

Short abstract
An analysis of genome-wide histone acetylation data using a few complementary statistical models gives support to a cumulative effect model for global histone acetylation.
Abstract
Background
Histone acetylation plays important but incompletely understood roles in gene regulation. A comprehensive understanding of the regulatory role of histone acetylation is difficult because many different histone acetylation patterns exist and their effects are confounded by other factors, such as the transcription factor binding sequence motif information and nucleosome occupancy.
Results
We analyzed recent genomewide histone acetylation data using a few complementary statistical models and tested the validity of a cumulative model in approximating the global regulatory effect of histone acetylation. Confounding effects due to transcription factor binding sequence information were estimated by using two independent motif-based algorithms followed by a variable selection method. We found that the sequence information has a significant role in regulating transcription, and we also found a clear additional histone acetylation effect. Our model fits well with observed genome-wide data. Strikingly, including more complicated combinatorial effects does not improve the model's performance. Through a statistical analysis of conditional independence, we found that H4 acetylation may not have significant direct impact on global gene expression.
Conclusion
Decoding the combinatorial complexity of histone modification requires not only new data but also new methods to analyze the data. Our statistical analysis confirms that histone acetylation has a significant effect on gene transcription rates in addition to that attributable to upstream sequence motifs. Our analysis also suggests that a cumulative effect model for global histone acetylation is justified, although a more complex histone code may be important at specific gene loci. We also found that the regulatory roles among different histone acetylation sites have important differences.
Background
Gene activities in eukaryotic cells are concertedly regulated by transcription factors and chromatin structure. The basic repeating unit of chromatin is the nucleosome, an octamer containing two copies each of four core histone proteins. Recent microarray based studies [1-8] have begun to uncover the global regulatory role of nucleosome positioning and modifications. While nucleosome occupancy in promoter regions typically occludes transcription factor binding, thereby repressing global gene expression [1-8], the role of histone modification is more complex [9-11]. Histone tails can be modified in various ways, including acetylation, methylation, phosphorylation, and ubiquitination. Even the regulatory role of histone acetylation, the best characterized modification to date, is still not fully understood [12,13].
Each of the four core histones contains several acetylable sites at their amino terminus tails. Genome-wide histone acetylation data from Saccharomyces cerevisiae [2,8] have offered new opportunities for us to evaluate the regulatory effects of histone acetylation at these lysine sites. In particular, both H3 and H4 acetylation levels were found to be positively correlated with gene transcription rates. However, a subtle but important issue in analyzing such data is that effects of other potentially important factors not included in the analysis, generally termed as confounding factors, cannot be revealed by simple correlation plots. It is unclear, for example, how much regulatory information associated with histone acetylation is redundant with the genomic sequence information. To gain insights into this, we conducted a statistical analysis by combining acetylation [2,4,8], nucleosome occupancy [1,3,8], gene upstream sequence information [14], and gene expression data [1,15,16] to investigate the effect of histone acetylation in the context of other regulatory factors in S. cerevisiae.
A related question is whether different histone acetylation sites play similar roles in gene regulation. It is commonly postulated that globally H3 and H4 acetylation are both associated with global gene activation. Indeed, the acetylation levels of H3 and H4 across gene promoters have been shown to be highly correlated [2,7,8]. However, other experimental studies have also suggested that H3 and H4 acetylations have different regulatory roles [17-20]. We investigated the validity of a cumulative model for the regulatory effect of histone acetylation and also compare the regulatory effects of H3 and H4 acetylation in a coherent statistical framework.
Another interesting question is whether combinatorial patterns of histone acetylation code for distinct regulatory information at a global level [9,11], with each pattern being recognized by a specific regulatory protein. If such codes exist, a large number of codes may result from combinations of different histone acetylation sites. On the other hand, if the effect is cumulative, multiple histone acetylation sites may be used to gradually control the interaction between nucleosomes or the stability of the regulatory proteins. Recent mutagenesis studies [5] have suggested that multiple H4 acetylation sites have a cumulative effect. Here we revisit this question using a statistical approach to combine available genome-wide data. Our analysis suggests that the simple additive-effect model is sufficient for fitting the available data.
Results
Effect of histone acetylation on gene transcription rate
Standard analysis
We analyzed two recent genome-wide histone acetylation datasets [2,8] (see Materials and methods for details about the data sources). Due to space limits, here we only present the results for Pokholok et al.'s data [8], with the discussion of Kurdistani et al.'s data [2] in Additional data file 1. Pokholok et al. measured acetylation levels at three different sites, H3K9, H3K14, and H4, with the last referring to non-specific acetylation on any of the four acetylable lysines on H4 tails.
A typical analysis, when both histone acetylation data on a single site (for example, H3K9) and transcription rate data are available, is to simply correlate the two sets of measurements and to report the apparent significant statistical correlation between the two. When data on multiple acetylation sites are available, a slightly more formal analysis is to fit a linear regression model of the form:
where yi is the transcription rate of gene i, and xij, for j = 1,2,3, is the histone acetylation level of H3K9, H3K14, and H4, respectively. All data were log-transformed before analysis. This model is highly statistically significant for both intergenic (p value < 2.0 × 10-16) and coding regions (p value < 2.0 × 10-16). The association between gene expression and intergenic histone acetylation is commonly interpreted as regulatory effects, whereas correlation between gene expression and coding histone acetylation is believed to be a result of passing of transcriptional machineries through active genes.
Significant confounding factors
Gene regulation is a complex process involving many contributing factors. Probably the best characterized factor for controlling gene transcription is the upstream sequence information. Although histone acetyltransferases (HATs) and histone deacetylases (HDACs) do not have obvious sequence specificity themselves, they may be recruited by transcription factors that recognize specific sequences. Thus, sequence information is an important confounding factor. Our main interest here is to delineate the roles of these factors and investigate whether histone acetylation provides any additional information on gene transcription. In the past decade, numerous computational methods have been developed to identify target sequences of transcription factors and to use such information to predict gene expression [21-26].
Another well-characterized property of the chromatin structure, the nucleosome occupancy, also plays an important role in gene regulation. Histone acetylation and nucleosome positioning are closely related events. Genome-scale, high-resolution nucleosome positioning data have led to the observation that transcription factor binding sites tend to be nucleosome-depleted [6]. Although genome-wide, high-resolution nucleosome positioning data are still unavailable, lower resolution data have already shown that gene expression levels are reciprocally correlated with nucleosome occupancy [1,3,8]. Therefore, nucleosome occupancy may also be an important confounding factor in explaining gene regulation.
Refined analysis
We tested using two different sequence motif based-methods to account for the cis regulatory information (see Materials and methods for details). As shown in Additional data file 1, the two methods gave remarkably consistent results. Here we present results from using MDscan [24], which infers sequence motif information de novo. The combined transcriptional control by transcription factor binding motifs (TFBMs), nucleosome occupancy, and histone acetylation is modeled as:
where the xij values are the three histone acetylation levels (corresponding to H3K9, H3K14, and H4, respectively), the zij values are the corresponding scores to the 33 selected motifs, and wi is the nucleosome occupancy level. Table 1 shows the R2 (referring to the adjusted R-square statistic, which measures the fraction of explained variance after an adjustment of the number of parameters fitted; see page 231 of [27]) of the various linear models. One can see that a simple regression of transcription rates against histone acetylation without considering any other factors gave an R2 of 0.1841 (Table 1), implying that about 18% of the variation of the transcription rates is attributable to histone acetylation. In contrast, the regression of transcription rates against motif scores and nucleosome density levels (no histone acetylation) gave an R2 of 0.1997. The comprehensive model with all the variables we considered (equation 2) bumped up the R2 to 0.3262, indicating that the histone acetylation does have a significant effect on the transcription rate, although not as high as that in the naïve model.
Table 1.
Model performance (adjusted R2) with different covariates
| Intergenic regions | Coding regions | |||||||
| Acetylation sites included | - | Seq | Nuc | Seq/Nuc | - | Seq | Nuc | Seq/Nuc |
| - | 0 | 0.1387 | 0.1145 | 0.1997 | 0 | 0.1315 | 0.1440 | 0.2185 |
| H3K9 and H3K14 | 0.1808 | 0.2700 | 0.2641 | 0.3208 | 0.1014 | 0.2059 | 0.2515 | 0.3068 |
| H4 | 0.0849 | 0.2086 | 0.2487 | 0.3085 | 0.0222 | 0.1522 | 0.2131 | 0.2774 |
| H3K9, H3K14, and H4 | 0.1841 | 0.2706 | 0.2704 | 0.3262 | 0.1957 | 0.2627 | 0.2619 | 0.3131 |
The adjusted R2 for the linear regression model (equation 2) containing different regulatory factors (Nuc, nucleosome occupancy; Seq, sequence information). (The adjusted R2 is related to the (unadjusted) R2 as , where n is the sample size, and p is the number of explanatory variables in the linear regression model.)
To confirm that the above results indicate intrinsic statistical associations rather than artifacts of the statistical procedure, we validated our model using two independent methods. First, we tested whether applying the above procedure to the random inputs would yield substantially worse performance than applying it to the real data. We generated 50 independent samples by random permutation (see Materials and methods). The R2 for these randomized data are much smaller than for the real data. For example, considering equation 2 fitted with sequence motif information only, the largest R2 for coding regions for the 50 randomly permutated samples is 0.0378 (Figure 1), compared to R2 of 0.1315 for the real data. The differences between the R2 values are even larger if we also include histone acetylation and nucleosome occupancy in the model. Therefore, our model is able to extract real statistical association. Secondly, we tested whether the model might overfit by a five-fold cross validation procedure (see Materials and methods). The root mean square (rms) errors for the training data are 1.500 (for intergenic regions) and 1.483 (for coding regions), whereas the rms errors for the testing data were 1.519 (for intergenic regions) and 1.498 (for coding regions). In both cases, the difference between the in-sample and out-of-sample errors is less than 2%, suggesting overfitting is not an issue here.
Figure 1.
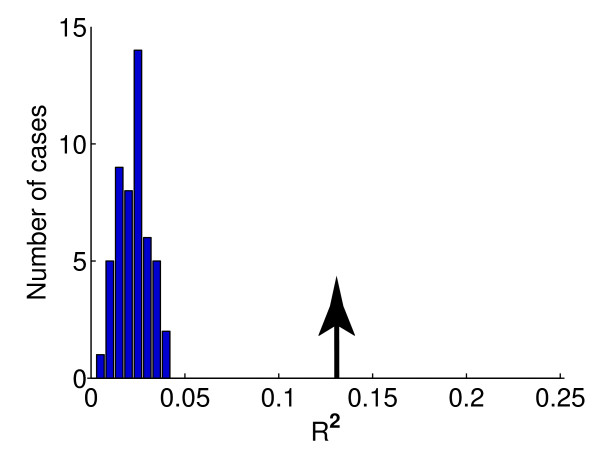
Model validation by comparing the R2 for the real versus randomly permutated datasets. The R2 obtained by applying the motif selection and fitting equation 2 (with sequence motif information only) procedures to randomly permutated and real data. The histogram is obtained based on 50 randomly permutated samples. The arrow on the right marks the R2 for the real data. Results for the coding regions are represented here. See the main text for details.
Multiple histone acetylation sites have cumulative regulatory effects
With the foregoing regression framework, we further investigated whether the combined effect of various histone acetylation sites could be approximated by a simple cumulative model, or a more complex 'combinatorial histone code' is needed. To gain a qualitative overview, we grouped genes according to their upstream histone acetylation patterns, each corresponding to a combination of high (greater than 60th percentile) or low (less than 40th percentile) histone acetylation levels at three acetylation sites. To avoid ambiguity due to measurement noise, the middle 20% of genes was not included in any groups. This coarse-grained partition method results in eight groups of genes with distinct upstream acetylation patterns. For example, one of these eight groups contains genes with high H3K9, high H3K14, and low H4 acetylation levels in their upstream intergenic regions. Increasing H3K9 acetylation level enhances gene transcription (Table 2), regardless of the acetylation level at other sites. A similar but weaker pattern can be seen for H3K14 acetylation. In contrast, the increase of H4 acetylation level is associated with both elevated and reduced transcription rates. A possible explanation is that the regulatory effect of H4 acetylation is dependent on acetylation level at other sites, while another explanation is that the H4 acetylation effect is weak overall. These relationships are not sensitive to the cutoff threshold for removing ambiguous genes.
Table 2.
Mean transcription rates (log-transformed) for genes with similar histone acetylation patterns
| H3K9ac Low | H3K9ac High | |
| H3K14ac Low | ||
| H4ac Low | -0.850 | 0.207 |
| H4ac High | -0.522 | 0.307 |
| H3K14ac High | ||
| H4ac Low | -0.454 | 0.816 |
| H4ac High | -0.126 | 0.460 |
Ac, acetylation.
As a quantitative validation of the above observation, we re-examined the validity of equation 2 in modeling the regulatory role of histone acetylation. We observed that the inclusion of all quadratic interaction terms among the three histone acetylation covariates in the regression model does not improve the model fitting (that is, R2 = 0.3262 and 0.3278, respectively, for intergenic regions, and R2 = 0.3131 and 0.3132, respectively, for coding regions). The same conclusion also holds when we do not include the sequence motif information and nucleosome occupancy data as covariates (R2 = 0.1841 and 0.1925, respectively, for intergenic regions). These observations suggest that the combinatorial effect is, at best, undetectable from the current data and the simple cumulative model (equation 2) is sufficient. Similar results were obtained using the acetylation data in Kurdistani et al. [2] (Additional data file 1).
H3 and H4 acetylation play different roles in gene regulation
A statistical measure of the effect of an individual factor/covariate on the response variable is the partial correlation (Materials and methods), which roughly reflects the 'pure' relationship between two variables while controlling other factors. As shown in Table 3, partial correlations between transcription rates and intergenic H3K9 and H3K14 acetylation levels, while controlling the sequence information and H4 acetylation levels, are 0.25 and 0.21, respectively; whereas that between transcription rates and H4 acetylation effect is insignificant (-0.03). In addition, the difference between the effects of H3 and H4 acetylations is visually evident (Figure 2). The same phenomenon can be observed by comparing different regression models. As shown by Table 1, the R2 (adjusted R-square) for the model without using the H4 acetylation information is comparable to the full model, whereas the performance of the model without H3 acetylation is significantly poorer. Interestingly, the transcription rate is negatively correlated with coding region H4 acetylation. These observations suggest that while H3 acetylation plays an important role in global gene activation, H4 acetylation in the intergenic region has little global effect. Similar results were also obtained for the acetylation data in Kurdistani et al. (Additional data file 1).
Table 3.
Partial correlation between covariate and transcription rates
| Intergenic regions | Coding regions | |||||||
| Covariate | Control variable | Partial correlation | Control variables | Partial correlation | Control variable | Partial correlation | Control variables | Partial correlation |
| H3K9 | H4 | 0.3015 | H4 and Seq | 0.2507 | H4 | 0.2439 | H4 and Seq | 0.2038 |
| H3K14 | H4 | 0.2359 | H4 and Seq | 0.2105 | H4 | 0.4070 | H4 and Seq | 0.3473 |
| H4 | H3K9, H3K14 | -0.0656 | H3K9, H3K14 and Seq | -0.0344 | H3K9, H3K14 | -0.3245 | H3K9, H3K14 and Seq | -0.2678 |
The partial correlation between transcription rates and H3 (or H4) acetylation levels while controlling for the effects of H4 (or H3) acetylation and sequence information (Seq).
Figure 2.
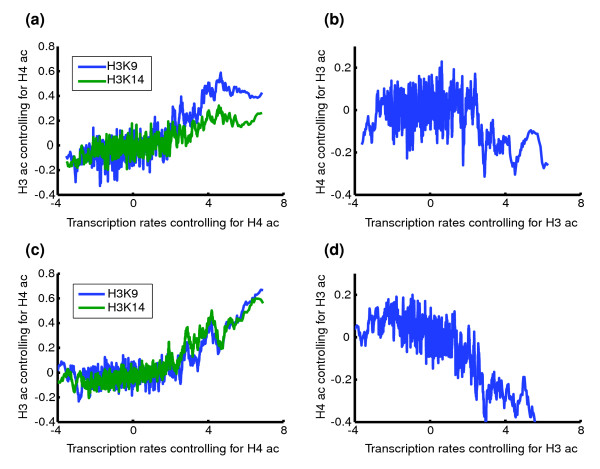
Dependency of transcription rates on histone acetylation levels (ac) after controlling for confounding effects. (a) Transcription rates versus intergenic H3K9 and K14 acetylation levels controlling for H4 acetylation levels. (b) Transcription rates versus intergenic H4 acetylation levels controlling for H3K9 and K14 acetylation levels. (c) Same as (a) except that coding region histone acetylation data are used. (d) Same as (b) except that coding region histone acetylation data are used. All data are log-transformed. Genes are sorted by transcription levels. A sliding smoothing window of 20 genes is applied to the transcription rates and histone acetylation data.
Gcn5 is the catalytic component of the SAGA complex and preferentially acetylates H3 lysines, including K9, K14, and K18. Esa1 is the catalytic component of the NuA4 complex and preferentially acetylates H4 lysines. Based on the above analysis, we predicted that the global gene expression was significantly affected by the abundance of Gcn5 but not of Esa1. Genome-wide occupancy of Gcn5 and Esa1 has been measured previously [4]. Both enzymes were found to be globally associated with active genes, and the Pearson correlation between the occupancy levels of the two is as high as 0.7890, probably because they share a common component, Tra1, for recognizing targets [28]. We modified equation 2 to estimate the association between Pol II occupancy [4] and these two HATs. In this case, the response variable yi is the log-ratio of Pol II binding, whereas the xij (j = 1,2) are the log-transformed binding ratios of Gcn5 and Esa1, respectively. Fitting the model using both Gcn5 and Esa1 data yields an R2 of 0.2365. If we remove Gcn5 from the model, the R2 is reduced to 0.1808. However, removing Esa1 causes little change in model performance (R2 = 0.2366). In addition, the partial correlation between the occupancy levels of Pol II and Gcn5 while controlling Esa1 occupancy is 0.2690, whereas the number is reduced to only 0.0154 if the order of Gcn5 and Esa1 is reversed. Taken together, these results show that, indeed, Esa1 only marginally affects the global association with Pol II binding.
Discussion
The global regulatory role of histone acetylation is still not fully understood and contradictory results have been reported in the literature [2,5,8]. Part of this inconsistency has been attributed to data analysis procedures [29]. In this paper, we analyzed two recent CHIP-chip datasets [2,8] in a statistically coherent framework. Our model isolates the regulatory role of individual acetylation sites and systematically controls for the effects of important confounding factors, thus resulting in a more detailed evaluation of the regulatory role of histone acetylation than previous studies [2,8]. Interestingly, our analyses of the two aforementioned datasets yielded similar results, even though the biological interpretations in the original papers were drastically different.
We found that the regulatory effect of histone acetylation can be well approximated by a simple regression model. In contrast to Kurdistani et al.'s claims [2], our results suggest that the currently available data supports a simple cumulative effect model instead of a combinatorial code model of histone modifications as originally proposed in [9], consistent with a recent mutagenesis study [5] showing that three of the four acetylable sites on H4 tails are functionally redundant. It is worth noting that these results do not exclude the possibility that combinatorial control is critical at specific gene loci, but it is unlikely that a fully combinatorial code regulates global gene expression.
We also quantitatively analyzed the regulatory effects due to individual acetylation sites. To our surprise, we found that the overall effects of H3 and H4 acetylation were quite different, at least statistically. In particular, while elevated H3 acetylation in promoter regions appears to be responsible for activating global gene expression, H4 acetylation seems to play a less important role. Levels of H3 and H4 acetylation in intergenic regions are closely coordinated by the binding of Gcn5 and Esa1, both of which have been found to bind to actively transcribed genes [4]. However, our analysis suggests that Esa1 may not be important for global regulation, consistent with previous experimental studies by Kevin Struhl's group [17,30,31]. In these studies, the authors show that depletion of Esa1 causes a global decrease of H4 acetylation, but only a small subset of the genes responds with significant transcription change [30]. They also found that the effect of H4 acetylation may be highly transcription factor specific [17]. It will be interesting to further investigate whether there is any biological benefit for the co-recruitment of Esa1 and Gcn5 to activate genes.
Histone modification in coding regions is often viewed as demarcating recent transcriptional events rather than playing a regulatory role. In this view, our analysis suggests that, along with methylation, acetylation also serves as a potent marker for transcription activities. On the other hand, H4 acetylation in coding regions may also have important regulatory roles. For example, the binding of the HDAC protein Hos2 to coding regions is important for active transcription [18,20]. The negative partial correlation between transcriptional activities and H4 acetylation levels is consistent with the aforementioned experimental results.
Materials and methods
Data sources
Datasets analyzed in this study include those for histone acetylation, nucleosome occupancy, gene expression, and genome sequence. In two recently published papers, genome-wide histone acetylation levels at eleven [2] and three sites [8] in yeast were measured using CHIP-chip. A major difference in experimental procedure between these two studies is that the acetylated DNA was hybridized against nucleosomal DNA on microarrays in Pokholok et al.'s study [8], but was hybridized against the genomic DNA in Kurdistani et al.'s study [2]. Since in the latter dataset histone acetylation was confounded with nucleosome occupancy, our discussion in the main text is focused on analyzing Pokholok et al.'s data. To compare the results from the two experiments, we repeated our analysis procedure on a normalized version of Kurdistani et al.'s data after removing its dependency on nucleosome occupancy. We found that the main conclusions remain the same. Detailed analysis of Kurdistani et al.'s data is presented in Additional data file 1.
In addition, several groups have measured genome-wide nucleosome occupancy in yeast [1,3,8]. We chose to utilize Pokholok et al.'s nucleosome occupancy data in our analysis as well, since nucleosome occupancy has a clear effect on gene regulation [6]. We used Bernstein et al.'s transcription rate data [1] as the response variable in our study of the relationship between gene transcription and histone acetylation. These transcription rates were estimated by dividing the transcription levels by half-life time [16]. Due to concern that measured microarray data may vary significantly among different microarray platforms or research groups [32-34], we repeated our analysis using an independent dataset [15]. The results obtained from the two gene transcription data are similar (Additional data file 1).
After removing genes (with their corresponding intergenic and coding regions) that have missing data in any of the above datasets, we merged all the data into a single dataset, which contains 3,049 intergenic and 3,384 coding regions. The genomic sequence of S. cerevisiae was downloaded from the Saccharomyces Genome Database [14]. The promoter sequences (up to 800 base pairs (bp) upstream of the translation start site of each gene) were extracted for cis regulatory signal analysis.
Delineating cis regulatory information
Transcription factors regulate genes by binding to transcription factor binding sites (TFBSs), which are short sequence segments (approximately 10 bp) located near genes' transcription start sites (TSSs). In yeast, these binding sites are mostly within 500 bp upstream of each gene's TSS. It has been shown that a gene's expression pattern can be predicted to a great extent by its upstream sequence information [26]. We took two different approaches to accommodate sequence information in our analysis of the histone acetylation effect.
In our first approach, we conducted de novo TFBS predictions using MDscan [24] among the upstream sequences of the genes that were transcribed at high rates [1]. In particular, this algorithm searched for enriched sequence motifs of widths 5 to 15 in the promoter sequences, resulting in 580 statistically significant, possibly overlapping, candidate TFBMs (p value < 0.05). We then used these motif patterns to scan all promoter regions for matches so as to compute a motif score for each TFBM at each promoter [24]. To avoid overfitting, we selected a subset of 33 functional motifs based on the association of the motif score of a promoter with the transcription rate of the corresponding gene. In particular, we used both a linear regression procedure, Motif Regressor [25], and a model-free method, regularized sliced inverse regression (RSIR) [35], as explained below.
Our second approach to account for the cis regulatory information was to directly use the 666 transcription factor binding motifs reported by Beer and Tavazoie [26], which is a combination of computational predictions using AlignACE [22] and 51 experimentally derived ones [36,37]. Since these motifs have been shown to have a high predictive power for gene expression patterns, they may also be informative for predicting transcription rate. Out of these 666 motifs, our linear regression and RSIR procedures (see below) found 15 that are highly relevant to predicting gene transcription rates.
Model free motif selection
RSIR [35] is a statistical method for dimension reduction and variable selection. It assumes that gene i's transcription rate yi and its sequence motif scores xi = (xi1,...,xiM)T are related as:
where f() is an unknown (and possibly nonlinear) function, βl = (βl1,...,βlM)T, (l = 1,...,k), are vectors of linear coefficients, and εi represents the noise. The number k is called the dimension of the model. A linear regression model is a special one-dimensional case of equation 3. RSIR estimates both k and the βl values without estimating f(). Since many entries of the βlj values are close to zero, which implies that the corresponding motif scores contribute very little in equation 2, we retain only those motifs whose coefficient βlj is significantly nonzero.
We applied RSIR to the 580 candidate motifs selected by MDscan and the 666 motifs from [26], with the transcription rate as the response variable. In both cases, k was estimated as 1, and f() showed a strong linear pattern. We found 104 and 69 motifs, respectively, that have significantly nonzero coefficients in our RSIR model.
Previous application suggests that RSIR is conservative in selecting variables [35]. We applied the stepwise regression algorithm (which is a recursive method commonly used for variable selection; see page 347 of [27]) to further reduce the number of motifs. In the end, a total of 33 motifs from MDscan and 15 motifs from [26] were retained for further use. These motifs represent our summary of sequence-specific information on gene transcription rates.
Model validation
To assess the significance of our model for controlling the confounding effects due to sequence information, we randomly permuted the transcription rates data 50 times and repeated the same statistical procedures: identifying motif candidates using MDscan, selecting the most significant motifs using RSIR, and fitting the linear regression model. The distribution of R2 obtained for these randomized data was used as a baseline to evaluate the significance of our statistical procedure.
We also performed a five-fold cross validation procedure to test whether equation 2 overfit data. In particular, the full set of genes (seethe Data sources section above) was randomly partitioned into five subsets of equal sizes. Each subset was used for testing in turn with the rest used for training. For each training subset, the sequence motifs were inferred using MDscan, RSIR, and stepwise regression methods. We fit the model equation 2 using the training data and then evaluated out-of-sample error by applying to the testing data. The in-sample and out-of-sample root mean square errors were then compared.
Partial correlation
Let X and Y represent two random variables and Z = (Z1,Z2,...,Zp) be a set of control random variables. The linear relationship between X and Z can be estimated via a linear regression model X = αX + ZβX + εX, similarly for that between Y and Z. The residues εX and ε Y contain the information left unexplained by Z. The partial correlation between X and Y while controlling Z is defined as the Pearson correlation between εX and εY.
Additional data files
The following additional data are available with the online version of this paper: Additional data file 1 contains supporting text, figures, and tables. The adequacy of the linear regression, normalization of the Kurdistani et al. data, and sensitivity issues are discussed in further detail in the text. The figures and tables included demonstrate and compare the Pokholok et al. and Kurdistani et al. data.
Supplementary Material
Supporting text, figures, and tables relating to the Kurdistani et al., Pokholok et al. and Kurdistani et al. data.
Acknowledgments
Acknowledgements
We thank Oliver Rando for helpful discussions. GY was partially supported by the Bauer Center for Genomics Research. PM was partially supported by a research board grant from University of Illinois. JSL acknowledges support from NSF DMS-0204674 and a grant (10228102) from NSF China.
Contributor Information
Guo-Cheng Yuan, Email: gyuan@cgr.harvard.edu.
Ping Ma, Email: pingma@uiuc.edu.
Wenxuan Zhong, Email: wenxuan@stat.harvard.edu.
Jun S Liu, Email: jliu@stat.harvard.edu.
References
- Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL. Global nucleosome occupancy in yeast. Genome Biol. 2004;5:R62. doi: 10.1186/gb-2004-5-9-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdistani SK, Tavazoie S, Grunstein M. Mapping global histone acetylation patterns to gene expression. Cell. 2004;117:721–733. doi: 10.1016/j.cell.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet. 2004;36:900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- Robert F, Pokholok DK, Hannett NM, Rinaldi NJ, Chandy M, Rolfe A, Workman JL, Gifford DK, Young RA. Global position and recruitment of HATs and HDACs in the yeast genome. Mol Cell. 2004;16:199–209. doi: 10.1016/j.molcel.2004.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion MF, Altschuler SJ, Wu LF, Rando OJ. Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci USA. 2005;102:5501–5506. doi: 10.1073/pnas.0500136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- Liu CL, Kaplan T, Kim M, Buratowski S, Schreiber SL, Friedman N, Rando OJ. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 2005;3:e328. doi: 10.1371/journal.pbio.0030328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Turner BM. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/S0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- Schreiber SL, Bernstein BE. Signaling network model of chromatin. Cell. 2002;111:771–778. doi: 10.1016/S0092-8674(02)01196-0. [DOI] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Kurdistani SK, Grunstein M. Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol. 2003;4:276–284. doi: 10.1038/nrm1075. [DOI] [PubMed] [Google Scholar]
- Saccharomyces Genome Database http://www.yeastgenome.org/
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/S0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert J, Struhl K. Histone acetylation at promoters is differentially affected by specific activators and repressors. Mol Cell Biol. 2001;21:2726–2735. doi: 10.1128/MCB.21.8.2726-2735.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Kurdistani SK, Grunstein M. Requirement of Hos2 histone deacetylase for gene activity in yeast. Science. 2002;298:1412–1414. doi: 10.1126/science.1077790. [DOI] [PubMed] [Google Scholar]
- Kurdistani SK, Robyr D, Tavazoie S, Grunstein M. Genome-wide binding map of the histone deacetylase Rpd3 in yeast. Nat Genet. 2002;31:248–254. doi: 10.1038/ng907. [DOI] [PubMed] [Google Scholar]
- Wiren M, Silverstein RA, Sinha I, Walfridsson J, Lee HM, Laurenson P, Pillus L, Robyr D, Grunstein M, Ekwall K. Genomewide analysis of nucleosome density histone acetylation and HDAC function in fission yeast. EMBO J. 2005;24:2906–2918. doi: 10.1038/sj.emboj.7600758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JS, Neuwald AF, Lawrence CE. Bayesian models for multiple local sequence alignment and gibbs sampling strategies. J Am Stat Assoc. 1995;90:1156–1170. doi: 10.2307/2291508. [DOI] [Google Scholar]
- Roth FP, Hughes JD, Estep PW, Church GM. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- Bussemaker HJ, Li H, Siggia ED. Regulatory element detection using correlation with expression. Nat Genet. 2001;27:167–171. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- Liu XS, Brutlag DL, Liu JS. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat Biotechnol. 2002;20:835–839. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- Conlon EM, Liu XS, Lieb JD, Liu JS. Integrating regulatory motif discovery and genome-wide expression analysis. Proc Natl Acad Sci USA. 2003;100:3339–3344. doi: 10.1073/pnas.0630591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer MA, Tavazoie S. Predicting gene expression from sequence. Cell. 2004;117:185–198. doi: 10.1016/S0092-8674(04)00304-6. [DOI] [PubMed] [Google Scholar]
- Neter J, Kutner MH, Wasserman W, Nachtsheim CJ. Applied Linear Statistical Models. 4. Singapore; Boston: McGraw-Hill; 1996. [Google Scholar]
- Brown CE, Howe L, Sousa K, Alley SC, Carrozza MJ, Tan S, Workman JL. Recruitment of HAT complexes by direct activator interactions with the ATM-related Tra1 subunit. Science. 2001;292:2333–2337. doi: 10.1126/science.1060214. [DOI] [PubMed] [Google Scholar]
- Schubeler D, Turner BM. A new map for navigating the yeast epigenome. Cell. 2005;122:489–492. doi: 10.1016/j.cell.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Reid JL, Iyer VR, Brown PO, Struhl K. Coordinate regulation of yeast ribosomal protein genes is associated with targeted recruitment of Esa1 histone acetylase. Mol Cell. 2000;6:1297–1307. doi: 10.1016/S1097-2765(00)00128-3. [DOI] [PubMed] [Google Scholar]
- Reid JL, Moqtaderi Z, Struhl K. Eaf3 regulates the global pattern of histone acetylation in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:757–764. doi: 10.1128/MCB.24.2.757-764.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bammler T, Beyer RP, Bhattacharya S, Boorman GA, Boyles A, Bradford BU, Bumgarner RE, Bushel PR, Chaturvedi K, Choi D, et al. Standardizing global gene expression analysis between laboratories and across platforms. Nat Methods. 2005;2:351–356. doi: 10.1038/nmeth0605-477a. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Warren D, Spencer F, Kim IF, Biswal S, Frank BC, Gabrielson E, Garcia JG, Geoghegan J, Germino G, et al. Multiple-laboratory comparison of microarray platforms. Nat Methods. 2005;2:345–350. doi: 10.1038/nmeth756. [DOI] [PubMed] [Google Scholar]
- Larkin JE, Frank BC, Gavras H, Sultana R, Quackenbush J. Independence and reproducibility across microarray platforms. Nat Methods. 2005;2:337–344. doi: 10.1038/nmeth757. [DOI] [PubMed] [Google Scholar]
- Zhong W, Zeng P, Ma P, Liu JS, Zhu Y. RSIR: regularized sliced inverse regression for motif discovery. Bioinformatics. 2005;21:4169–4175. doi: 10.1093/bioinformatics/bti680. [DOI] [PubMed] [Google Scholar]
- Hughes JD, Estep PW, Tavazoie S, Church GM. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting text, figures, and tables relating to the Kurdistani et al., Pokholok et al. and Kurdistani et al. data.