

Abstract
Quantitative real-time PCR (qPCR) is a commonly used validation tool for confirming gene expression results obtained from microarray analysis; however, microarray and qPCR data often result in disagreement. The current study assesses factors contributing to the correlation between these methods in five separate experiments employing two-color 60-mer oligonucleotide microarrays and qPCR using SYBR green. Overall, significant correlation was observed between microarray and qPCR results (ρ=0.708, p<0.0001, n=277) using these platforms. The contribution of factors including up- vs. down-regulation, spot intensity, ρ-value, fold-change, cycle threshold (Ct), array averaging, tissue type, and tissue preparation was assessed. Filtering of microarray data for measures of quality (fold-change and ρ-value) proves to be the most critical factor, with significant correlations of ρ>0.80 consistently observed when quality scores are applied.
Keywords: Polymerase Chain Reaction, Microarray Analysis, Gene Expression, Nucleic Acid Amplification Techniques, Reverse Transcriptase Polymerase Chain Reaction, RNA
Introduction
DNA microarrays provide an unprecedented capacity for whole genome profiling. However, the quality of gene expression data obtained from microarrays can vary greatly with platform and procedures used. Quantitative real-time PCR (qPCR) is a commonly used validation tool for confirming gene expression results obtained from microarray analysis; however, microarray and qPCR data often result in disagreement. Presently, no standard definition of validation exists, correlations of qPCR and microarray data are seldom presented in the literature, and non-agreeing data are rarely explained. It is well documented that both qPCR and microarray analysis have inherent pitfalls (1-5) that may significantly influence the data obtained from each method. Additionally, many different platforms exist for both microarray and qPCR analyses that have led to debate over which methods produce the most accurate measurements of gene expression (6-12). In this study we compiled data from five independent experiments to establish the degree of correlation between two-color inkjet printed 60-mer oligonucleotide microarrays and qPCR using SYBR green. Using this compiled data set we sought to identify factors that influence the correlation between these two techniques.
Variability in both biological and technical procedures can have a great impact on both microarray and qPCR results (2, 4) and, as biological variability cannot be controlled, care must therefore be taken in the experimental design to minimize irregularities and ensure adequate replication to eliminate “noise” in the experiment. The quality of RNA is essential to accurate results, as gene expression can be affected by carry-over of contaminating factors (e.g., different tissues, airborne particles, etc.), and salts, alcohols, and phenol, which can affect reverse transcriptases used in both qPCR and RNA amplification procedures for microarray labeling (3). Furthermore, different efficiencies of reverse transcriptases and varied priming methods can also affect the results of qPCR and microarray experiments (3). The effects of dye biases (due, in part, to the physical properties of various dyes that affect efficiencies of incorporation) (5) and non-specific and/or cross hybridizations of labeled targets to array probes (2) are unique to microarray procedures. Likewise, qPCR has its own sources of error including amplification biases (2), the exponential amplification of errors (3), mispriming or the formation of primer dimers (1), and the changing efficiency of qPCR at later cycles (3, 13). In addition, data normalization fundamentally differs between microarray analysis and qPCR, the former requiring global normalization, while the latter generally utilizes the expression of one or more reference genes against which all other gene expression is calibrated. Therefore, selection and appropriate application of normalization criteria may also play a major role in the correlations found between these methods. While the above mentioned list of the potential pitfalls in microarray and qPCR methodologies is long, most sources of error can be controlled through robust experimental designs, good laboratory practices, and rigorous normalization of the data.
A survey of the literature reveals widely ranging correlations between microarray and qPCR data of -0.48 to 0.94 (14-16 and others). Moreover, rarely are these correlations presented with statistical analyses and few authors define the criteria they used to determine acceptable validation of microarray results. Rajeevan et al. (17) considered a result valid if the fold change measured by both qPCR and microarray were greater than or equal to 2-fold. They did not consider the magnitude of difference between the measurements, which Svaren et al. (18) found to vary significantly. More commonplace in the literature is simply the statement that results were validated, often with no, or extremely low, reported correlations.
Several studies have attempted to determine what factors contribute to the variation in results obtained by microarray versus qPCR. Lower correlations were consistently reported for genes exhibiting small degrees of change, generally less than 2-fold, as compared to those showing greater than 2-fold change (4, 15, 19). In addition, Etienee et al. (15) found that increased distance between the location of the PCR primers and microarray probes on a given gene also decreased the correlation between the two methods. Beckman et al. (14) investigated the effects of array spot intensity on correlation, finding that low intensity spots (intensities less than the highest intensity of negative controls) and spots with one high intensity and one low intensity (for 2-color arrays) had considerably lower correlations with qPCR data than high intensity spots (intensities greater than the highest intensity of negative controls). While these studies provide insight into some sources of variation in qPCR and array correlation, we chose to additionally investigate parameters of tissue type, sample preparation, and data quality in the context of the particular platforms used in a gene expression study.
Oligonucleotide microarrays have become a widely used alternative to cDNA microarray because of their superior specificity, reproducibility, and ease of design (20). However, their ability to accurately report changes in gene expression has been debated (10-12, 21 and others). Furthermore, given the variation in reported correlations between microarray and qPCR results, we wanted to assess which aspects of each method may influence correlation and to determine if we can define array responses that, given specific data parameters, will consistently yield significant correlations of 0.80 or greater. To this end we have conducted an analysis of the correlations observed between two-color oligonucleotide microarrays and qPCR results, using SYBR Green, from five independent experiments. In studies 1 and 2, adult mice were exposed to the excitatory neurotoxin, domoic acid (DA), and brain transcriptional response was determined over an acute time course or dose response in freshly prepared tissue (22). In a third experiment, the time course of blood transcriptional response to DA was investigated using Qiagen’s PaxGene tubes. Frozen tissue was used to investigate the transcriptional response in brain from mice exposed to the potent neurotoxin brevetoxin (PbTx) was investigated in the fourth experiment. Experiment 5 investigated the transcriptional response of a human T-lymphocyte cell line to the phycotoxin azaspiracid (AZA). The data sets resulting from microarray and qPCR analyses were first compared to determine at what frequencies the general trends of up- or down-regulation were conserved. The effects of several parameters on the correlation of data were also investigated, including up- vs. down-regulation, spot intensity, p-value from microarray analysis, fold change, cycle threshold (Ct) values, the use of individual or composite array data, tissue type, and the use of fresh or frozen tissue.
Methods
Domoic acid studies
All studies were conducted in accordance with NIH guidelines for the ethical care and use of laboratory animals.
Time course in brain: Studies on brain transcriptional profiles were carried out as described in Ryan et al. (22). Briefly, female mice were dosed intraperitoneally (IP) with 4 mg·kg-1 DA, the LD50, (cat. # D6152, Sigma, St. Louis, MO) in PBS while the control group was dosed with volumetric equivalents of PBS. Three animals per treatment or control group were sacrificed at 30 min, 60 min, and 240 min post-injection by cervical dislocation and the brain from each mouse was immediately dissected and prepared for RNA extraction. Brains from control animals were pooled prior to RNA extraction while RNA was extracted from the brains of experimental animals individually.
Dose response in brain: Studies on brain transcriptional profiles were carried out as described in Ryan et al. (22). Briefly, male animals were dosed by IP injection with 1 or 4 mg·kg-1 DA in PBS, while the controls were dosed with volumetric equivalents of PBS. All mice were sacrificed at 60 min by cervical dislocation, the brains dissected immediately and the tissue prepared for RNA extraction. As in the time course study, brains from three control animals were pooled prior to RNA extraction while RNA was extracted from the brains of experimental animals individually.
Dose response in blood: Adult female ICR mice (19-22g) were maintained as described in Ryan et al. (22). The experimental animals were dosed by IP injection with 2.5 mg·kg-1 DA in PBS, while the controls were dosed with volumetric equivalents of PBS. At 12, 24, or 48 h 3 mice per treatment or control group were deeply anesthetized with isoflurane (Baxter, Deerfield, IL) and blood was collected via cardiac puncture and stored in Paxgene (Qiagen, Valencia, CA) tubes at room temperature until the RNA extraction procedure was completed the following day. The blood from three control animals was pooled and split between two Paxgene tubes prior to extraction while RNA was extracted from the blood of each experimental animal individually.
Brevetoxin studies
Adult female ICR mice (19-22g) were maintained as described in Ryan et al. (22). The experimental animals were dosed by IP injection with an acute dose of 130 μg·kg-1 brevetoxin-3 (PbTx-3) in PBS with 4% methanol, while the controls were dosed with volumetric equivalents of methanolic vehicle. At 30, 60, or 240 min 6 mice per treatment group and 3 controls were sacrificed by cervical dislocation; the brains dissected immediately and flash frozen until RNA preparation. Prior to RNA extraction, 3 brains per control or treatment group were pooled.
Azaspiracid studies
Human Jurkat E6-1 lymphocyte T cells (ATCC # TIB-152) were grown in RPMI medium supplemented with 10% (v/v) fetal bovine serum (FBS) and maintained in humidified 5%:95% CO2:air at 37°C. Azaspiracid (AZA-1) extracted from mussels (Mytilus edulis) was determined to be > 93% pure by NMR and showed < 1% impurity of other AZA subtypes/cogners by liquid chromatograph-mass spectrometry (LC-MS) (23). For each experimental replicate (n=2), 60 mL of Jurkat cells were centrifuged at 1000xg for 7 min and resuspended in 40 mL of fresh RPMI medium supplemented with FBS. Freshly resuspended cells were inoculated into 35 mm Petri dishes containing 2 mL total volume. Total cell numbers per dish ranged from 4.4 x 106 to 10.6 x 106 cells for the separate replicates. Cells were allowed to grow for at least 12 h prior to addition of AZA-1 (10 nM final concentration) or equivalent amounts of methanolic vehicle (0.1% v/v final). Dishes were harvested for RNA extractions at 1, 4, and 24 h and cells were flash frozen in liquid nitrogen and stored at -80°C until RNA was extracted.
RNA extraction
Mouse brain and Jurkat cells: Following tissue dissection or cell harvesting, total RNA was extracted using Tri-Reagent according to the manufacturer’s protocol (Molecular Research Center, Inc., Cincinnati, OH). After re-suspension in nuclease-free water, RNA was purified using RNeasy columns (Qiagen), quantified by UV-Vis spectroscopy, and qualified on a 2100 Bioanalyzer (Agilent, Palo Alto, CA).
Blood: Upon deposition to the Paxgene tube (Qiagen) blood cells are lysed and RNA is stabilized. RNA extraction and purification was carried out according to the manufacturer's protocol, which includes a RNA clean-up step. Following processing, total RNA was quantified by UV-Vis spectroscopy and qualified on a 2100 Bioanalyzer (Agilent).
In all studies, the same RNA was used for both microarray and real-time PCR analyses.
Microarray
Total RNA was amplified and labeled with Cy3-dCTP or Cy5-dCTP (Perkin Elmer, Boston, MA) using the Agilent Low Input Linear Amplification kit according to manufacturer’s protocols. Following labeling and clean-up, cRNA was quantified by UV-Vis spectroscopy and 0.8-1 μg each of Cy3 and Cy5 labeled targets were combined and hybridized to Agilent arrays. The mouse brain DA time course and dose response experiments utilized an Agilent mouse 22K feature oligonucleotide microarray, while the mouse blood dose response and brain PbTx TC utilized the 44K mouse whole genome array. For all DA experiments, triplicate arrays were run, including a dye reversal to account for any dye bias. Because 3 experimental samples at each timepoint were pooled (final n=2) for the PbTx study, duplicate arrays were run which included a dye swap. The azaspiracid time course used the human whole genome 44K microarray, which was also run in duplicate with a dye swap. All arrays were hybridized and processed using a SSPE wash according to manufacturer’s protocols. Microarrays were imaged using an Agilent microarray scanner. Images were extracted with Agilent Feature Extraction version A7.5.1 and data analyzed with Rosetta Luminator 2.0 gene expression analysis system (Rosetta Informatics, Seattle, WA). Using a rank consistency filter, features were subjected to a combination linear and LOWESS normalization algorithm, the recommended algorithm for this microarray platform. This normalization allows non-linear corrections at intensities where dye chemistries introduce artifactual signal and allows linear corrections where signal intensities are linear in behavior. Based on the Rosetta error model designed for the Agilent platform, a composite array was generated at each time point from replicate arrays, in which the data for each feature underwent a weighted averaging based on feature quality from the individual array.
Quantitative real-time PCR
One microgram of total RNA was reverse transcribed using Ambion’s RETROscript kit with oligo(dT) primers for the 2-step qRT-PCR assays. Gene specific primers were used to amplify message by qPCR on a Cepheid Smart Cycler (Sunnyvale, CA) using the Qiagen SYBR Green master mix or on an ABI 7500 using the ABI SYBR Green master mix (Foster City, CA). Primer sets were designed against the complete nucleotide sequence, as deposited on GenBank, using Vector NTI 9.0.0 (InforMax, Frederick, MD). The optimum annealing temperature for each primer set was determined prior to the analysis of experimental samples. The specificity of each primer set and molecular weight of the amplicon were monitored by dissociation curve analysis and further verified by analysis using Agilent’s Bioanalyzer 2100. A sample volume of 25 μl was used for all assays, which contained a 1X final concentration of SYBR green PCR master mix, 400 nM gene specific primers, and 1 μl template. All samples and standards were run in triplicate, except for the azaspiracid time course which was run in duplicate. Assays were run using the following protocol: 95°C for 15 min or 10 min (Qiagen or ABI master mix, respectively), 94°for 15 sec, gene specific annealing temperature (55°-64°C) for 40 sec, 72°C for 1 min for 40 cycles, followed by a gradual increase in temperature from 60°C to 95°C during the dissociation stage. Table 3 (in the Supplemental information) details the genes validated by qPCR and assay conditions.
Following amplification, the instrument software was used to set the baseline and threshold for each reaction. A cycle threshold (Ct) was assigned at the beginning of the logarithmic phase of PCR amplification and the difference in the Ct values of the control and experimental samples were used to determine the relative expression of the gene in each sample. Prior to quantitative analysis, a standard curve was constructed using serial dilutions of RT product (species and tissue specific) and the efficiency of each primer set was determined using the equation [(10 (-1/-slope)-1)·100]. Efficiencies of 90-110% were required to include the qPCR assay in array validation. Relative expression levels between samples were then calculated as fold changes, where each PCR cycle represents a two-fold change. Therefore, the assay-specific efficiency was not used in the calculation of relative expression levels. For each experiment, a specific gene was chosen for normalization that did not exhibit any significant change in expression via microarray. All mouse experiments used tubulin, alpha 4 (NM_009447) for normalization while the human AZA study utilized an alpha tubulin-like gene (NM_145042). Statistical analysis was performed using a Wilcoxon/Kruskal-Wallis nonparametric test or a one-way ANOVA in JMP version 5.1.2 (SAS Institute Inc., Cary, NC).
Analysis of correlation between microarray and qPCR
Subsequent to microarray data analysis a set of genes was chosen for validation by qPCR based on their degree of expression change, p-value, and/or known effects of the toxin studied. Correlation between the microarray and qPCR results for this gene set was then performed for each experiment, and the statistical significance of the correlations determined. For the microarray, the data input into the correlation analysis was the Log2 ratio value of the weighted average for each gene on the composite array representing all replicate animals. For qPCR, we used the mean Log2 ratio value reported by qPCR from all replicate animals. Prior to performing correlation analyses, the data were tested for normality using the Shapiro-Wilk test. Because the data was not normally distributed, Spearman’s Rho was used. Spearman’s Rho is the rank-based non-parametric equivalent of the more commonly used Pearson’s correlation calculation. The effects of Ct, array spot p-value, degree of change, direction of change, and array spot intensity on correlation were investigated by binning subsets of genes according to these criteria. One-way ANOVAs were then used to determine the relationship between the observed correlations. To determine if the use of the weighted average from composite arrays influenced the correlation between microarray and qPCR, the correlations from the DA time course experiment were also calculated using the array and qPCR data from individual animals. As the RNA for the DA studies was extracted from fresh tissues while the RNA for the PbTx-3 study was extracted from frozen tissue, these studies were compared to determine any effects on data correlation. All statistical analyses utilized an alpha value of 0.05 and were performed using JMP version 5.1.2.
Results and Discussion
Data from five different gene expression profiling experiments outlined above, using three different inkjet printed Agilent 60-mer array designs in a 2-color format and SYBR Green qPCR, were analyzed both individually and combined to form a single large data set (Table 1). These five studies utilized RNA from several different sources; mouse brain, both fresh and frozen, mouse blood, and a human cell line. Of the 5 data sets, 3 were analyzed strictly to validate microarray results for publication, in which a selection of genes of biological interest identified as substantially up- or down-regulated by the array were verified by qPCR. In the mouse brain DA time course (TC) and dose response (DR) several genes exhibiting either non-significant, very minor, or no changes by microarray were also analyzed by qPCR in order to provide insight into the effects of fold change and data quality on the correlation between these two methods.
Table 1.
Correlations of microarray and qPCR data.
| Data Set | # Genes Verified | Correlation | p-value | n |
| Mouse brain DA TC | 28 | 0.686 | <0.0001 | 84 |
| Mouse brain DA DR | 29 | 0.676 | <0.0001 | 58 |
| Mouse blood DA TC | 12 | 0.748 | <0.0001 | 36 |
| Mouse brain PbTx TC | 13 | 0.633 | <0.0001 | 39 |
| Human AZA TC | 20 | 0.727 | <0.0001 | 60 |
| All Data Sets | 68 | 0.708 | <0.0001 | 277 |
| Five data sets were analyzed, both individually and combined to form a sixth large data set referred to as "all data." The number of individual genes verified for each data set is shown as well as the resulting total number of data points used for the calculations of correlation (n). All correlations were calculated using Spearman’s Rho. DA, domoic acid; PbTx, brevetoxin; AZA, azaspiracid; TC, time course; DR, dose response. | ||||
Overall, a significant correlation of 0.708 was observed in the combined data set (Spearman’s Rho, p<0.0001, n=277). Correlations for the individual data sets ranged from 0.633 – 0.748 (p<0.0001, Table 1). The direction of change in expression was in agreement by both qPCR and microarray for 72.9% of samples (202 of 277). In 59 of the 75 samples (78.7%) where the reported direction of change differed, the changes reported by both methods were minor (<1.4 fold). The samples that did not report the same direction of change by both methods had similar distributions of spot intensity, p-values, and Ct as the samples that did yield agreement in direction of change. This lack of concurrence between methods for genes exhibiting low levels of change (<1.4 fold) has been commonly reported (4, 19, 24).
Up- vs. down-regulation
A correlation of 0.700 (Spearman’s Rho, p<0.0001, n=169) was observed among genes exhibiting up-regulation by microarray and was significantly different than the correlation of 0.356 (Spearman’s Rho, p=0.0002, n=108) observed among down-regulated genes (ANOVA, p=0.0042, n=10). This trend was observed in all data sets except the mouse blood DA TC, in which down-regulated genes showed a slightly higher correlation than up-regulated genes (Fig. 1a). It is interesting to note, the mouse blood DA TC is the only data set that included a greater number of down-regulated genes than up-regulated genes exhibiting 1.4 fold change or greater. Overall, 72.2% of down regulated genes exhibited less than 1.4 fold change whereas only 60.4% of upregulated genes exhibited these low levels of changes in expression. The influence of the degree of fold change on data correlation will be discussed later.
Fig. 1. Analysis of data correlation categorized by direction of regulation, spot intensity, and cycle threshold.
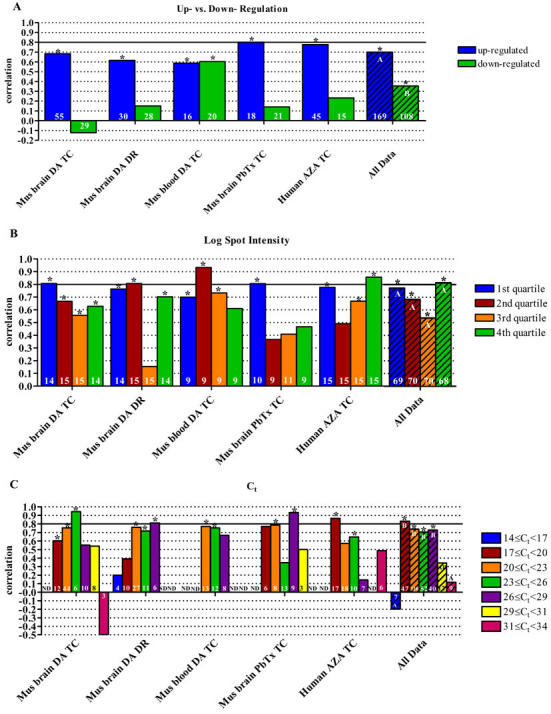
Correlation of microarray and qPCR data as it relates to (A) direction of regulation, (B) Log (spot intensity), and (C) cycle threshold. Spot intensity data was binned by quartiles and thus, as the intensities from each experiment differed slightly, actual intensities are not indicated in the legend. Asterisks indicate a statistically significant correlation of array and qPCR data (p<0.05). The hatched bars represent the compilation of the five individual data sets, referred to as "all data." Statistical differences of correlations, determined by ANOVA, are indicated by different letters. All correlations were calculated using Spearman’s Rho. The number of samples included in each correlation is shown in the base of the bar. ND, no data (insufficient sample size, n≤2); DA, domoic acid; PbTx, brevetoxin; AZA, azaspiracid; TC, time course; DR, dose response.
A similar trend of higher correlations among up-regulated genes was observed by Beckman et al. (%R[14}%), who proposed that this effect may be due to the increased variability observed in low-intensity array spots, i.e. down-regulated genes. However, in the current study, no trend was apparent in the effects of average array spot intensity on correlation of data (Fig. 1b). While the current study does not support the results of the study by Beckman et al. (14) this may be due to the fact that our genes were selected for verification following initial microarray analysis. Analysis using Agilent Feature Extraction and Rosetta Luminator software identifies spots likely to introduce errors due to signal strength, high background, and/or poor spot morphology, etc. Therefore, the problems introduced by low signal to noise ratios that were investigated by Beckman et al. (14) were likely excluded from analysis in our study and the observed effects are most likely influenced by the degree of change exhibited.
The lower correlation between the array and qPCR for down-regulated genes may alternatively be due to the effects of greater variability associated with decreased reaction efficiencies found in qPCR measurements at later cycles, where genes with low expression levels respond. In general, we observed significantly lower correlations at early and late cycle thresholds, especially in samples with Ct < 17 or Ct ≥ 31 (Fig. 1c, Kruskal-Wallis, p=0.0237, n=31). While we only investigated the effects of average signal intensity, the low correlations observed at early Cts (i.e. highly expressed genes or markedly up-regulated genes) may be attributed to the effects of large intensity ratios due to large differences in expression between the treatments compared as examined by Beckman et al. (14). In general, genes exhibiting late Ct values corresponded to low intensity microarray spots: 75.6% of genes with Ct values above the median exhibited below-median spot intensity, and thus likely represent genes with low expression levels. Likewise, 75.6% of genes with Ct values below the median exhibited above-median spot intensity and thus were likely genes with high levels of expression.
Fold change
In general, correlations increased with increasing degree of change as measured by both microarray and qPCR. Wurmbach et al. (4) reported 100% validation of results for genes exhibiting at least 1.6 fold change; however, they defined validation as directional confirmation only and large discrepancies in the amount of change were not addressed in their study. Dallas et al. (25) reported decreased correlations for genes expressing less than 1.5 fold change using probe based qPCR and oligonucleotide microarrays. More commonly, a 2 fold change is reported as the cutoff below which microarray and qPCR data begin to lose correlation. In the current study we consistently observed significant correlations of at least 0.75 where genes exhibited 1.4 fold change or higher (Figs. 2a and 2b). Genes exhibiting at least 1.4 fold change had significantly higher correlations than those demonstrating less change by both microarray (ANOVA, p<0.0001, n=12) and qPCR (ANOVA, p=0.0005, n=12).
Fig. 2. Analysis of data correlation categorized by fold change.
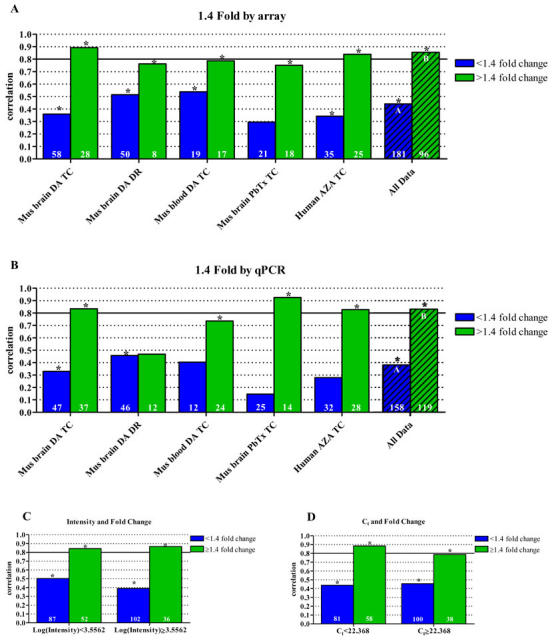
Correlation of microarray and qPCR data as it relates to (A) fold change measured by microarray and (B) fold change measured by qPCR. The combined data set of "all data" was queried first by microarray fold change (1.4 fold cut-off) and then by (C) spot intensity and (D) Ct values to determine the overall impact of fold change on array and qPCR data correlations. Asterisks indicate a statistically significant correlation (p<0.05). The hatched bars represent the compilation of the five individual data sets, referred to as "all data." Statistical differences of correlations, determined by ANOVA, are indicated by different letters. All correlations were calculated using Spearman’s Rho. The number of samples included in each correlation is shown in at the base of the bar. ND, no data (insufficient sample size, n≤2); DA, domoic acid; PbTx, brevetoxin; AZA, azaspiracid; TC, time course; DR, dose response.
As fold change in expression is commonly used to filter microarray data, we queried the combined data set to determine the limitations of our system based on fold change measured by microarray. Figures 2c and 2d show the combined effects of fold change and spot intensity or cycle threshold and illustrate the prevailing impact of fold change on data correlation. Genes exhibiting less than 1.4 fold change had data correlations of 0.40-0.50, regardless of spot intensity or Ct (Figs. 2c and 2d). However, when fold change was 1.4 or greater, again regardless of spot intensity or Ct, significant data correlations of at least 0.80 were observed. Further, the correlations presented in Table 1 have a nearly direct relationship with fold change. The mouse blood DA TC, which exhibited the highest correlation of 0.748, has the highest percentage (50%) of data points exhibiting 1.4 fold change or greater. The mouse brain DA DR, exhibiting the 2nd lowest correlation (0.676), has only 13.8% of data points exhibiting 1.4 fold change or greater.
Microarray spot p-value
Overall, microarray spot p-value appeared to have a considerable effect on the correlations between array and qPCR results (Fig. 3a). Among the genes assayed by qPCR, those that were labeled as “signature” (genes with a composite p≤0.01) by the Luminator gene expression analysis software package had a correlation of 0.847 (Spearman’s Rho, p<0.0001, n=107, data not shown), whereas genes that were not called “signature” only exhibited a correlation of 0.435 (Spearman’s Rho, p<0.0001, n=170, data not shown). However, as indicated by the mouse blood DA TC and Human AZA TC data sets, a p-value of 0.01 or less does not always yield high correlations (Fig. 3a). Genes with a p-value of 0.0001 or less exhibited a statistically significantly higher correlation than genes with greater p-values (ANOVA, p=0.0007, n=22). The calculation of a composite p-value includes measurements of signal strength, background levels, spot morphology, and fold change from all replicate arrays (http://www.rosettabio.com). Therefore, the smaller the p-value reported, the more confidence in the accuracy of the microarray results, as the errors discussed by Chuaqui et al. (2 and others), including experimental noise and non-specific or cross hybridization, are discounted. As many microarray data analysis programs do not report p-values, a stringent filtering of array data using signal to noise ratios and the coefficient of variation from replicate arrays may yield a final data set of high quality and increased accuracy, and thus, increased correlations with qPCR results.
Fig. 3. Analysis of data correlation categorized by p-values from microarray analyses. (A).
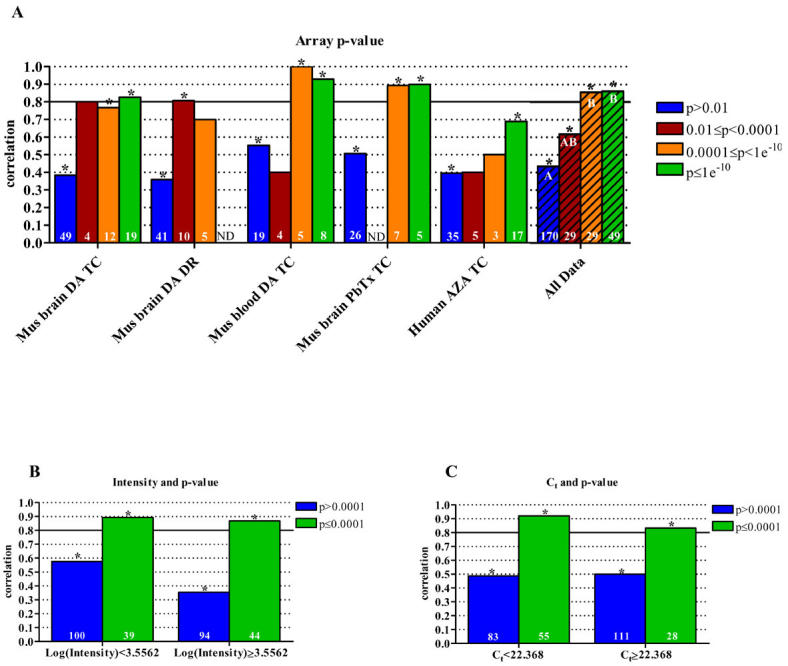
Correlation of microarray and qPCR data as it relates to microarray spot p-values. The combined data set of "all data" was analyzed by (B) spot intensity and (C) Ct values to determine the effect of p-values (0.0001 cutoff) on array and qPCR data correlation. P-values are based on calculations including signal strength, background values, spot morphology, fold change, variation between replicates, etc. Asterisks indicate a statistically significant result (p<0.05). The hatched bars represent the compilation of the five individual data sets, referred to as "all data." Statistical differences of correlations, determined by ANOVA, are indicated by different letters. All correlations were calculated using Spearman’s Rho. The number of samples included in each correlation is shown in the base of the bar. ND, no data (insufficient sample size, n≤2); DA, domoic acid; PbTx, brevetoxin; AZA, azaspiracid; TC, time course; DR, dose response.
Again, we queried the combined data set based on array p-values to determine the limitations of our system, as p-values (or some other measure of data quality such as background to noise ratios) are commonly used to filter microarray data. As shown in Figures 3b and 3c, significant correlations of at least 0.80 are observed for genes with a p-value of 0.0001 or less. As with fold change, this analysis demonstrates the predominant effect of array data p-value on microarray and qPCR correlations, as genes yielding highly significant array results generated high correlations regardless of spot intensity or Ct values (Fig. 3b and 3c). Given that fold change is a component of the p-values generated by Agilent and Rosetta data analysis software, the overall effect of fold change on data correlation is likely to be significant as shown in Figure 4. However, genes with a p-value greater than 0.0001 exhibiting a fold change of 1.4 or greater, only yielded a correlation of 0.676 (Spearman’s Rho, p=0.0003, n=24), whereas genes with a p-value of 0.0001 or less and exhibiting at least a 1.4 fold change resulted in a significant correlation of 0.905 (Spearman’s Rho, p<0.0001, n=72). This increase in correlation between microarray and qPCR for highly significant genes demonstrates the importance of microarray data quality, demonstrated here as composite p-values, and not only fold change on the accuracy of results.
Fig. 4. Combined effects of array fold change and p-value on data correlation.
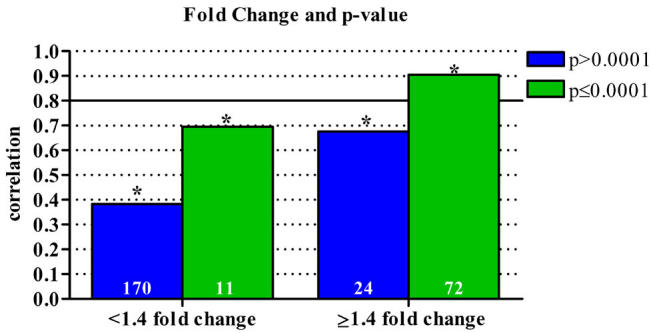
Correlation of microarray and qPCR data as it relates to both array fold change and p-value. Analyses of the combined data set of "all data" indicate that fold change has the greatest impact on array and qPCR data correlation. However, array data quality, measured here as a p-value, is essential to predicting reliable data. Asterisks indicate a statistically significant correlation (p<0.05). All correlations were calculated using Spearman’s Rho. The number of samples included in each correlation is shown in the base of the bar.
Composite array data
The correlation analyses presented above are based on a single array value for each gene. This value is derived from the composite array produced by the analysis software, in which each feature of replicate arrays underwent a weighted averaging based on feature quality. In contrast, the data reported for qPCR was the un-weighted average of qPCR results from replicate animals. Thus, the microarray and qPCR results may not directly correlate as they were averaged in a different manner. To determine if the common practice of using the composite array biased our results, we next calculated the correlation of the DA time course experiment in brain in two ways, comparing the composite array value to the average qPCR value or comparing the individual array and qPCR values for each animal. For all queries of the data set, the correlation between composite array data and qPCR data was very similar to the correlation of individual array data and qPCR data and all trends previously discussed were maintained (Fig. 5). Overall, the composite array and average qPCR values resulted in a correlation of 0.686 (Spearman’s Rho, p<0.0001, n=84), while the individual array and qPCR values resulted in a slightly lower correlation of 0.607 (Spearman’s Rho, p<0.0001, n=244). Thus, while minor differences were observed, it does not appear that the use of the composite array appreciably influenced the observed correlations with qPCR data (Wilcoxon, p=0.2014, n=56). If anything, the composite array yielded a higher correlation, possibly by minimizing the contribution of poorer quality array spots.
Fig. 5. Effects of composite array use on array and qPCR data correlation.
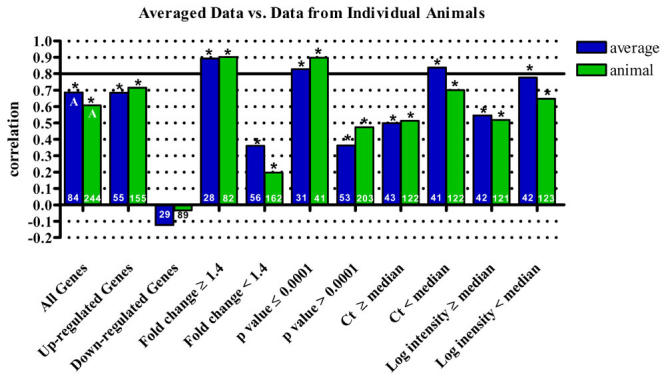
Correlation of microarray and qPCR results based on data from individual animals versus the use of weighted averages from composite array data. While minor differences were observed depending on the data set used, one data set did not consistently yield higher correlations. It does not appear that the use of composite arrays appreciably influence the observed correlations with qPCR data. Asterisks indicate a statistically significant correlation (p<0.05). All correlations were calculated using Spearman’s Rho. Statistical differences of correlation, determined by Wilcoxon’s test, are indicated by different letters in the "all genes" data set. The number of samples included in each correlation is shown in the base of the bar.
Fresh tissue vs. frozen tissue
It is well documented that RNA quality may be severely impacted by handling and storage conditions (1, 26-28, and others). As RNA was extracted from fresh tissue for the DA TC and DR studies in brain and from frozen tissue for the PbTx TC study in brain, we have compared these results to determine if flash freezing tissue impacts the correlation of microarray and qPCR results (Fig. 6). Five genes were validated by identical qPCR assays in all three studies. These genes yielded a correlation of 0.807 (Spearman’s Rho, p<0.0001, n=25) from fresh tissues and a correlation of 0.868 from frozen tissues (Spearman’s Rho, p<0.0001, n=15). The slight increase in correlation observed in frozen tissues was not statistically different from the correlation observed in fresh tissues (ANOVA, p=0.133, n=10).
Fig. 6. Correlation of microarray and qPCR data from fresh vs. frozen tissue.
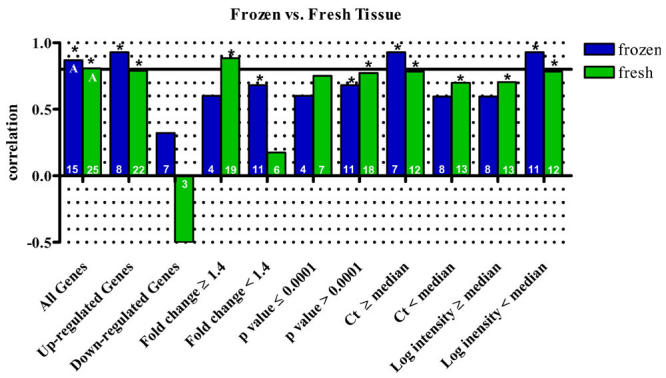
Correlation of microarray and qPCR results based on data from RNA extracted from fresh mouse brains in the DA TC and DR versus the use of RNA extracted from flash frozen brains in the PbTx TC. While minor differences were observed depending on the data set used, one data set did not consistently yield higher correlations. It does not appear that the use of frozen tissue, rather than fresh, appreciably influences the observed correlations with qPCR data. Asterisks indicate a statistically significant correlation (p<0.05). All correlations were calculated using Spearman’s Rho. Statistical differences of correlation, determined by ANOVA, are indicated by different letters in the "all genes" data set. The number of samples included in each correlation is shown in the base of the bar.
Limitations of validation by qPCR
The genes most commonly selected for microarray validation are those exhibiting large degrees of change, which are those of biological interest because of their response to some challenge or change in condition. Given that it is not practical to confirm by qPCR the tens of thousands of genes spotted on an array, the current study provides insight into the limitations under which qPCR might be expected to agree well with expression data generated using inkjet printed 60-mer oligonucleotide arrays. Microarray data from the mouse brain DA dose response and time course experiments were initially screened for genes of interest, based upon their significant biological response and inclusion in a trend set requiring differential expression of at least 1.7 fold in at least one time point and a composite p-value of 0.0001 or less (22). Fourteen genes were selected for further analysis. Tubulin, alpha 4 was used to normalize the qPCR data set, because it exhibited no change from microarray data at all times and doses investigated. The normalized data yielded a correlation of 0.882 (Table 2, Spearman’s Rho, p<0.0001, n=45) for the time course and 0.811 (data not shown, Spearman’s Rho, p<0.0001, n=30) for the dose response. However, in light of the range of correlations reported in the literature, we sought to determine how well these correlations represented the microarray results as a whole. Consequently, we selected 13 additional genes that were excluded from the array trend analysis for verification by qPCR, including genes of interest given the known effects of DA that exhibited little change on the array as well as additional genes selected at random. Again, tubulin, alpha 4 was used to normalize the qPCR data set.
Table 2.
Correlations of genes of included in microarray trend analysis versus genes excluded from microarray trend analysis.
| Genes included in array trend analysis | Genes excluded from array trend analysis | |||||
| Data Set | Correlation | p-value | n | Correlation | p-value | n |
| All Primers | 0.882 | <0.0001 | 45 | 0.306 | 0.049 | 42 |
| Up-regulated on array | 0.898 | <0.0001 | 33 | 0.048 | 0.8278 | 23 |
| down-regulated on array | -0.140 | 0.6641 | 12 | -0.243 | 0.3155 | 19 |
| Fold Change ≥ ± 1.4 by array | 0.893 | <0.0001 | 28 | ND | ND | 0 |
| Fold Change < ± 1.4 by array | 0.586 | 0.0134 | 17 | 0.306 | 0.049 | 42 |
| Fold Change ≥ ± 1.4 by qPCR | 0.893 | <0.0001 | 25 | 0.562 | 0.0366 | 14 |
| Fold Change < ± 1.4 by qPCR | 0.537 | 0.0147 | 20 | -0.009 | 0.9629 | 28 |
| Ct > median | 0.870 | <0.00011 | 22 | 0.465 | 0.0339 | 21 |
| Ct < median | 0.895 | <0.0001 | 23 | 0.004 | 0.9866 | 21 |
| Log(Intensity) > median | 0.843 | <0.0001 | 22 | -0.072 | 0.7557 | 21 |
| Log(Intensity) < median | 0.934 | <0.0001 | 23 | 0.443 | 0.0444 | 21 |
| p-value ≤ 0.0001 | 0.910 | <0.0001 | 26 | 0.872 | 0.0539 | 5 |
| p-value > 0.0001 | 0.708 | 0.0007 | 19 | 0.180 | 0.2875 | 37 |
| Up or down-regulated conserved | 80.00%* | 36 | 76.19% | 32 | ||
| For verification of the mouse brain DA time course 14 genes included in the array trend set (≥1.7 fold change in at least one time point and a composite p-value ≤0.0001) determined to be of biological interest, given their known responses to DA or the degree of change exhibited by microarray, were selected for validation by qPCR. In addition, 13 genes of lesser confidence that were excluded from array trend analysis were verified by qPCR. Both data sets were normalized to tubulin, alpha 4. The effects of direction of regulation, degree of fold change, Ct value, spot intensity, and array p-value on correlation were examined. All correlations were calculated using Spearman’s Rho. *This value is the percentage of genes for which the direction of change was determined to be the same by both array and qPCR analyses. ND: no data (insufficient sample size, n≤2). | ||||||
When the combined set of 28 genes is considered, the correlation of the time course dropped to 0.686 (Spearman’s Rho, p<0.0001, n=84) and the dose response to 0.676 (Spearman’s Rho, p<0.0001, n=58) (Table 1). The correlation of the data was severely negatively skewed following the addition of genes showing no significant change in expression or genes with poor p-values on the array. Table 2 shows an analysis of the 2 subsets of genes from the mouse brain DA time course. While the correlation between the array and qPCR results was 0.882 (Spearman’s Rho, p<0.0001) for the differentially expressed genes initially considered, genes showing no significant change or genes with poor p-values on the array exhibited only 0.306 correlation (Spearman’s Rho, p=0.049). In general, similar trends were observed in both data sets, regardless of the biological interest of the included genes; correlations were higher among up-regulated genes, genes exhibiting greater degrees of change, earlier Cts, and lower p-values.
Summary
In summary, this study demonstrates both the utility and the limitations of qPCR as a validation tool for oligonucleotide microarray studies. As both microarrays and qPCR have inherent pitfalls, the correlation of gene expression results between the two methods is influenced by data quality parameters, presented here as array p-values, and the amount of change in expression reported. Correlation between the two methods is affected by direction of regulation and qPCR Ct, but not spot intensity, the use of composite array data, or the use of frozen tissues. This analysis has determined a threshold of reliability based on fold change and p-value for the platform of Agilent inkjet printed 60-mer oligonucleotide microarrays and qPCR using SYBR green. Using this platform, genes exhibiting at least 1.4 fold change and a p-value of 0.0001 or less in microarray analyses consistently yielded significant correlations of at least 0.80 for array and qPCR data. Data below these thresholds need not be discarded, but rather, approached cautiously before time and resources are expended for further investigation. The pairing of microarray and qPCR is common in gene expression studies. However, the two methods require and utilize vastly different normalization procedures. While the current study did not address the issues of normalization, it has demonstrated that data from the two different technologies, if properly filtered, will yield comparable results. Here we used both qualitative (direction) and quantitative agreement between the two methods to define “validation.” Until a standard definition of validation of microarray results is established, data quality characteristics must be thoroughly presented in the literature to allow for individual assessment of the results.
Disclaimer
This publication does not constitute an endorsement of any commercial product or intend to be an opinion beyond scientific or other results obtained by the National Oceanic and Atmospheric Administration (NOAA). No reference shall be made to NOAA, or this publication furnished by NOAA, to any advertising or sales promotion which would indicate or imply that NOAA recommends or endorses any proprietary product mentioned herein, or which has as its purpose an interest to cause the advertised product to be used or purchased because of this publication.
Supplemental Information
Table 3.
Genes selected for validation by qPCR from microarray analyses (supplemental table).
| Gene Name | Accession # | Bases Spanned | Annealing Temperature | Experiments* |
| 7-dehydrocholesterol reductase | NM_001360 | 127-277 | 58°C | 5 |
| Activator of heat shock 90kDa protein ATPase homolog 1 | NM_012111 | 494-621 | 60°C | 5 |
| Adrenomedullin | NM_001124 | 40-148 | 55°C | 5 |
| Aldolase C, fructose-biphosphate | NM_005165 | 60-252 | 60°C | 5 |
| Alpha tubulin-like | NM_145042 | 686-848 | 62°C | 5 |
| bHLH-PAS type transcription factor NXF | NM_153553.1 | 388-578 | 58°C | 1 & 2 |
| Calpain 10 | NM_011796 | 844-1626 | 64°C | 1 & 2 |
| Calponin 1 | NM_009922 | 303-771 | 62°C | 1 & 2 |
| CCAAT/enhancer binding protein, beta | NM_009883 | 1174-1305 | 58°C | 1 & 2 |
| CCAAT/enhancer binding protein, delta | NM_007679 | 1278-1434 | 60°C | 3 |
| CD52 antigen | NM_013706 | 52-234 | 62°C | 3 |
| c-FOS | NM_010234 | 36-390 | 62°C | 1, 2, 3, & 4 |
| Chemokine (C-X-C motif) receptor 4 | NM_009911 | 158-288 | 58°C | 4 |
| Cofilin 1 | BB097704 | 55-273 | 58°C | 3 |
| Cold inducible RNA binding protein | NM_007705 | 221-349 | 58°C | 1 & 2 |
| Cyclin G2 | NM_004354 | 124-271 | 60°C | 5 |
| Cytochrome P450, family 1, subfamily a, polypeptide 2 | NM_009993 | 489-609 | 62°C | 4 |
| Cytochrome P450, family 2, subfamily a, polypeptide 4 | NM_009997 | 458-628 | 55°C | 4 |
| Cytochrome P450, family 2, subfamily f, polypeptide 2 | NM_007817 | 236-408 | 55°C | 4 |
| Cytochrome P450, family 51, subfamily a, polypeptide 1 | NM_000786 | 10-172 | 60°C | 5 |
| DNA damage inducible transcript 4 | NM_019058 | 365-499 | 60°C | 5 |
| Dystrobrevin beta | NM_007886 | 1496-1838 | 62°C | 1 & 2 |
| Fatty acid desaturase 1 | NM_013402 | 359-514 | 60°C | 5 |
| FK506 binding protein 11 | NM_024169 | 253-644 | 62°C | 1 & 2 |
| G protein-coupled receptor 114 | NM_153837 | 150-284 | 58°C | 5 |
| G1p2, Isg15 ubiquitin-like modifier | NM_015783 | 107-234 | 58°C | 3 |
| Glucocorticoid induced gene 1 | NM_133218 | 1236-1566 | 55°C | 1 & 2 |
| Glutamate receptor, ionotropic, kainate 5 | NM_008168 | 911-1769 | 62°C | 1 & 2 |
| Glutamine Synthetase | X16314.1 | 309-829 | 62°C | 1 & 2 |
| Granzyme A | NM_010370 | 187-475 | 62°C | 3 |
| Growth arrest and DNA-damage-inducible 45 beta | NM_008655 | 351-506 | 60°C | 1, 2, & 3 |
| Growth arrest and DNA-damage-inducible 45 gamma | NM_011817 | 225-348 | 55°C | 1 & 2 |
| Growth Hormone | NM_008117.2 | 347-447 | 58°C | 1 |
| Intercellular adhesion molecule 2 | NM_000873 | 85-207 | 60°C | 5 |
| Interferon regulatory factor 7 | AK079685 | 703-837 | 60°C | 3 |
| Interleukin 21 receptor | NM_021798 | 213-406 | 58°C | 5 |
| Internexin neuronal intermediate filament protein | NM_010563 | 1022-1281 | 55°C | 1 & 2 |
| Jun dimerization protein 2 | NM_130469 | 44-211 | 60°C | 5 |
| Jun-B | NM_008416 | 994-1214 | 62°C | 1 & 2 |
| KRAB zinc finger protein KR18 | XM_139814 | 108-206 | 60°C | 4 |
| Mitogen-activated protein kinase kinase kinase 6 | NM_016693 | 677-791 | 62°C | 1, 2, & 4 |
| Neural Proliferation, differentiation and control gene 1 | NM_008721 | 200-586 | 62°C | 1 & 2 |
| Neurotransmitter transporter, creatine Slc6a8 | NM_133987 | 2203-2499 | 60°C | 1 & 2 |
| Neurotransmitter transporter, glycine Slc6a9 | NM_008135 | 67-285 | 58°C | 3 |
| Nuclear factor of kappa light chain gene enhancer in B-cells inhibitor, alpha | NM_010907 | 292-467 | 58°C | 1 & 2 |
| Nucleoporin 88kDa | NM_002532 | 65-218 | 60°C | 5 |
| pEL98 protein | D00208 | 231-341 | 60°C | 3 |
| Peroxiredoxin 3 | NM_007452 | 86-488 | 62°C | 1 & 2 |
| Pigpen protein | AK086587 | 529-722 | 60°C | 1 & 2 |
| Placenta-specific 8 | NM_139198 | 88-196 | 58°C | 3 |
| Programmed cell death 10 | NM_019745 | 508-791 | 58°C | 1 & 2 |
| Prostaglandin-endoperoxide synthase 2 | NM_011198 | 1231-1410 | 62°C | 1 & 2 |
| RAB1 | NM_008996 | 81-237 | 58°C | 1 & 2 |
| RAB33A | NM_004794 | 344-510 | 58°C | 5 |
| S100 calcium binding protein A8 | NM_013650 | 245-348 | 55°C | 3 |
| S100 calcium binding protein A9 | NM_009114 | 35-174 | 62°C | 3 |
| Serum/glucocorticoid regulated kinase | NM_011361 | 158-329 | 60°C | 1, 2, & 4 |
| Spindle pole body component 24 homolog | NM_182513 | 381-486 | 60°C | 5 |
| Squalene epoxidase | NM_003129 | 609-785 | 60°C | 5 |
| SRY-box containing gene 2 | NM_011443 | 633-756 | 55°C | 4 |
| Thioredoxin 1 | NM_011660 | 88-335 | 55°C | 1 & 2 |
| Transcription factor 7-like 2 | NM_030756 | 345-496 | 60°C | 5 |
| Transformation related protein 53 inducible nuclear protein 1 | NM_021897 | 236-416 | 58°C | 4 |
| Tubulin, alpha 4 | NM_009447 | 94-310 | 60°C | 1, 2, 3, & 4 |
| Tumor necrosis factor (ligand) superfamily, member 13b | NM_033622 | 661-1056 | 65°C | 1 & 2 |
| Tumor protein p53 inducible nuclear protein 1 | NM_033285 | 1883-2040 | 58°C | 5 |
| UDP-glucuronic acid decarboxylase | AK005536 | 570-706 | 58°C | 4 |
| X-box binding protein 1 | NM_005080 | 610-715 | 60°C | 5 |
| Following microarray analyses, genes were chosen based on fold change and p-value for validation by qPCR. GenBank accession numbers are listed. *1: Mouse brain DA TC, 2: Mouse brain DA DR, 3: Mouse blood DA TC, 4: Mouse brain PbTx TC, 5: Human AZA TC. DA: domoic acid. PbTx: brevetoxin. AZA: azaspiracid. TC: time course. DR: dose response. | ||||
Acknowledgments
We would like to thank Dr. Michael Twiner for the use of array data from the human AZA experiment. This work was funded by NOAA programmatic research funds. The authors have no conflicts of interest to declare related to this publication.
Abbreviations
- AZA
azaspiracid
- Ct
cycle threshold
- DA
domoic acid
- DR
dose response
- IP
intraperitoneally
- PbTx
brevetoxin
- qPCR
quantitative real-time polymerase chain reaction
- RT
reverse transcription
- TC
time course
References
- Bustin S. Invited review: quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol. 2002;29:23–39. doi: 10.1677/jme.0.0290023. [DOI] [PubMed] [Google Scholar]
- Chuaqui RF, Bonner RF, Best CJM, Gillespie JW, Flaig MJ, Hewitt SM, Phillips JL, Krizman DB, Tangrea MA, Ahram M, Linehan WM, Knezevic V, Emmert-Buck MR. Post-analysis follow-up and validation of microarray experiments. Nat Genet. 2002;32:509–514. doi: 10.1038/ng1034. [DOI] [PubMed] [Google Scholar]
- Freeman WM, Walker SJ, Vrana KE. Quantitative RT-PCR: pitfalls and potential. BioTechniques. 1999;26:112–125. doi: 10.2144/99261rv01. [DOI] [PubMed] [Google Scholar]
- Wurmbach E, Yuen T, Sealfon SC. Focused microarray analysis. Methods. 2003;31:306–316. doi: 10.1016/S1046-2023(03)00161-0. [DOI] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucl Acids Res. 2002;30(4):e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Kawasaki ES. Microarrays: the use of oligonucleotides and cDNA for the analysis of gene expression. Drug Discov Today. 2003;8:134. doi: 10.1016/S1359-6446(02)02578-3. [DOI] [PubMed] [Google Scholar]
- Brazeau DA. Combining genome-wide and targeted gene expression profiling in drug discovery: microarrays and real-time PCR. Drug Discov Today. 2004;9:838–845. doi: 10.1016/S1359-6446(04)03231-3. [DOI] [PubMed] [Google Scholar]
- Bustin S. Review: absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol. 2000;25:169–193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- Mah N, Thelin A, Lu T, Nikolaus S, Kuhbacher T, Gurbuz Y, Eickhoff H, Kloppel G, Lehrach H, Mellgard B, Costello CM, Schreiber S. A comparison of oligonucleotide and cDNA-based microarray systems. Physiol Genomics. 2004;16:361–370. doi: 10.1152/physiolgenomics.00080.2003. [DOI] [PubMed] [Google Scholar]
- Tan PK, Downey TJ, Spitznagel EL Jr, Xu P, Fu D, Dimitrov DS, Lempicki RA, Raaka BM, Cam MC. Evaluation of gene expression measurements from commercial microarray platforms. Nucl Acids Res. 2003;31:5676–5684. doi: 10.1093/nar/gkg763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauk CL, Berndt ML, Williams A, Douglas GR. Comprehensive comparison of six microarray technologies. Nucl Acids Res. 2004;32:e124. doi: 10.1093/nar/gnh123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Ping G, Shinohara Y, Zhang Y, Baba Y. Comparison of gene expression measurements from cDNA and 60-mer oligonucleotide microarrays. Genomics. 2005;85:657–665. doi: 10.1016/j.ygeno.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Bustin SA. Quantification of nucleic acids by PCR. In Bustin SA, editor. A-Z of Quantitative PCR. La Jolla, CA: International University Line; 2004. p. 3-46.
- Beckman KB, Lee KY, Golden T, Melov S. Gene expression profiling in mitochondrial disease: assessment of microarray accuracy by high-throughput Q-PCR. Mitochondrion. 2004;4:453. doi: 10.1016/j.mito.2004.07.029. [DOI] [PubMed] [Google Scholar]
- Etienne W, Meyer MH, Peppers J, Meyer RA Jr. Comparison of mRNA gene expression by RT-PCR and DNA microarray. BioTechniques. 2004;36:618–621. doi: 10.2144/04364ST02. [DOI] [PubMed] [Google Scholar]
- Larkin JE, Frank BC, Gaspard RM, Duka I, Gavras H, Quackenbush J. Cardiac transcriptional response to acute and chronic angiotensin II treatments. Physiol Genomics. 2004;18:152–166. doi: 10.1152/physiolgenomics.00057.2004. [DOI] [PubMed] [Google Scholar]
- Rajeevan MS, Ranamukhaarachchi DG, Vernon SD, Unger ER. Use of real-time quantitative PCR to validate the results of cDNA array and differential display PCR technologies. Methods. 2001;25:443–451. doi: 10.1006/meth.2001.1266. [DOI] [PubMed] [Google Scholar]
- Svaren J, Ehrig T, Abdulkadir SA, Ehrengruber MU, Watson MA, Milbrandt J. EGR1 target genes in prostate carcinoma cells identified by microarray analysis. J Biol Chem. 2000;275:38524–38531. doi: 10.1074/jbc.M005220200. [DOI] [PubMed] [Google Scholar]
- Rajeevan MS, Vernon SD, Taysavang N, Unger ER. Validation of array-based gene expression profiles by real-time (kinetic) RT-PCR. J Mol Diagn. 2001;3:26–31. doi: 10.1016/S1525-1578(10)60646-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Mao M, Jones AR, Burchard J, Marton MJ, Shannon KW, Lefkowitz SM, Ziman M, Schelter JM, Meyer MR, Kobayashi S, Davis C, Dai H, He YD, Stephaniants SB, Cavet G, Walker WL, West A, Coffey E, Shoemaker DD, Stoughton R, Blanchard AP, Friend SH, Linsley PS. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotech. 2001;19:342–347. doi: 10.1038/86730. [DOI] [PubMed] [Google Scholar]
- Li J, Pankratz M, Johnson JA. Differential gene expression patterns revealed by oligonucleotide versus long cDNA arrays. Toxicol Sci. 2002;69:383–390. doi: 10.1093/toxsci/69.2.383. [DOI] [PubMed] [Google Scholar]
- Ryan JC, Morey JS, Ramsdell JS, Van Dolah FM. Acute phase gene expression in mice exposed to the marine neurotoxin domoic acid; excititoxicity stimulates ischemia. Neuroscience. 2005;136:1121–1132. doi: 10.1016/j.neuroscience.2005.08.047. [DOI] [PubMed] [Google Scholar]
- Twiner MJ, Hess P, Bottein Dechraoui M-Y, McMahon T, Ramsdell JS, Samons MS, Satake M, Yausumoto T, Doucette GJ. Cytotoxic and cytoskeletal effects of azaspiracid-1 on multiple mammalian cell lines. Toxicon. 2005;45:891–900. doi: 10.1016/j.toxicon.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Williams TD, Gensberg K, Minchin SD, Chipman JK. A DNA expression array to detect toxic stress response in European flounder (Platichthys flesus). Aquat Toxicol. 2003;65:141–157. doi: 10.1016/S0166-445X(03)00119-X. [DOI] [PubMed] [Google Scholar]
- Dallas PB, Gottardo NG, Firth MJ, Beesley AH, Hoffmann K, Terry PA, Freitas JR, Boag JM, Cummings AJ, Kees UR. Gene expression levels assessed by oligonucleotide microarray analysis and quantitative real-time PCR - how well do they correlate? BMC Genomics. 2005;6(1):59. doi: 10.1186/1471-2164-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Morgan DG, Finch CE. Extensive postmortem stability of RNA from rat and human brain. J Neuroscience Res. 1986;16:267–280. doi: 10.1002/jnr.490160123. [DOI] [PubMed] [Google Scholar]
- Karsten SL, Van Deerlin VM, Sabatti C, Gill LH, Geschwind DH. An evaluation of tyramide signal amplification and archived fixed and frozen tissue in microarray gene expression analysis. Nucl Acids Res. 2002;30(2):e4. doi: 10.1093/nar/30.2.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin VM, Gill LH, Nelson PT. Optimizing Gene Expression Analysis in Archival Brain Tissue. Neurochemical Research. 2002;27:993–1003. doi: 10.1023/A:1020996519419. [DOI] [PubMed] [Google Scholar]