

Abstract
Cavities and clefts are frequently important sites of interaction between natural enzymes or receptors with their corresponding substrate or ligand molecules and exemplify the types of molecular surfaces that would facilitate engineering artificial catalysts and receptors. Even so, structural characterizations of designed cavities are rare. To address this issue, we performed a systematic study of the structural effects of single amino acid substitutions within the hydrophobic cores of tetrameric coiled-coil peptides. Peptides containing single glycine, serine, alanine, or threonine amino acid substitutions at the buried L9, L16, L23, and I26 hydrophobic core positions of a GCN4-based sequence were synthesized and studied by solution-phase and crystallographic techniques. All peptides adopt the expected tetrameric state and contain tunnels or internal cavities ranging in size from 80 Å3 to 370 Å3. Two closely-related sequences containing an L16G substitution, one of which adopts an antiparallel configuration and one of which adopts a parallel configuration, illustrate that cavities of different volumes and shapes can be engineered from identical core substitutions. Finally, we demonstrate that two of the peptides (L9G and L9A) bind the small molecule iodobenzene when present during crystallization, leaving the general peptide quaternary structure intact but altering the local peptide conformation and certain superhelical parameters. These high-resolution descriptions of varied molecular surfaces within solvent-occluded internal cavities illustrate the breadth of design space available in even closely-related peptides and offer valuable models for the engineering of de novo helical proteins.
A significant challenge frustrating the de novo design of functional molecules is that of precisely constructing molecular surfaces able to suitably participate in desired molecular recognition events. Internal cavities, relatively buried clefts, and tunnels are frequently important sites of interaction between natural enzymes or receptors with their corresponding substrate or ligand molecules, and the ability to engineer such surfaces would facilitate the successful design of highly-selective artificial catalysts and receptors. Mutagenesis of natural proteins to create or alter internal cavities has been used for such purposes as redesigning enzyme specificities, creating novel binding sites, and studying protein stability (1–12), but despite the diverse roles of natural cavities in biology, high-resolution structural investigations of model cavities in the aqueous milieu are rare. We believe studying designed cavities within synthetic peptides may provide models for understanding natural binding sites as well as designing novel receptors or biocatalysts.
The most-studied motif in de novo protein engineering is the coiled-coil (13), a naturally-derived peptide assembly composed of two or more α-helices wrapped around each other to bury a hydrophobic core. The homotetrameric GCN4-pLI coiled-coil (14) is an attractive scaffold for engineering cavities because a) it is amenable to backbone and amino acid substitution without significant perturbation of the overall structure (15, 16), presumably because it is one of the most stable reported GCN4-derived peptides; b) its crystal structure is known (14), allowing structure-based design of internal cavities; and c) the interhelical distance is greater in the tetramer than in other naturally-derived dimeric or trimeric coiled-coils (14), allowing the formation of large internal cavities.
Most previous work involving hydrophobic core substitutions in coiled-coil peptides has focused on the structural effects of buried polar residues (especially Asn and Gln) in specifying oligomerization state or strand orientation (17, 18), although several reports of observed or designed cavities have also been made. Benzene binding in the designed cavity of a coiled-coil trimer was found to control the peptide’s configuration (19, 20), and internal cavities were more recently described in the crystal structures of peptides containing Ser or Thr core substitutions (21). Apart from these examples, however, engineered cavities in coiled-coil systems have gone without high-resolution characterization. A template-assembled trimeric bundle containing Leu→Ala core substitutions was shown to bind a fluorescent ligand (22), and similarly tri- and tetrameric bundles containing core substitutions have been show to bind a volatile anaesthetic (23–26). The avoidance of cavity formation, or steric matching, has recently been described as a means of controlling quaternary structure in trimeric peptide assemblies (27, 28), whereas pairs of Ala core substitutions have been shown capable of controlling the parallel/antiparallel configuration and the aggregation state of coiled-coil peptides (29, 30), reportedly due to thermodynamic preferences for avoiding large internal cavities. These several examples illustrate not only the utility of cavities in protein engineering, but also the need for structural characterizations of such surfaces to more fully understand the means by which they alter protein folding and function.
We initially set out to perform a systematic study of the structural effects of single amino acid substitutions within the hydrophobic core of parallel coiled-coil tetramers, with the specific goal of generating discrete internal cavities by replacing bulky hydrophobic residues at core a and d positions with smaller amino acids (Figure 1). After subsequently coming to appreciate the plasticity of the scaffold to amino acid substitution, and considering the recently reported structural characterization of antiparallel tetrameric coiled-coils (31), we expanded our initial goal to include investigation of internal cavities within peptides likely to adopt the antiparallel configuration. Along with solution-phase biophysical data, we report here crystal structures of one antiparallel and 11 parallel GCN4-pLI variants containing tunnels or internal cavities ranging from 80 Å3 to 370 Å3 (total volume occupied by a probe of radius 1.4 Å), a subset of which (L9G and L9A) bind iodobenzene in the crystalline state with conformational changes reminiscent of induced fit binding events in natural receptors. These high-resolution snapshots of varied molecular surfaces in peptides adopting nearly identical quaternary structures illustrate the wide breadth of design space available in even closely-related peptides and may offer valuable models for the engineering of de novo helical proteins.
Figure 1.
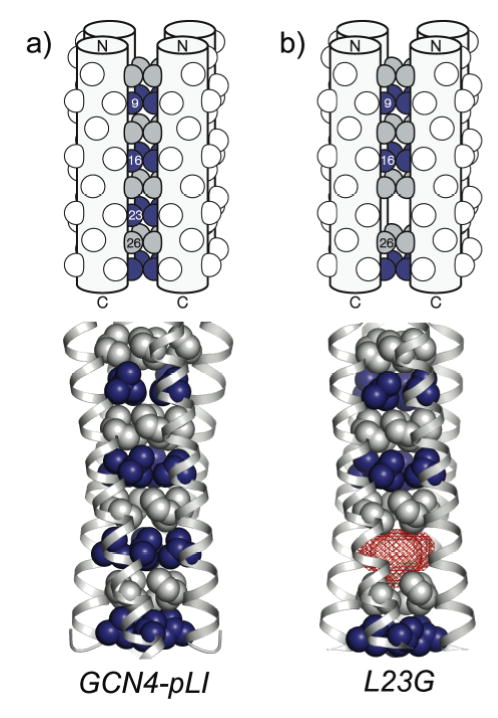
Engineering internal cavities in tetrameric coiled-coils. Upon assembly of the noncovalent tetramer, the spatial localization of one large→small amino acid substitution from each strand creates a buried cavity. Representative schematic diagrams (top) and crystal structures (bottom) are shown for a) GCN4-pLI and b) the L23G variant, emphasizing hydrophobic core a position Leu (blue) and d position Ile (gray) residues. The sites of large-to-small core substitutions described in this paper are numbered in the schematic diagrams. In the L23G crystal structure, the 280 Å3 cavity (probe-occupied volume using a probe of radius 1.4 Å) is colored red.
Materials and Methods
Peptide sequence and nomenclature
The sequences of the GCN4-pLI variants described herein are listed in Table 1. The peptides are named by their core substitutions (for example, the variant containing the Leu→Ala substitution at position 9 is termed L9A), with the two peptides containing the L16G substitution named L16G E20C and L16G E20C Y17H, denoting all substitutions from GCN4-pLI.
Table 1.
Peptide sequences and nomenclature.
| Peptide | Sequence |
|---|---|
| GCN4-pLI | Ac-R-MKQIEDK-LEEILSK-LYHIENE-LARIKKL-LGER-OH |
| L9G | Ac-R-MKQIEDK-GEEILSK-LYHIENE-LARIKKL-LGER-OH |
| L9S | Ac-R-MKQIEDK-SEEILSK-LYHIENE-LARIKKL-LGER-OH |
| L9A | Ac-R-MKQIEDK-AEEILSK-LYHIENE-LARIKKL-LGER-OH |
| L9T | Ac-R-MKQIEDK-TEEILSK-LYHIENE-LARIKKL-LGER-OH |
| L16G | Ac-R-MKQIEDK-LEEILSK-GYHICNE-LARIKKL-LGER-OH |
| E20C | |
| L16G | ABAa-R-MKQIEDK-LEEILSK-GHHICNE-LARIKKL-LGER-OH |
| E20C | |
| Y17H | |
| L23G | Ac-R-MKQIEDK-LEEILSK-LYHIENE-GARIKKL-LGER-OH |
| L23S | Ac-R-MKQIEDK-LEEILSK-LYHIENE-SARIKKL-LGER-OH |
| L23A | Ac-R-MKQIEDK-LEEILSK-LYHIENE-AARIKKL-LGER-OH |
| L23T | Ac-R-MKQIEDK-LEEILSK-LYHIENE-TARIKKL-LGER-OH |
| I26G | Ac-R-MKQIEDK-LEEILSK-LYHIENE-LARGKKL-LGER-OH |
| I26S | Ac-R-MKQIEDK-LEEILSK-LYHIENE-LARSKKL-LGER-OH |
| I26A | Ac-R-MKQIEDK-LEEILSK-LYHIENE-LARAKKL-LGER-OH |
| I26T | Ac-R-MKQIEDK-LEEILSK-LYHIENE-LARTKKL-LGER-OH |
ABA = acetamidobenzoic acid
Peptide synthesis
Peptide synthesis was typically performed on Fmoc-Arg(Pbf)-Wang resin using an Advanced ChemTech 348Ω automated synthesizer, with diisopropyl carbodiimide (DIC)/N-hydroxy benzotriazole (HOBt) in N-methyl pyrrolidinone (NMP) for couplings, and 30% piperidine in NMP for deprotection. N-termini of peptides were acetylated with solutions of 2:2:1 NMP/acetic anhydride/N,N-diisopropylethylamine (DIEA). Side chain protecting groups were Arg(Pbf), Ser(tBu), Lys(Boc), Glu(tBu), Asp(tBu), Tyr(tBu), Gln(Trt), Asn(Trt), Cys(Trt), and His(Trt). Cleavage of peptides was typically effected with a mixture of 94:2.5:2.5:1 TFA/ethanedithiol/water/triisopropylsilane for 4 hours. Peptides were precipitated with ether (50 mL), centrifuged, and washed with ether. Following drying in vacuo, peptides were purified on a C18 column (Vydac 218TP) eluting with a water/acetonitrile/TFA gradient, and lyophilized. Molecular weights of peptides were verified using a PerSeptive Biosystems Voyager DE MALDI-TOF mass spectrometer.
Circular Dichroism Spectroscopy
Peptide stock solutions in water for CD spectroscopy were prepared at a concentration of 20 mg/mL and standardized by measurement of tyrosine absorbance. Typically, a 10 μL aliquot of stock solution was diluted with 1.0 mL of buffer (50 mM phosphate, 150 mM NaCl, pH 7.0) and absorbance was measured at 280 nm. Measurements were made in triplicate. The concentration was determined assuming ε280nm = 1280 m−1cm−1. Peptide stock solutions were stored at −80 °C, and diluted to an appropriate concentration with buffer or water before use. CD thermal melts were recorded in stirred 0.5 cm path length cuvettes with 10 μM peptide in buffer (50 mM phosphate, 150 mM NaCl, pH 7.0, 1 M guanidine hydrochloride). Added dithiothreitol (0.5 mM) was included as reducing agent for the peptides containing the E20C substitution. The temperature was increased in 2 °C intervals, with an equilibration time of 90 sec before recording the CD signal at 222 nm. The CD signal vs. temperature was fit to a fourth order polynomial over the melting transition, and the melting temperature (Tm) was calculated as the point at which the second derivative of this function was zero.
Size exclusion chromatography
Peptide stock solutions were prepared as described above for CD experiments. Size exclusion chromatography (SEC) was performed at room temprature on a Superdex 75 10/30 column eluted with buffer (50 mM phosphate, 150 mM NaCl, pH 7.0) at a flow rate of 0.5 mL/min. Samples for calibration were bovine serum albumin (2.5 mg/mL, MW 67000), bovine erythrocyte carbonic anhydrase (2 mg/mL, MW 29000), horse skeletal muscle myoglobin (1 mg/mL, MW 17600), horse heart cytochrome C (1 mg/mL, MW 12384), aprotinin (2 mg/mL, MW 6500), bovine oxidized insulin B chain (2 mg/mL, MW 3496) and cyanocobalamin (1 mg/mL, MW 1355). 20 μL aliquots of each of the calibrant solutions were mixed and injected. Elution volume was plotted against MW and fit using a nonlinear regression with the program Mathematica. The peptide stock solutions in water were diluted with elution buffer to final concentrations of 250 μM, of which 50 μL was injected. Monitoring was carried out by ultraviolet (UV) absorption at 214 nm. Apparent aggregation states (Nagg) were calculated from the elution volume by first using the calibration plot to determine the apparent MW of the bundle, and then dividing this value by the calculated MW of an individual peptide. A control peptide reported to form a coiled-coil trimer in solution (14) had an observed Nagg of 3.2.
Crystallization and Data Collection
Peptide stock solutions for crystal growth were prepared at a concentration of approximately 20 mg/mL in 50 mM Tris HCl buffer (pH 7.0 prior to addition of peptide) or in water alone. Stock solutions were filtered through a 0.22 μm membrane filter before use. The final concentration as measured by UV was typically about 2 mM. Crystal screens were performed using Crystal Screen 2, Cryo Screen (Hampton Research), or Wizard II (Emerald Biostructures) kits. Hanging drop volumes of 1 – 2 μL of peptide stock with 0.1–1 μL of added buffer were used with buffer reservoir volumes of 0.5 mL–1 mL. Peptide L16G E20C Y17H was crystallized by screening using an Innovadyne spotting robot, employed due to the difficulty in obtaining crystals for this peptide. Crystallization buffers consisted of 10% w/v PEG 6000, 2 M NaCl, no cryoprotectant (peptides L9G, L9S, L9A, L9T, L9A+IB, L9G+IB, L16G E20C); 10% w/v PEG 8000, 100 mM Tris, pH 7.0, 200 mM MgCl2, with glycerol and ethylene glycol as cryoprotectant (peptide L23G); 2.5 M NaCl, 100 mM NaOAc, pH 4.5, 200 mM Li2SO4, with glycerol and ethylene glycol as cryoprotectant (peptides GCN4-pLI, L23S, I26G, I26A, I26S, I26T); and 20% PEG 3350, 200 mM KSCN (peptide L16G E20C Y17H). We subsequently found that the 10% w/v PEG 6000, 2 M NaCl buffer was sufficient for all peptides crystallized in the P4132 lattice. The crystals from this condition displayed characteristic cubic or rhombododechedral shapes, and exhibited no birefringence (characteristic of a cubic unit cell). The antiparallel peptide (L16G E20C Y17H) crystallized in the trigonal P31 spacegroup.
Crystals were mounted on nylon loops, immersed in the cryoprotectant, and frozen at ~100 K in the diffractometer cryostream. Data for L9G and L9G+IB crystals were collected at Stanford Sychrotron Radiation Laboratory (SSRL), and L16G E20C Y17H data was collected at Advanced Photon Source (APS) beamline GM/CA-CAT. Data for the remaining crystals were collected using MSC R-Axis IV or Mar345 image plate detectors (wavelength 1.54 Å, I(f',f'')=(−0.31,6.90)). Integration and scaling were carried out with the CrystalClear software (MSC) (32), Mosflm (33) and HKL2000 (34), and the CCP4 suite of programs (35, 36). The initial structure determination was performed on a small cubic crystal (0.15 mm on an edge) of the L9G variant using experimental phases obtained from single isomorphous replacement (SIR) heavy atom phasing of xenon diffused into the crystal. The data were collected at SSRL beam line 9-1 (wavelength 0.97 Å) using the SSRL pressure cell to pressurize the crystal for 3 min. at 300 psi of xenon (37). A single ordered Xe atom was found to occupy the small cavity at the I26 position, and not the large cavity at the L9G site. This structure was subsequently used as an initial model to solve the remaining parallel structures via molecular replacement using Molrep (35). The structure of L16G E20C Y17H was determined with Phaser (38) using a truncated (residues 5–30) single strand from L16G E20C as a model for molecular replacement. Structural refinements were carried out using Refmac5 (35) and XtalView (39).
We took care to ensure that our spacegroup assignment for the cubic crystals was correct. After scaling and averaging complete data in the maximal non-isomorphic subgroup P213 and performing molecular replacement and refinement, we did not observe improvement in the R and Rfree (while doubling the number of fitted parameters). The iodine anomalous difference maps for the L9G+IB and L9A+IB complexes independently demonstrated two-fold symmetry when the 4n for all structures also data were processed in P213. The systematic absences at h00=0k0=00l≠support the P4132 assignment. The first three N-terminal and the last three C-terminal residues generally provided very weak electron density and proved difficult to fit in most structures. Residues that could not be fit (either manually or using ARP/warp (40)), were omitted from the structure, which is reflected in the higher Rfree values in structures such as I26G. All peptides exhibit higher B factors towards the ends of the chain and lower B factors for central residues, particularly those in the hydrophobic core, which is likely because both termini of the tetramer are effectively free of crystal contacts in the P4132 lattice.
Iodobenzene complexes were prepared by crystallizing peptides L9G and L9A placing 2 μL of iodobenzene liquid (Aldrich) on top of the aqueous drop. Reflection data for L9G+IB were collected at SSRL beam line 11-1 (wavelength 0.82 Å, I(f',f'')=(−0.22,2.34)). A highly redundant data set was collected for L9G+IB (20-fold) in order to ensure observation of the anomalous signal from the iodine atom in iodobenzene. The L9A+IB crystal data set was not collected to such a high redundancy as L9G+IB due to the stronger anomalous scattering factor at the Cu-Kα wavelength.
Superhelical parameters and cavity volumes were determined using the programs FITCC (Mark Sales, personal communication; http://ucxray.berkeley.edu/~mark/fitcc.html) and VOIDOO (41), respectively. Based on the 2.0 Å resolution data obtained for most crystals, we estimate errors in atomic positions of the refined structures to be ±0.2 Å, and the resulting errors for cavity volumes to be ~15 %. The volume of iodobenzene was also calculated using VOIDOO. Root mean square (rms) deviations were calculated using the McLachlan algorithm (42) as implemented in the program ProFit (Andrew C. R. Martin, http://www.rfcgr.mrc.ac.uk/Registered/Help/profit/).
Results and Discussion
By replacing bulky hydrophobic residues with smaller residues in the buried core of a homotetrameric coiled-coil, we hoped to create internal cavities (Figure 1). Terminal hydrophobic core a (sites 9 and 23) and d (site 26) heptad positions of the 33-residue GCN4-pLI were initially chosen as amino acid substitution sites, since we thought that making substitutions near the termini would likely leave much of the peptide’s association energy intact and drive folding of the desired parallel tetrameric state. Variants containing single Gly, Ala, Ser, or Thr substitutions (chosen to sample a range of side chain sizes and polarities) at each of these three sites were thus prepared. The study was later expanded with two additional peptides, both containing an E20C substitution (which has recently been shown to switch the crystal structure of GCN4-pLI from parallel to antiparallel (31)) and an L16G substitution, to characterize internal cavities within tetramers adopting an antiparallel crystallographic configuration. In an antiparallel homotetramer, a large single cavity can only be created at the center of peptide (position 16), since the more terminal regions of the assembly juxtapose two N-terminal residues and two C-terminal residues. All peptides are named for their respective substitution, and sequences are listed in Table 1.
Solution Studies
Each peptide, with the exceptions of I26G and I26S, exhibited the expected tetrameric aggregation state at peptide concentrations of 250 μM in neutral phosphate-buffered aqueous solutions, and all peptides except L16G E20C Y17H eluted as single, sharp peaks from the size exclusion column, discrediting the possibility of multiple aggregation states in solution (Table 2). We attribute the larger Nagg of I26G and I26S to C-terminal fraying which increases the apparent size of the aggregate. This explanation is supported by the crystallographic observation of C-terminal fray and disorder (described below) and is probably most pronounced at the I26 position since it is the most terminal of the four substitution sites. The L16G E20C Y17H peptide is predominantly tetrameric in solution, but a small proportion (roughly 20%) elutes with an Nagg of 3.0. This result demonstrates that hydrophobic core substitutions near the center of the assembly are more structurally perturbative than substitutions at the helix termini.
Table 2.
Apparent aggregation states, thermal denaturation values, and probe-occupied cavity volumes for GCN4-pLI and variants, along with surface areas and volumes for relevant amino acids.
| Cavity volume (Å3)e | Cavity volume (Å3)e | ||||||
|---|---|---|---|---|---|---|---|
| Peptide | Nagga | Tm (°C)c | 1.4 Å probe radius | 1.8 Å probe radius | Amino acid | Side chain nonpolar surface area (43) (Å2) | Side chain volume (37) (Å3) |
| GCN4-pLI | 4.3 | 94d | 20–40 | 0 | Gly | 47 | ~ |
| L9G | 4.1 | 64 | tunnelf | 220 | Ala | 86 | 26.3 |
| L9G+IB | ~ | ~ | tunnelf | 240 | Ser | 56 | 30.4 |
| L9S | 4.1 | 68 | 100 | 80 | Thr | 90 | 56.2 |
| L9A | 4.0 | 70 | 220 | 190 | Ile | 155 | 101.1 |
| L9A+IB | ~ | ~ | tunnelf | 210 | Leu | 164 | 100.8 |
| L9T | 4.0 | 95 | ~g | ~g | |||
| L16G E20C | 3.9 | 37 | 330 | 260 | |||
| L16G E20C Y17H | 3.7b | 37 | 370 | 300 | |||
| L23G | 3.8 | 51 | 280 | 230 | |||
| L23S | 4.0 | 66 | 150 | 120 | |||
| L23A | 4.1 | 80 | ~ h | ~h | |||
| L23T | 4.0 | 84 | ~ h | ~ h | |||
| I26G | 5.0 | 75 | ~g | ~g | |||
| I26S | 4.7 | 78 | tunnelf | 520 | |||
| I26A | 4.3 | 88 | 200 | 110 | |||
| I26T | 4.2 | 87 | 80 | 0 |
Apparent aggregation number as measured by SEC. Estimated errors are ± 0.1.
~20% of the sample eluted with an Nagg of 3.0.
Thermal denaturation curves determined by CD in neutral solutions containing 1 M guanidinium hydrochloride. Estimated errors are ± 2 °C.
Performed in 3 M guanidinium hydrochloride.
Based on the reported high resolution values of ~2.0 Å, we estimate errors in cavity volume to be ~15%.
The cavity was not enclosed on all sides, creating a tunnel through the peptide assembly.
Electron density could not be modeled beyond the site of the cavity, preventing determination of the cavity volume.
Crystals were not obtained for this peptide.
All peptides displayed circular dichroism (CD) spectra typical of α-helical coiled-coils with minima at 222 nm and 208 nm. As shown in Table 2, thermal stabilities of the pLI variants (10 μM peptide, pH 7.0 phosphate buffer containing 1 M guanidine hydrochloride) generally correlate with the non-polar surface area of the substituted residue (43), where the more stable assemblies are those containing the more non-polar amino acids (Table 2). An important consequence of this stability profile is that peptides containing larger cavities created by an Ala core substitution are more stable than the peptides containing smaller cavities created by Ser substitutions (see below for a discussion of cavity size). The core residues nearer to the center of the assembly contribute more to the thermal stability of the bundle, accounting for the lower melting temperatures of the variants containing the substitution in the central L16 position relative to peptides with more terminal substitutions.
Crystal Structures
Here we describe the crystal structure of GCN4-pLI in a different spacegroup than originally reported (14) (Figure 1a), and report structures of 11 peptides in the parallel configuration and one peptide in the antiparallel configuration containing hydrophobic core substitutions (Figure 2, Table 3). All crystals in which peptides adopted the parallel configuration were processed in the P4132 spacegroup, where the four-helix bundle is generated by a crystallographic two-fold symmetry axis acting on the two-strand asymmetric unit, so that 12 four-helix bundles comprise the unit cell contents. Peptide L16G E20C Y17H was processed in the P31 spacegroup, in which the asymmetric unit is an all-antiparallel coiled-coil tetramer. The observation of an antiparallel configuration for this peptide, which contains an E20C substitution, is consistent with our recent report describing crystallization of several pLI variants containing the E20C substitution in the antiparallel assembly (31). Somewhat unexpectedly, peptide L16G E20C adopted the parallel configuration and crystallized in the P4132 spacegroup. It is unlikely that the N-terminal acetamidobenzoic acid (ABA) moiety causes the change in configuration, since we have determined both parallel and antiparallel structures of GCN4-pLI variants containing this N-terminal group (31). Computational studies are underway to address these observed changes in configuration. Nonetheless, the two closely-related peptides, both containing L16G substitutions but adopting different quaternary structures, illustrate that at least two cavities of different shapes and sizes can be engineered from identical core substitutions.
Figure 2.
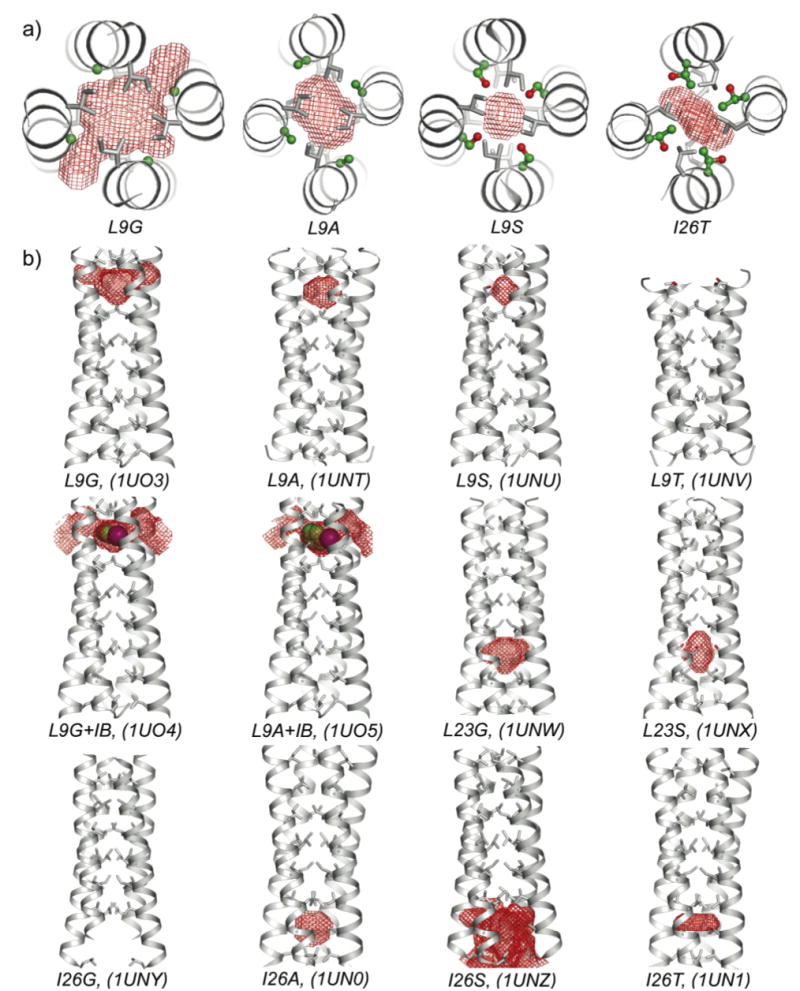
Crystal structures and cavities of GCN4-pLI variants. a) Cross-sections and b) side views depicting red mesh surfaces for the calculated 1.4 Å radius probe-occupied cavities or tunnels. The substituted residues are shown using ball-and-stick representations, while other core residues are shown with sticks. VDW spheres are shown for iodobenzene. PDB ID numbers are shown in parentheses.
Table 3.
Data collection and refinement statistics.a
| Peptide | Edge Length (Å) | Resol. (Å) | Total Obs. | Unique Refl. | I/σ (average) | I/σ (outer) | Rsym | R | Rfree (~5%) |
|---|---|---|---|---|---|---|---|---|---|
| (P4132) | |||||||||
| GCN4-pLI | 79.6 | 1.99 | 37822 | 6330 | 6.8 | 1.6 | 0.064 | 0.242 | 0.286 |
| L9G | 79.2 | 1.91 | 85641 | 6964 | 7.5 | 1.3 | 0.059 | 0.235 | 0.281 |
| L9S | 78.5 | 2.07 | 53164 | 5435 | 17.0 | 3.1 | 0.030 | 0.231 | 0.270 |
| L9A | 78.4 | 2.07 | 34573 | 5427 | 13.8 | 2.1 | 0.035 | 0.227 | 0.286 |
| L9T | 78.1 | 2.07 | 34996 | 5363 | 9.1 | 1.6 | 0.055 | 0.230 | 0.290 |
| L9G+IB | 79.1 | 1.70 | 188225 | 9782 | 5.5 | 2.0 | 0.080 | 0.225 | 0.257 |
| L9A+IB | 78.5 | 2.07 | 71214 | 5435 | 14.6 | 3.2 | 0.030 | 0.260 | 0.280 |
| L23G | 78.5 | 2.30 | 50281 | 4599 | 8.3 | 1.1 | 0.059 | 0.250 | 0.284 |
| L23S | 78.8 | 2.03 | 38975 | 3607 | 3.4 | 1.7 | 0.140 | 0.249 | 0.314 |
| I26G | 79.3 | 2.30 | 57910 | 4127 | 8.8 | 1.9 | 0.063 | 0.256 | 0.322 |
| I26S | 79.3 | 1.80 | 85655 | 8233 | 7.1 | 1.0 | 0.066 | 0.259 | 0.312 |
| I26A | 79.3 | 2.40 | 45238 | 3676 | 5.8 | 1.3 | 0.123 | 0.229 | 0.299 |
| I26T | 79.4 | 2.22 | 40086 | 3283 | 7.5 | 1.9 | 0.094 | 0.259 | 0.312 |
| L16G E20C | 78.4 | 2.17 | 35342 | 4747 | 5.7 | 1.9 | 0.078 | 0.269 | 0.292 |
| (P31) | |||||||||
| L16G E20C Y17H | 25.8, 25.8, 148.6 | 1.50 | 89229 | 17250 | 25.7 | 4.7 | 0.068 | 0.238 | 0.276 |
All datasets were > 97% complete.
A number of differences are apparent between the GCN4-pLI tetramer in the P4132 spacegroup reported here relative to the originally-reported structure in the P212121 spacegroup (14). Most notable is the crystallographic two-fold symmetry axis along the helix axis in the P4132 spacegroup, which results in each four-helix bundle being generated from just two unique chains, rather than four. The Glu6-Arg1 interhelical salt bridge is not observed in most cases, possibly because the N-terminal Arg residue is often not resolvable. Other ionic lattice interactions in the original structure appear to be largely retained in the variants reported here. The final two C-terminal residues are not reported in the original structure and could only sometimes be modeled in the structures presented here. An α-carbon superposition of the two tetrameric structures for residues 2–29 resulted in a root mean square (rms) deviation of 1.06 Å. Because the termini of the tetrameric peptide assembly are essentially free of crystal contacts in the P4132 lattice, B factors increase from the center of the assembly to the termini.
Cavities
Coiled-coils generally contain small internal cavities between the a and d layers. These cavities are competent for occupation by small guests, as evidenced by the xenon binding used for the initial structure determination of the peptides reported here. In GCN4-pLI, there are three such inter-layer cavities ranging in volume from ~20–40 Å3 (using a probe of radius 1.4 Å). Substituting single hydrophobic core residues with Gly, Ser, Ala or Thr creates cavities that are much larger than these inter-layer cavities since the tetrameric assembly juxtaposes the four substitutions sites, creating an entire level of vacancy within the aggregate. Cavity sizes are reported as probe-occupied volumes for probes of radius 1.4 Å or 1.8 Å (representing a water molecule or a chloride ion, respectively) (Table 2).
The engineered cavities demonstrate a significant span of volumes and range from hydrophobic to moderately polar (Figure 2). In some cases, the internal cavities are not completely closed off from bulk solvent (using a 1.4 Å probe), generating tunnels through the interior of the peptide assemblies. As expected from individual side chain volumes (44) (Table 2), the observed cavity sizes are greatest for Gly substitutions, followed by Ala, Ser, and finally Thr. The I26S peptide deviates from this trend, containing a much larger cavity than the other Ser variants due to a pronounced C-terminal fray. While present within the L23G and the more polar I26S cavities, structured water molecules are somewhat surprisingly not observed within any of the other polar cavities. Although the L16G E20C Y17H and L16G E20C peptides contain the same core substitution, the peptides demonstrate cavities of different volumes and shapes because of their contrasting antiparallel and parallel configurations (Figure 3). In the antiparallel L16G E20C Y17H, Ile19 and Cys20 pairs encloses the cavity on two sides, but this elongated cavity’s volume is still greater than that in the parallel L16G E20C. The cavities within the L16G peptides are the largest fully enclosed cavities using a 1.4 Å radius probe, although these peptides are also the least thermally stable.
Figure 3.
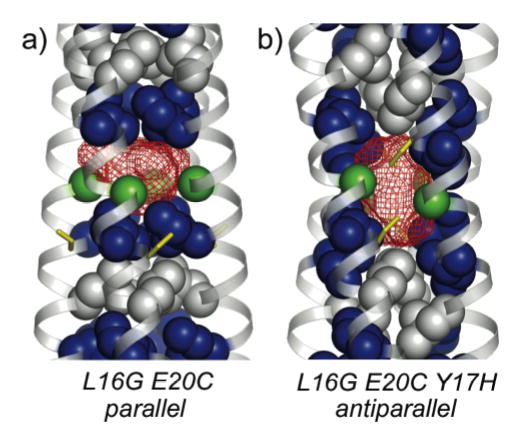
Crystal structures of L16G variants of GCN4-pLI. a) L16G E20C adopts a parallel tetrameric configuration, creating a 330 Å3 cavity (using a probe of radius 1.4 Å). Hydrophobic core a position Leu (blue) and d position Ile (gray) residues are shown with van der Waals (VDW) surfaces, as are α-carbons of the Gly16 residues (green). The Cys20 residues are shown as yellow sticks. b) L16G E20C Y17H adopts an antiparallel tetrameric configuration, creating a vertically elongated 370 Å3 cavity. The coloring is identical to panel a).
Polar contacts are formed in the variants containing Ser or Thr core substitutions (21) that likely compensate somewhat for the destabilizing placement of polar groups in the hydrophobic core (Figure 4). Based on distance criteria (interatomic distances of > 4 Å), each side chain hydroxyl group in the Ser and Thr variants forms backbone hydrogen bonds to the carbonyl oxygen of the i, i-4 residue, where the i-4 residue can be resolved (L9S, L23S, I26S, and I26T). In the L23S variant, one of the unique Ser residues forms a polar interaction with Glu22, thereby participating in an interhelical polar network (interatomic distances of 3.1–3.8 Å) between residues Glu22, His18, and Glu20 (Figure 4a). Along with the i, i-4 hydrogen bond, the chain B Ser of I26S forms a backbone hydrogen bond with the i, i-3 carbonyl, while the chain A Ser interacts with a structured water molecule bound inside the cavity (Figure 4b).
Figure 4.
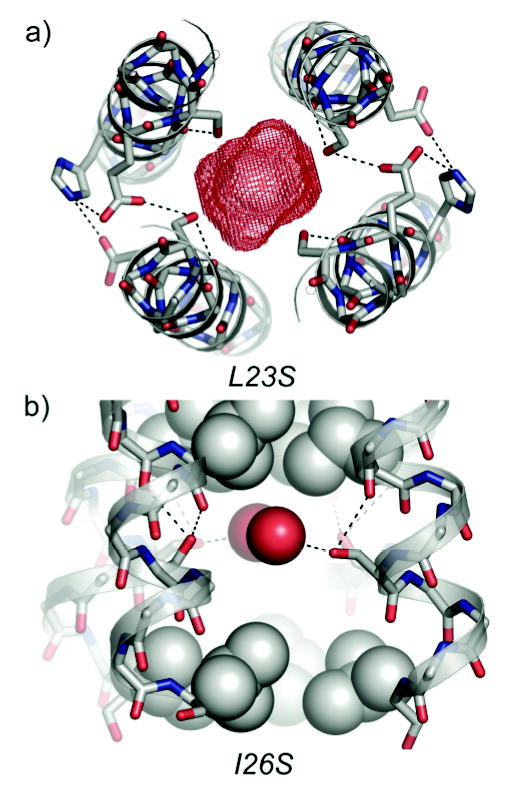
Electrostatic interactions in GCN4-pLI variants containing polar core substitutions. a) A cross-section of the L23S variant, illustrating polar contacts between the Ser23 hydroxyl groups and backbone or side chain atoms of adjacent residues. Sticks are shown for Ser23, Glu22, Glu20, and His18, and dotted black lines indicate potential contacts. The 150 Å3 cavity is shown in red. b) A side view of the I26S variant, depicting polar contacts between the Ser26 hydroxyl groups and backbone atoms or water molecules inside the cavity. Both unique Ser residues form at least two polar contacts (based on interatomic distances of > 4 Å). VDW spheres are shown for water molecules (red) and hydrophobic core residues (gray), while sticks are show for Ser26 and backbone atoms. Dotted black lines indicate potential contacts.
The polar side chains of the Ser and Thr variants pack differently depending on the heptad positioning of the substitution. In contrast to the Ser substitutions at a heptad positions (L9S and L23S) in which the side chains adopt the same rotamer and the hydroxyls are projected toward the center of the peptide assembly (Figure 4a), the d position Ser hydroxyls in I26S are skewed away from the cavity and the two unique side chains adopt different rotameric conformations (Figure 4b). Similarly for the Thr variants, the d position I26T hydroxyl groups project away from the center of the peptide assembly such that the side chain methyl group is oriented toward the cavity. In the a position L9T variant, the hydroxyl groups are contrastingly projected toward the center of the cavity with the side chain methyl groups directed toward bulk solvent. As the closest hydroxyl groups in L9T are separated by 4.7 Å, these groups appear not to be forming polar contacts, and the presence of these four polar groups within the core might explain the observed lack of N-terminal electron density beyond this substitution site.
Iodobenzene Binding
We reasoned that a relatively small hydrophobic molecule might bind the more hydrophobic cavities formed by Gly or Ala substitutions, so we co-crystallized several of these peptides with iodobenzene. The presence of an iodine atom bound within the cavities of L9A and L9G was independently confirmed by the strong 5σ peak apparent at the same position in both the |Fo|-|Fc| difference Fourier map (Figure 5) and in the anomalous difference Fourier map for both peptides when crystallized in the presence of iodobenzene (L9A+IB and L9G+IB). There were no comparable peaks observed in any of the anomalous difference maps for any of the structures crystallized without iodobenzene. These results are consistent with an iodobenzene molecule bound inside the cavities of the peptides, with a crystallographic C2 axis running through the benzene ring (Figures 5, S1). The P4132 space group imposes two symmetry equivalent conformations of iodobenzene in the tetrameric coiled-coil. While it is likely that the cavities described here will bind other molecules, the heavy atom anomalous signal made characterization of the iodobenzene complexes unequivocal.
Figure 5.
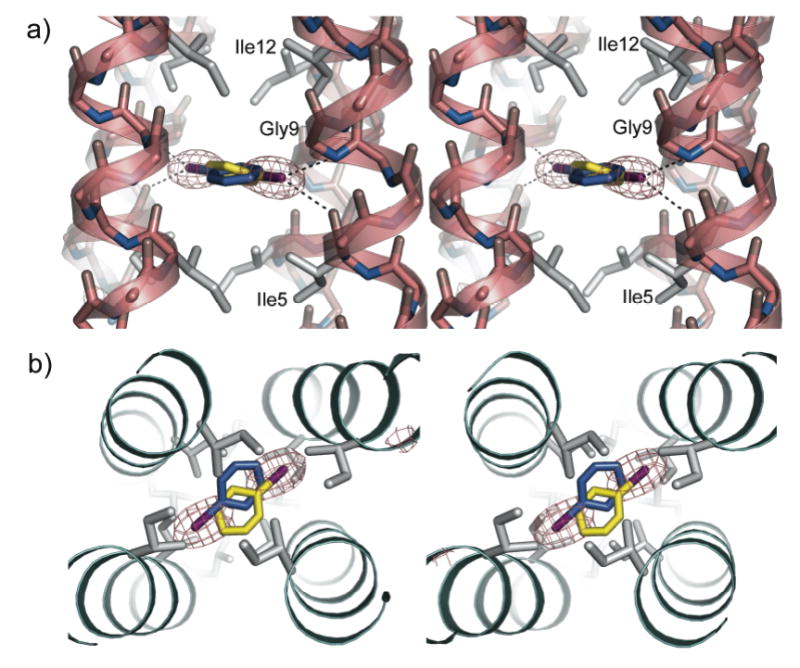
Stereo views of iodobenzene bound within the hydrophobic cavity created in the L9G variant of GCN4-pLI. a) Side view illustrating the potential polar contacts (dotted black lines) between the iodine atom and backbone nitrogen (interatomic distance of 3.9 Å) or carbonyl oxygen atoms (interatomic distance of 3.8 Å). The Fo-Fc electron density (gray mesh, contoured at 4σ) indicates the positioning of the iodine atoms from the two symmetry-equivalent iodobenzene molecules (blue and yellow). Side chains of hydrophobic core residues are shown as gray sticks. b) Top view of the iodobenzene-bound L9G using the same representations as panel (a).
The molecular volume of iodobenzene is 170 Å3, which fills about 75% of the calculated cavities (1.8 Å radius probe) in L9G+IB and L9A+IB. However, these fractional occupation values are overestimates since there is a significant amount of void space not included in the calculated cavity when a 1.8 Å radius probe is used to measure the cavity. Although the cavities are not fully enclosed using a more reasonable 1.4 Å probe, we can estimate a more realistic cavity volume for L9G+IB using values from L16G E20C, L16G E20C Y17H, and L23G, all of which contain Leu→Gly mutations that are fully enclosed using both probe sizes. Applying the average difference in calculated volume for the two probe sizes for these three peptides (60 Å3) to L9G+IB cavity, we estimate a cavity volume for L9G+IB of 300 Å3, which is about 60% occupied by the molecular volume of iodobenzene. A similar calculation for L9A+IB again results in an occupation value of about 60%. These fractional occupation values agree closely with the ideal encapsulation value of 55 ± 9% reported by Rebek and coworkers (45). The cavities within the L9A and L9G structures increase in volume by about 20 Å3 when occupied by iodobenzene (Table 2). These volume increases result from greater N-terminal interhelical distances (described below), and likely take place to allow the most enthalpically favorable packing interactions between the host assembly and guest molecule. The plane of the iodobenzene aromatic ring is more tilted with respect to the long bundle axis in L9A+IB than in L9G+IB, which is likely due to the tighter packing in the smaller cavity.
The polarizable iodine atom of the bound iodobenzene is projected toward and potentially forms polar contacts with the backbone nitrogen of Gly9 (3.90 Å interatomic distance) and the backbone carbonyl oxygen of Ile5 (3.82 Å interatomic distance) (Figures 5a, S1a), consistent with previous reports of polar interactions between halogen atoms and proteins (46, 47). The L9G+IB and L9A+IB structures also contain new water molecules inside and near the periphery of the channel between the ligand and bulk solvent, which may be explained by the increased size of the cavity (Figure S1b). The side chains of Glu10 have distinctly different conformations in the unbound and bound forms. In the ligand-free L9G, both unique Glu10 residues form interhelical salt bridges with adjacent Lys8 residues and effectively occlude the empty cavity (Figure S1c). In contrast, these salt bridges are not present in the ligand-bound structure, which may be due to, or which may contribute to, the increased interhelical distance and cavity size. In the free L9A structure, one of these Glu10-Lys8 interactions forms and is similarly absent in the bound structure. This conformational change can be clearly perceived in the "morph" movie (48) that has been generated between the bound and unbound forms (49).
Superhelical Parameters
Apart from the L9G and L9S peptides, GCN4-pLI and all variants that crystallized in the P4132 spacegroup display an increase in interhelical distance from the N-termini to the C-termini (Figure 6), as determined by calculating local supercoil parameters by fitting residues 5–29 in discrete bins of seven residues at a time (see supporting information for further superhelical parameters data). This drift in interhelical distance in not present in the originally reported GCN4-pLI structure, and may therefore result from effects imposed by the cubic lattice. The notable exceptions of L9G and L9S, located ~25 Å from the C-termini, demonstrate that core substitutions can affect the overall structure at relatively large distances; these exceptions might result from local disorder or solvent accessibility at the N-termini leading to tighter compensatory packing near the C-termini for these peptides.
Figure 6.
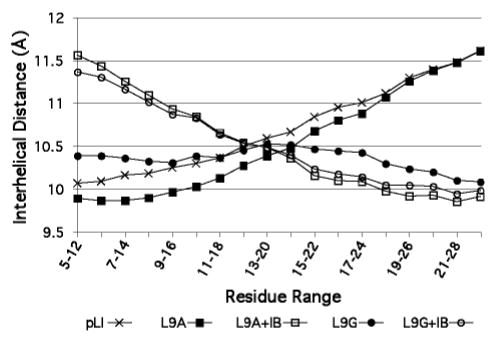
Interhelical distance profiles for GCN4-pLI (crosses), L9A (closed squares), L9A+IB (open squares), L9G (closed circles), and L9G+IB (open circles). The indicated heptads were used to determine the local interhelical distance values.
There is a substantial increase in N-terminal interhelical distance for the L9G+IB and L9A+IB assemblies compared with their ligand-free forms (Figure 6). This increased interhelical distance results in an enlarged, more solvent-accessible cavity presumably better able to accommodate the bound iodobenzene guest. Although only two examples are presented here, interhelical distance profiles may thus be diagnostic of guest binding in coiled-coils. Together, the observed changes in interhelical distance profile, increases in cavity volume, and rotamer alterations in solvent-exposed side chains for the L9G+IB and L9A+IB suggest that induced fit binding events (50, 51) must be taken into consideration when designing artificial receptors or enzymes. These conformational changes associated with ligand binding complicate the rational design aspects of peptide and protein engineering, but also make more likely the potential of binding several molecules with different shapes in the same host assembly.
Conclusion
In summary, we have characterized internal cavities of different sizes and polarities in a series of parallel and antiparallel coiled-coil tetramers and have shown that these cavities in some cases can bind an exogenous small molecule. The three-dimensional structures illustrate some of the varied surfaces that can be constructed in closely-related peptides with very similar overall folds and may provide a basis for the future design of de novo helical proteins. This study was only possible because of the substantial plasticity of the hydrophobic core in the GCN4-pLI coiled-coil tetramer to substitution. Mixtures of peptides like those reported here might be used for such purposes as the dynamic combinatorial formation of peptide-based receptors, and such heteromeric peptide assemblies containing non-symmetric cavities may augment the scope of the system described here.
Supplementary Material
Acknowledgments
We thank the staff at SSRL and APS, and professors Peter Kuhn and Raymond C. Stevens for their generous assistance. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
We thank the National Institutes of Health for financial support (GM52190). JER, JMAG, and LJL thank the Wellcome Trust (061454/Z/00/Z), the Spanish MCYT, and NSF, respectively, for fellowships.
Coordinates have been deposited in the RCSB Protein Data Bank: GCN4-pLI, 1UO2; L9G, 1UO3; L9S, 1UNU; L9A, 1UNT; L9T, 1UNV; L9G+IB, 1UO4; L9A+IB, 1UO5; L23G, 1UNW; L23S 1UNX; I26G, 1UNY; I26S, 1UNZ; I26A, 1UN0; I26T, 1UN1; L16G E20C, 1W5I; L16G E20C Y17H, 2BNI.
Abbreviations: CD, circular dichroism; SEC, size exclusion chromatography; ABA, acetamidobenzoic acid; MW, molecular weight; Nagg, apparent aggregation number; UV, ultraviolet; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight, SSRL, Stanford Synchrotron Radiation Laboratory; APS, Advanced Photon Source; SIR, single isomorphous replacement; VDW, van der Waals.
Supporting Information Available. “Morph” movie illustrating observed conformational changes for L9G and L9A upon iodobenzene binding, Figure S1, and tabulated local superhelical parameters for each peptide. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Xu J, Baase WA, Baldwin E, Matthews BW. The response of T4 lysozyme to large-to-small substitutions within the core and its relation to the hydrophobic effect. Protein Sci. 1998;7:158–177. doi: 10.1002/pro.5560070117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo Z, Zhou D, Schultz PG. Designing small-molecule switches for protein-protein interactions. Science. 2000;288:2042–5. doi: 10.1126/science.288.5473.2042. [DOI] [PubMed] [Google Scholar]
- 3.Takano K, Yamagata Y, Yutani K. Buried water molecules contribute to the conformational stability of a protein. Protein Eng. 2003;16:5–9. doi: 10.1093/proeng/gzg001. [DOI] [PubMed] [Google Scholar]
- 4.Machicado C, Bueno M, Sancho J. Predicting the structure of protein cavities created by mutation. Protein Eng. 2002;15:669–675. doi: 10.1093/protein/15.8.669. [DOI] [PubMed] [Google Scholar]
- 5.Xu J, Baase WA, Quillin ML, Baldwin EP, Matthews BW. Structural and thermodynamic analysis of the binding of solvent at internal sites in T4 lysozyme. Protein Sci. 2001;10:1067–1078. doi: 10.1110/ps.02101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quillin ML, Breyer WA, Griswold IJ, Matthews BW. Size versus Polarizability in Protein-Ligand Interactions: Binding of Noble Gases within Engineered Cavities in Phage T4 Lysozyme. J Mol Biol. 2000;302:955–977. doi: 10.1006/jmbi.2000.4063. [DOI] [PubMed] [Google Scholar]
- 7.Mulder FAA, Hon B, Muhandiram DR, Dahlquist FW, Kay LE. Flexibility and Ligand Exchange in a Buried Cavity Mutant of T4 Lysozyme Studied by Multinuclear NMR. Biochemistry. 2000;39:12614–12622. doi: 10.1021/bi001351t. [DOI] [PubMed] [Google Scholar]
- 8.Vlassi M, Cesareni G, Kokkinidis M. A correlation between the loss of hydrophobic core packing interactions and protein stability. J Mol Biol. 1999;285:817–27. doi: 10.1006/jmbi.1998.2342. [DOI] [PubMed] [Google Scholar]
- 9.Buckle AM, Cramer P, Fersht AR. Structural and Energetic Responses to Cavity-Creating Mutations in Hydrophobic Cores: Observation of a Buried Water Molecule and the Hydrophilic Nature of Such Hydrophobic Cavities. Biochemistry. 1996;35:4298–305. doi: 10.1021/bi9524676. [DOI] [PubMed] [Google Scholar]
- 10.Eriksson AE, Baase WA, Wozniak JA, Matthews BW. A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature. 1992;355:371–3. doi: 10.1038/355371a0. [DOI] [PubMed] [Google Scholar]
- 11.Penning TM, Jez JM. Enzyme redesign. Chem Rev. 2001;101:3027–3046. doi: 10.1021/cr000049n. [DOI] [PubMed] [Google Scholar]
- 12.Harris JL, Craik CS. Engineering enzyme specificity. Curr Op Chem Biol. 1998;2:127–32. doi: 10.1016/s1367-5931(98)80044-6. [DOI] [PubMed] [Google Scholar]
- 13.Crick FHC. The packing of alpha-helices: simple coiled-coils. Acta Cryst. 1953;6:689–97. [Google Scholar]
- 14.Harbury PB, Zhang T, Kim PS, Alber T. A switch between two-, three, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–7. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 15.Horne WS, Yadav MK, Stout CD, Ghadiri MR. Heterocyclic Peptide Backbone Modifications in an alpha-Helical Coiled Coil. J Am Chem Soc. 2004;126:15366–15367. doi: 10.1021/ja0450408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilcoxen KM. PhD thesis. The Scripps Research Institute; La Jolla, CA: 2002. [Google Scholar]
- 17.Gonzalez L, Jr, Woolfson DN, Alber T. Buried polar residues and structural specificity in the GCN4 leucine zipper. Nat Struct Biol. 1996;3:1011–1018. doi: 10.1038/nsb1296-1011. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez L, Jr, Brown RA, Richardson D, Alber T. Crystal structures of a single coiled-coil peptide in two oligomeric states reveal the basis for structural polymorphism. Nat Struct Biol. 1996;3:1002–1010. doi: 10.1038/nsb1296-1002. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez L, Jr, Plecs JJ, Alber T. An engineered allosteric switch in leucine-zipper oligomerization. Nat Struct Biol. 1996;3:510–515. doi: 10.1038/nsb0696-510. [DOI] [PubMed] [Google Scholar]
- 20.Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci U S A. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akey DL, Malashkevich VN, Kim PS. Buried Polar Residues in Coiled-Coil Interfaces. Biochemistry. 2001;40:6352–6360. doi: 10.1021/bi002829w. [DOI] [PubMed] [Google Scholar]
- 22.Obataya I, Sakamoto S, Ueno A, Mihara H. Design and synthesis of 3 alpha-helix peptides forming a cavity for a fluorescent ligand. Biopolymers. 2001;59:65–71. doi: 10.1002/1097-0282(200108)59:2<65::AID-BIP1006>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 23.Johansson JS, Gibney BR, Rabanal F, Reddy KS, Dutton PL. A Designed Cavity in the Hydrophobic Core of a Four-alpha-Helix Bundle Improves Volatile Anesthetic Binding Affinity. Biochemistry. 1998;37:1421–1429. doi: 10.1021/bi9721290. [DOI] [PubMed] [Google Scholar]
- 24.Johansson JS, Scharf D, Davies LA, Reddy KS, Eckenhoff RG. A designed four-alpha-helix bundle that binds the volatile general anesthetic halothane with high affinity. Biophys J. 2000;78:982–993. doi: 10.1016/S0006-3495(00)76656-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manderson GA, Michalsky SJ, Johansson JS. Effect of Four-alpha-Helix Bundle Cavity Size on Volatile Anesthetic Binding Energetics. Biochemistry. 2003;42:11203–11213. doi: 10.1021/bi034623b. [DOI] [PubMed] [Google Scholar]
- 26.Manderson GA, Johansson JS. Towards a three-alpha-helix bundle protein that binds volatile general anesthetics. Biopolymers. 2004;75:338–354. doi: 10.1002/bip.20138. [DOI] [PubMed] [Google Scholar]
- 27.Schnarr NA, Kennan AJ. Strand orientation by steric matching: a designed antiparallel coiled-coil trimer. J Am Chem Soc. 2004;126:14447–14451. doi: 10.1021/ja047496v. [DOI] [PubMed] [Google Scholar]
- 28.Schnarr NA, Kennan AJ. Peptide Tic-Tac-Toe: Heterotrimeric Coiled-Coil Specificity from Steric Matching of Multiple Hydrophobic Side Chains. J Am Chem Soc. 2002;124:9779–9783. doi: 10.1021/ja0174940. [DOI] [PubMed] [Google Scholar]
- 29.Monera OD, Zhou NE, Lavigne P, Kay CM, Hodges RS. Formation of parallel and antiparallel coiled-coils controlled by the relative positions of alanine residues in the hydrophobic core. J Biol Chem. 1996;271:3995–4001. doi: 10.1074/jbc.271.8.3995. [DOI] [PubMed] [Google Scholar]
- 30.Monera OD, Soennichsen FD, Hicks L, Kay CM, Hodges RS. The relative positions of alanine residues in the hydrophobic core control the formation of two-stranded or four-stranded alpha-helical coiled-coils. Protein Eng. 1996;9:353–363. doi: 10.1093/protein/9.4.353. [DOI] [PubMed] [Google Scholar]
- 31.Yadav MK, Leman LJ, Stout CD, Ghadiri MR. Crystal Structure of an Antiparallel Homotetrameric Coiled-Coil. 2005 Unpublished results. [Google Scholar]
- 32.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 33.Leslie AGW. Joint CCP4/ESF-EAMCB Newsletter on Protein Crystallography. 1992;26 [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Anonymous The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 36.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 37.Cohen A, Ellis P, Kresge N, Soltis SM. MAD phasing with krypton. Acta Crystallogr D. 2001;57:233–238. doi: 10.1107/s0907444900014670. [DOI] [PubMed] [Google Scholar]
- 38.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 39.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–65. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 40.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–63. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 41.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr D. 1994;50:178–85. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 42.McLachlan AD. Rapid comparison of protein structures. Acta Crystallogr A. 1982;38:871–3. [Google Scholar]
- 43.Karplus PA. Hydrophobicity regained. Protein Sci. 1997;6:1302–7. doi: 10.1002/pro.5560060618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harpaz Y, Gerstein M, Chothia C. Volume changes on protein folding. Structure. 1994;2:641–9. doi: 10.1016/s0969-2126(00)00065-4. [DOI] [PubMed] [Google Scholar]
- 45.Mecozzi S, Rebek J., Jr The 55% solution: a formula for molecular recognition in the liquid state. Chem Eur J. 1998;4:1016–1022. [Google Scholar]
- 46.Auffinger P, Hays FA, Westhof E, Ho PS. Halogen bonds in biological molecules. Proc Natl Acad Sci U S A. 2004;101:16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ippolito JA, Christianson DW. The contribution of halogen atoms to protein-ligand interactions. Int J Biol Macromol. 1992;14:193–7. doi: 10.1016/s0141-8130(05)80026-1. [DOI] [PubMed] [Google Scholar]
- 48.Echols N, Milburn D, Gerstein M. MolMovDB: analysis and visualization of conformational change and structural flexibility. Nucleic Acids Res. 2003;31:478–482. doi: 10.1093/nar/gkg104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. See Supplementary Information or MolMovDB ID# 929950–12297, http://molmovdb.mbb.yale.edu/cgi-bin/morph.cgi?ID=929950–12297.
- 50.Koshland DE., Jr The lock-and-key principle and the induced-fit theory. Angew Chem, Int Ed Engl. 1994;33:2375–78. [Google Scholar]
- 51.Davis AM, Teague SJ. Hydrogen bonding, hydrophobic interactions, and failure of the rigid receptor hypothesis. Angew Chem, Int Ed. 1999;38:736–749. doi: 10.1002/(SICI)1521-3773(19990315)38:6<736::AID-ANIE736>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.