, which is specific to a given baseline physiological state and pulse sequence, is equivalent to the fractional BOLD signal increase that would occur if all deoxyhemoglobin were eliminated from the tissue sample. The constants α and β, which respectively reflect the influences of blood volume and deoxyhemoglobin concentration, were assigned values of α = 0.38 and β = 1.5 (10, 52, 53). Iso-CMRO2 contours in the BOLD vs. perfusion plane were then calculated at 10% intervals by using the experimentally determined M value of 0.22 ± 0.03. Solution of the fitted equation for CMRO2/CMRO2|0 permitted calculation of fractional CMRO2 changes from the measured BOLD and perfusion data.
Figure 4.

Coupling relationships between CBF and CMRO2 during graded visual stimulation. (a) Plot of BOLD vs. perfusion increases (± standard error) during different types of stimulation, with iso-CMRO2 contours at 10% intervals. (b) Relative CMRO2 responses to different stimuli, calculated by using the BOLD-CBF data in a. The data reveal a coupling Δ%CBF:Δ%CMRO2 ratio of ≈2:1.
RESULTS
Fig. 3a shows perfusion as a function of time, averaged over twelve subjects, for four levels of hypercapnia (black) and for visual stimulation with the high-spatial frequency squarewave grating (red) at contrast levels adjusted to match the hypercapnia-induced perfusion increases. The corresponding BOLD signals (Fig. 3b) reveal significant attenuation of the visually evoked responses compared with those produced by hypercapnia. The degree of attenuation increased systematically with perfusion, indicating that CBF and CMRO2 underwent coupled increases.
Figure 3.

Perfusion and BOLD signals as a function of time during graded hypercapnia and visual stimulation (n = 12; stimulation intervals indicated by grey background). (a) Relative perfusion as a function of time during graded hypercapnia (black curve) and graded visual stimulation (red curve) with contrasts adjusted to match hypercapnia-induced CBF increases. (b) BOLD signal as a function of time during perfusion increases shown in a. BOLD signals during visual stimulation are significantly lower than those observed during hypercapnia at matched perfusion levels, revealing graded increases in oxygen consumption.
It is unlikely that differences in blood volume during hypercapnia and visual stimulation caused these signal reductions. The BOLD signal depends exclusively on the volume fraction of deoxygenated blood in tissue, which can be increased only by distension of venous vessels. Because available evidence (54–57) indicates that this is a passive biomechanical process, venous blood volume can be considered a simple correlate of perfusion. Matching perfusion levels therefore implies matching of blood volume.
It is also unlikely that shifts in the hemoglobin O2 saturation curve caused by alterations in pCO2 could account for the observed BOLD signal differences. The change in hemoglobin saturation produced by a change in partial pressure of blood carbon dioxide of 5 mmHg (the increase in end-tidal CO2 observed at the highest level of hypercapnia) is likely to be very small, based on published curves (e.g., ref. 58). This is especially true for arterial hemoglobin, because of the shallow slope of the sigmoidal saturation curve for highly oxygenated blood (our pulse oximetry measurements confirmed that there was no change in arterial O2 saturation during hypercapnia). Even if the O2 affinity of venous hemoglobin were decreased significantly because of higher pCO2 during hypercapnia, this would actually lead to underestimation of the CMRO2-related attenuating effect during visual stimulation. Moreover, increases in venous pCO2 during hypercapnia were at least partially balanced by increases in CO2 production because of acceleration of aerobic metabolism during visual stimulation.
The other visual stimuli also produced strong attenuation of the BOLD signal within the range of perfusion levels attained with hypercapnia, and the trend of systematic BOLD attenuation (compared with the extrapolated hypercapnia relationship) was maintained at higher perfusion levels. Fig. 4a shows average increases in the oxygenation-dependent MRI signal plotted as a function of perfusion increase for the different visual stimuli used in this study, as well as hypercapnia. Every combination of BOLD and perfusion values corresponds to a specific rate of oxygen consumption, with the hypercapnia data points tracing out a baseline iso-CMRO2 contour. Nonbaseline contours, calculated by fitting Eq. 1 to the hypercapnia data, are shown at 10% intervals.
Fig. 4b shows percent changes in CMRO2, computed by solving the fitted model function at measured BOLD and CBF values, plotted as a function of Δ%CBF. All visual stimuli produced linearly (r > 0.98) coupled CBF and CMRO2 responses with an average slope of 0.51 ± 0.02. Slopes for the different stimulus types, equivalent to the relative change in oxygen extraction fraction (ΔOEF/OEF0), are listed in Table 1 [see supplementary table on the PNAS web site (www.pnas.org) for percent changes in CBF, BOLD, and CMRO2].
Table 1.
Slopes of Δ%CMRO2 vs. Δ%CBF plots for different stimuli
| Stimulus | Symbol in Fig. 4 | Subjects | Slope |
|---|---|---|---|
| Hypercapnia (four CO2 concentrations) | ● | 12 | 0 |
| High spatial-frequency black/white grating (four contrast levels)* | ◃ | 12 | 0.5 ± 0.1 |
| High spatial-frequency black/white grating (four contrast levels)† | — | 12 | 0.5 ± 0.1 |
| High spatial-frequency black/white grating (four contrast levels)† | — | 12 | 0.4 ± 0.1 |
| High spatial-frequency black/white grating (four contrast levels)† | — | 12 | 0.5 ± 0.1 |
| High spatial-frequency black/white grating (four contrast levels)†‡ | — | 1 | 0.5 ± 0.1 |
| 3-Hz red uniform field to isoluminant grey (four color saturations)§ | ▹ | 12 | 0.51 ± 0.08 |
| 4-Hz yellow/blue radial checkerboard (four low contrast levels)§ | ○ | 12 | 0.52 ± 0.06 |
| 4-Hz yellow/blue radial checkerboard (four high contrast levels)§‡ | ▿ | 1 | 0.52 ± 0.04 |
| 8-Hz yellow/blue radial checkerboard (four intermediate contrast levels)¶ | ▵ | 6 | 0.50 ± 0.02 |
| Yellow/blue radial checkerboard (four frequencies: 2, 4, 6, 8 Hz)¶ | □ | 6 | 0.51 ± 0.08 |
Hypercapnic calibration performed in some session.
Grating replication experiment, not calibrated directly against hypercapnia (data plotted in Fig. 5a).
This experiment was repeated six times in a single subject and averaged.
These experimental sessions also included the high spatial-frequency grating as a reference stimulus.
Stimuli presented in the same session—variable frequency runs were at the highest contrast of variable contrast runs.
Slopes of Δ%CMRO2 vs. Δ%CBF plots for the grating reproducibility tests did not vary significantly from that of the directly calibrated curve (Table 1). This stability supports use of the hypercapnically derived calibration factor M in subsequent experiments, as long as experimental conditions are equivalent and sufficient averaging is performed. Recalculation of Δ%CMRO2 vs. Δ%CBF plots for all stimuli by using M values two standard errors above and below the best-fit value of 0.22 (0.15 and 0.29) did not affect the overall linearity of the relationship and altered the slope only modestly (recalculated slopes were 0.43 ± 0.03 and 0.56 ± 0.01). The effects of variations in β were largely counteracted by β-dependent shifts in M, minimizing sensitivity to uncertainty in β.
At maximal contrast settings, the nonspecific radial checkerboard stimuli generally produced larger responses in V1 than stimuli, such as the red/grey field and high spatial-frequency grating, selective for specific subpopulations of neurons. This is consistent with activation of larger numbers of neurons by the nonspecific patterns. The CMRO2 increase of 25 ± 4% observed during 4 Hz radial checkerboard stimulation at maximum contrast was in agreement with the value of 25 ± 5% measured for identical stimulation at our laboratory by using positron emission tomography (9, 11).
DISCUSSION
During graded physiological stimulation, fractional changes in oxygen consumption and blood flow in V1 were coupled through an invariant linear relationship that did not depend on stimulus type. Interestingly, the invariance of the CMRO2 vs. CBF function held even during presentation of patterns believed to stimulate tissues with different levels of cytochrome oxidase. This consistency, along with the finding of monotonic increases in CMRO2 with perfusion, argues against the enzymatic limitation theory put forward by Fox (5). The CBF/CMRO2 coupling relationship observed is instead consistent with the general notion of diffusion-limited oxygen delivery and supports the quantitative formulation of Hyder et al. (19) as the more realistic of the alternate diffusion models described above. Under the specific condition of very high baseline oxygen extraction fraction (≈60%), the model of Buxton and Frank also predicts results similar to ours (see Fig. 3 in ref. 18).
Computation of putative ATP yields from the observed fractional changes and previously reported absolute baseline measurements of large changes in glucose uptake (CMRGlu) and CMRO2 (37, 59, 60) suggests that, even if fractional changes in CMRGlu and CBF are equal, over 90% of additional ATP produced during brain activation is generated aerobically. This is so because the aerobic ATP yield, per mol of glucose, is 18 times higher than that of anaerobic glycolysis.
In conclusion, we note that our results lend support to the use of BOLD contrast as a generally applicable marker for neuronal activation. One possible outcome of this study was that cytochrome oxidase-dependent variations in CBF/CMRO2 coupling might have led to stimulus-specific transduction of perfusion increases into BOLD signals. Variations of this kind were not observed, however. Regardless of the stimulus type, fractional BOLD signal changes in V1 were consistently proportional to Δ%CBF, which was in turn linearly correlated with Δ%CMRO2 (an index of increased energy utilization). This should be true in general for specific tissue volumes if systemic physiological parameters, such as arterial pCO2, are stabilized. The proportionality constants involved may not be the same for different tissues samples, however, because of regional variations in blood volume and baseline CMRO2. It is hoped that the present study will provide a useful basis for future CMRO2 mapping efforts.
Supplementary Material
Acknowledgments
We thank Pamela Rabbitz for technical assistance. We are also grateful to Curtis Baker, Gareth Barnes, Cathy Bushnell, Ken Kwong, Joe Mandeville, Bruce Rosen, Eric Shoubridge, and Gary Zaharchuk for discussion and critical reading of the manuscript. This work was supported by the Killam Foundation, Le Fonds de la Recherche en Santé du Québec (Canada), National Sciences and Engineering Research Council (Ottawa, Canada), and the Whitaker Foundation (Rosslyn, VA).
ABBREVIATIONS
- BOLD
blood oxygenation level-dependent
- CBF
cerebral blood flow
- CMRO2
cerebral metabolic rate of oxygen consumption
- FAIR
flow-sensitive alternating inversion recovery
- fMRI
functional MRI
- T*2
transverse relaxation time constant
- V1
primary visual cortex
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Barinaga M. Science. 1997;276:196–198. doi: 10.1126/science.276.5310.196. [DOI] [PubMed] [Google Scholar]
- 2.Raichle E. Proc Natl Acad Sci USA. 1998;95:765–772. doi: 10.1073/pnas.95.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brodersen P, Paulson O, Bolwig T. Arch Neurol (Chicago) 1973;28:334–338. doi: 10.1001/archneur.1973.00490230070010. [DOI] [PubMed] [Google Scholar]
- 4.Roland P, Eriksson L, Stone-Elander S, Widen L. J Neurosci. 1987;7(8):2373–2389. [PMC free article] [PubMed] [Google Scholar]
- 5.Fox P T, Raichle M E, Mintun M A, Dence C. Science. 1988;241:462–464. doi: 10.1126/science.3260686. [DOI] [PubMed] [Google Scholar]
- 6.Widén L. In: Brain Work and Energy Metabolism. Lassen N, Ingvar D, Raichle M, Friberg L, editors. Copenhagen: Munskgaard; 1991. pp. 127–139. [Google Scholar]
- 7.Malonek D, Grinvald A. Science. 1996;272:551–554. doi: 10.1126/science.272.5261.551. [DOI] [PubMed] [Google Scholar]
- 8.Kim S-G, Uĝurbil K. Magn Reson Med. 1997;38:59–65. doi: 10.1002/mrm.1910380110. [DOI] [PubMed] [Google Scholar]
- 9.Marrett S, Gjedde A. Adv Exp Med Biol. 1997;413:205–208. doi: 10.1007/978-1-4899-0056-2_22. [DOI] [PubMed] [Google Scholar]
- 10.Davis T, Kwong K, Weisskoff R, Rosen B. Proc Natl Acad Sci USA. 1998;95:1834–1839. doi: 10.1073/pnas.95.4.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vafaee M, Marrett S, Meyer E, Evans A, Gjedde A. Acta Neurol Scand. 1998;98:85–89. doi: 10.1111/j.1600-0404.1998.tb01724.x. [DOI] [PubMed] [Google Scholar]
- 12.Prichard J, Rothman D, Novotny E. Proc Natl Acad Sci USA. 1991;88:5829–5831. doi: 10.1073/pnas.88.13.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sappey-Marinier D, Calabrese G, Fein G, Hugg J, Biggins C, Weiner M. J Cereb Blood Flow Metab. 1992;12:584–592. doi: 10.1038/jcbfm.1992.82. [DOI] [PubMed] [Google Scholar]
- 14.Frahm J, Krüger G, Merboldt K-D, Kleinschmidt A. Magn Reson Med. 1996;35:143–148. doi: 10.1002/mrm.1910350202. [DOI] [PubMed] [Google Scholar]
- 15.Magistretti P, Pellerin L. Cereb Cortex. 1996;6:50–61. doi: 10.1093/cercor/6.1.50. [DOI] [PubMed] [Google Scholar]
- 16.Gjedde A, Ohta S, Kuwabara H, Meyer E. In: Brain Work and Energy Metabolism. Lassen N, Ingvar D, Raichle M, Friberg L, editors. Copenhagen: Munskgaard; 1991. pp. 177–184. [Google Scholar]
- 17.Gjedde A. In: Cerebrovascular Disease. Bajter H, Caplan L, editors. Philadelphia: Lippincott-Raven; 1997. pp. 23–40. [Google Scholar]
- 18.Buxton R B, Frank L R. J Cereb Blood Flow Metab. Jan 1997;17(1):64–72. doi: 10.1097/00004647-199701000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Hyder F, Shulman R, Rothman D. J Appl Physiol. 1998;85:554–564. doi: 10.1152/jappl.1998.85.2.554. [DOI] [PubMed] [Google Scholar]
- 20.Hudetz A. Brain Res. 1999;817:75–83. doi: 10.1016/s0006-8993(98)01200-1. [DOI] [PubMed] [Google Scholar]
- 21.Kety S, Woodford R, Harmel M, Freyhan F, Appel K, Schmidt C. Am J Psychiatry. 1947;104:765–770. [Google Scholar]
- 22.Pierce E, Lambersten C, Deutsch S, Chase P, Linde H, Dripps R, Price H. J Clin Invest. 1962;41:1664–1671. doi: 10.1172/JCI104623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith A, Wollman H. Anesthesiology. 1972;36:378–400. doi: 10.1097/00000542-197204000-00015. [DOI] [PubMed] [Google Scholar]
- 24.Fox P T, Raichle M E. Proc Natl Acad Sci USA. Feb 1986;83(4):1140–1144. doi: 10.1073/pnas.83.4.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seitz R, Roland P. Acta Neurol Scand. 1992;86:60–67. doi: 10.1111/j.1600-0404.1992.tb08055.x. [DOI] [PubMed] [Google Scholar]
- 26.Marrett S, Fujita H, Meyer E, Ribeiro L, Evans A, Kuwabara H, Gjedde A. In: Quantification of Brain Function. Uemura K, Lassen N, Jones T, Kanno I, editors. Amsterdam: Elsevier; 1993. pp. 217–224. [Google Scholar]
- 27.Ribeiro L, Kuwabara H, Meyer E, Fujita H, Marrett S, Evans A, Gjedde A. In: Quantification of Brain Function. Uemura K, Lassen N, Jones T, Kanno I, editors. Amsterdam: Elsevier; 1993. pp. 229–236. [Google Scholar]
- 28.Schwarzbauer C, Heinke W. Magn Reson Med. 1999;41:537–543. doi: 10.1002/(sici)1522-2594(199903)41:3<537::aid-mrm16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 29.Ohta S, Reutens D, Gjedde A. J Cereb Blood Flow Metab. 1999;19:260–265. doi: 10.1097/00004647-199903000-00003. [DOI] [PubMed] [Google Scholar]
- 30.Fujita H, Kuwabara H, Reutens D, Gjedde A. J Cereb Blood Flow Metab. 1999;19:266–271. doi: 10.1097/00004647-199903000-00004. [DOI] [PubMed] [Google Scholar]
- 31.Fox P, Raichle M. Ann Neurol. 1985;17(3):303–305. doi: 10.1002/ana.410170315. [DOI] [PubMed] [Google Scholar]
- 32.Sadato N, Ibanez V, Deiber M-P, Campbell G, Leonardo M, Hallett M. J Cereb Blood Flow Metab. 1996;16:23–33. doi: 10.1097/00004647-199601000-00003. [DOI] [PubMed] [Google Scholar]
- 33.Blinkenberg M, Bonde C, Holm S, Svarer C, Andersen J, Paulson O, Law I. J Cereb Blood Flow Metab. 1996;16:794–803. doi: 10.1097/00004647-199609000-00004. [DOI] [PubMed] [Google Scholar]
- 34.Rees G, Howseman A, Josephs O, Frith C, Friston K, Frackowiak R, Turner R. Neuroimage. 1997;6(4):270–278. doi: 10.1006/nimg.1997.0300. [DOI] [PubMed] [Google Scholar]
- 35.Zhu X, Kim S, Andersen P, Ogawa S, Uĝurbil K, Chen W. Magn Reson Med. 1998;40(5):703–711. doi: 10.1002/mrm.1910400510. [DOI] [PubMed] [Google Scholar]
- 36.Vafaee M, Meyer E, Marrett S, Paus T, Evans A, Gjedde A. J Cereb Blood Flow Metab. 1999;19:272–277. doi: 10.1097/00004647-199903000-00005. [DOI] [PubMed] [Google Scholar]
- 37.Alexander S, Colton E, Smith A, Wollman H. Anesthesiology. 1970;32(3):236–245. doi: 10.1097/00000542-197003000-00013. [DOI] [PubMed] [Google Scholar]
- 38.Hoge R, Atkinson J, Gill B, Crelier G, Marrett S, Pike G. Neuroimage. 1999;9:573–585. doi: 10.1006/nimg.1999.0443. [DOI] [PubMed] [Google Scholar]
- 39.Kety S, Schmidt C. J Clin Invest. 1948;27:484–492. doi: 10.1172/JCI101995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang S-P, Krasney J. J Cereb Blood Flow Metab. 1995;15:115–123. doi: 10.1038/jcbfm.1995.13. [DOI] [PubMed] [Google Scholar]
- 41.Ogawa S, Tank D W, Menon R, Ellermann J M, Kim S G, Merkle H, Uĝurbil K. Proc Natl Acad Sci USA. 1992;89:5951–5955. doi: 10.1073/pnas.89.13.5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jezzard P, Heineman F, Taylor J, DesPres D, Wen H, Balaban R, Turner R. NMR Biomed. 1994;7:35–44. doi: 10.1002/nbm.1940070107. [DOI] [PubMed] [Google Scholar]
- 43.Punwami S, Ordidge R J, Cooper C, Amess P, Clemence M. NMR Biomed. 1998;11:281–289. doi: 10.1002/(sici)1099-1492(199810)11:6<281::aid-nbm529>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 44.Ogawa S, Menon R S, Tank D W, Kim S G, Merkle H, Ellermann J M, Uĝurbil K. Biophys J. 1993;64(3):803–812. doi: 10.1016/S0006-3495(93)81441-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tootell R, Silverman M, Hamilton S, De Valois R, Switkes E. J Neurosci. 1988;8(5):1569–1593. doi: 10.1523/JNEUROSCI.08-05-01569.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tootell R, Silverman M, Hamilton S, De Valois R, Switkes E. J Neurosci. 1988;8(5):1610–1624. doi: 10.1523/JNEUROSCI.08-05-01610.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim S G. Magn Reson Med. 1995;34(3):293–301. doi: 10.1002/mrm.1910340303. [DOI] [PubMed] [Google Scholar]
- 48.Crelier G R, Hoge R D, Munger P, Pike G B. Magn Reson Med. 1999;41(1):132–136. doi: 10.1002/(sici)1522-2594(199901)41:1<132::aid-mrm18>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 49.Sereno M, Dale A, Reppas J, Kwong K, Belliveau J, Brady T, Rosen B, Tootell R. Science. 1995;268:889–893. doi: 10.1126/science.7754376. [DOI] [PubMed] [Google Scholar]
- 50.Tootell R B, Mendola J, Hadjikhani N, Ledden P, Liu A, Reppas J, Sereno M, Dale A. J Neurosci. 1997;17(18):7060–7078. doi: 10.1523/JNEUROSCI.17-18-07060.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Collins D L, Neelin P, Peters T M, Evans A C. J Comput Assist Tomogr. 1994;18(2):192–205. [PubMed] [Google Scholar]
- 52.Grubb R, Phelps M, Eichling J. Stroke (Dallas) 1974;5:630–639. doi: 10.1161/01.str.5.5.630. [DOI] [PubMed] [Google Scholar]
- 53.Boxerman J L, Bandettini P A, Kwong K K, Baker J R, Davis T L, Rosen B R, Weisskoff R M. Magn Reson Med. 1995;34(1):4–10. doi: 10.1002/mrm.1910340103. [DOI] [PubMed] [Google Scholar]
- 54.Orlov R, Isakov B, Ketkin A, Plekhanov I. Brain Blood Supply. Budapest: Akadémiai Kiadó; 1977. [Google Scholar]
- 55.Rovainen C, Woolsey T, Blocher N, Wang D-B, Robinson O. J Cereb Blood Flow Metab. 1993;13:359–371. doi: 10.1038/jcbfm.1993.49. [DOI] [PubMed] [Google Scholar]
- 56.van Zijl P, Eleff S, Ulatowski J, Oja J, Uluĝ A, Traystman R, Kauppinen R. Nat Med. 1998;4(2):159–167. doi: 10.1038/nm0298-159. [DOI] [PubMed] [Google Scholar]
- 57.Estrada C, DeFilipe J. Cereb Cortex. 1998;8:1047–3211. doi: 10.1093/cercor/8.3.193. [DOI] [PubMed] [Google Scholar]
- 58.Weibel E. The Pathway for Oxygen: Structure and Function in the Mammalian Respiratory System. Cambridge, MA: Harvard Univ. Press; 1984. [Google Scholar]
- 59.Sokoloff L. In: Handbook of Physiology-Neurophysiology. Field J, Magoun H, Hall V, editors. Washington, DC: Am. Physiol. Soc.; 1960. pp. 1843–1864. [Google Scholar]
- 60.Madsen P, Hasselbalch S, Hagemann L, Olsen K, Bülow J, Holm S, Wildschiødtz G, Paulson O, Lassen N. J Cereb Blood Flow Metab. 1995;15:485–491. doi: 10.1038/jcbfm.1995.60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.