

Abstract
Microbes frequently live within multicellular, solid surface-attached assemblages termed biofilms. These microbial communities have architectural features that contribute to population heterogeneity and consequently to emergent cell functions. Therefore, three-dimensional (3D) features of biofilm structure are important for understanding the physiology and ecology of these microbial systems. This paper details several protocols for scanning electron microscopy and confocal laser scanning microscopy (CLSM) of biofilms grown on polystyrene pegs in the Calgary Biofilm Device (CBD). Furthermore, a procedure is described for image processing of CLSM data stacks using amira™, a virtual reality tool, to create surface and/or volume rendered 3D visualizations of biofilm microorganisms. The combination of microscopy with microbial cultivation in the CBD – an apparatus that was designed for high-throughput susceptibility testing – allows for structure-function analysis of biofilms under multivariate growth and exposure conditions.
Keywords: Biofilms; Imaging, Three-Dimensional
Introduction
Life in a biofilm is part of the ecological cycle for the vast majority of bacteria and yeasts found in the environment and implicated in chronic disease (1, 2). Growth in a biofilm is a developmental process that is in some regards analogous to differentiation in tissues of multicellular organisms, and likewise involves cell-to-cell signals that regulate growth and coordinate cell behaviour (3-5). Biofilm formation occurs when microorganisms stick to a surface and become permanently attached, eliciting a change in physiology. These microbes then grow and divide to form highly ordered, matrix encased assemblages, all under the control of biofilm specific genes (6). The structure and development of mature biofilms is correlated to emergent biological properties of the adherent microbial community, such as metabolic stratification (7), antimicrobial resistance (8, 9), active dispersal processes (10) and virulence (11). Biofilm architecture is further influenced by environmental conditions, such as nutrient status (12), heavy metals (J. J. Harrison, H. Ceri and R. J. Turner, unpublished data), material composition and roughness of the substratum (13), as well as hydrodynamic shear force (14). Therefore, laboratory systems for imaging microbial biofilms as well as computer algorithms for analyzing this data are valuable research tools in microbiology.
Our group has previously described the Calgary Biofilm Device (CBD) for high-throughput susceptibility testing of microbial biofilms to antibiotics (15), disinfectants and metals (16, 17). This system consists of a polystyrene lid with 96 downwards protruding pegs that can be fitted into a standard 96-well microtiter plate. Through the use of this method, one batch culture apparatus allows single or multiple species biofilms to be tested against an 8 × 12 matrix of controlled variables. These variables may include growth medium formulations, exposure times, as well as antimicrobials at varying concentrations - alone or in combination. Here we report microscopy and image processing techniques to evaluate the biofilm community structures produced by bacteria and fungi cultivated in the CBD. The project to create these tools was undertaken with a single, specific aim: to provide a means to examine microbial biofilm structures under multivariate growth and/or exposure conditions.
This study presents several protocols for use in scanning electron microscopy (SEM), confocal laser scanning microscopy (CLSM), and three-dimensional (3D) visualization of CBD biofilms. For the purpose of computer rendering, virtual reality (VR) technology (amira™) was used to dynamically visualize CLSM data. This process functions in a manner analogous to 3D reconstruction of organs or tissues from two-dimensional (2D) stacks of medical images. As illustrative examples, these various techniques were used to examine the biofilm structures produced by several strains of Escherichia coli, Pseudomonas aeruginosa, Pseudomonas chlororaphis, Pseudomonas fluorescens, Burkholderia cenocepacia, Staphylococcus aureus and Candida tropicalis. The ability of the CBD to generate a large number of biofilm replicates was also used for the evaluation of a variety of fixing protocols and combinations of fluorescent stains. In this latter category, we describe the use of acridine orange, viability staining with Syto-9 and propidium iodide (components of the Live/Dead® bacterial cell viability kit), as well as the use of fluorophore-conjugated lectins for the staining of biofilm extracellular polysaccharides (EPS).
To validate the CBD as a tool for studying biofilm organization, three proof-in-principle experiments were carried out. First, the biofilm structures of several bacterial strains were evaluated in rich and minimal media, and the images presented here show that bacteria and fungi adopted a diverse range of structural conformations that were dependent on strain genetics as well as growth conditions. Second, Live/Dead® staining was used to illustrate that dead cells were a frequent, albeit variable, component of biofilms. Last of all, the staining of extracellular polymers using fluorophore-conjugated lectins was used to show that this component of the biofilm matrix was unevenly distributed throughout the surface-adherent communities. Collectively, these proof-in-principle experiments were designed to simultaneously illustrate important caveats in the interpretation of microscopy data but also to validate the CBD as a potential tool for the study of biofilm structure-function relationships.
Materials and Methods
Strains, growth media and buffers
All of the strains used in this study are summarized in Table 1. Bacterial and fungal strains were stored in Micobank™ vials at -70°C as described by the manufacturer (ProLab Diagnostics, Richmond Hill, ON, Canada). The following growth media were used to culture these microorganisms as indicated throughout this study: trypticase soy agar (TSA) and trypticase soy broth (TSB) (EMD Chemicals Inc., Gibbstown, NJ, USA); Miller Luria-Bertani broth (LB, EMD Chemicals Inc.) or LB that was amended with 1.5% w/v granulated agar (LBA); King’s Broth (KB) that was prepared as previously described (18) or KB that was amended with 1.5% w/v agar (KBA); minimal salts vitamins pyruvate (MSVP) broth (19) and minimal salts dextrose (MSD) broth (20), which were also prepared as previously described. Lastly, MSD plus yeast extract and casamino acids (MSD-YC) broth was prepared by enriching MSD with 2.0 g L-1 yeast extract and 1.0 g L-1 casamino acids (Sigma-Aldrich, St. Louis, MO, USA). The incubation temperatures required for microbial cultures varied from strain to strain and are indicated in Table 1. All rinse steps were performed by using either 0.9% saline or phosphate buffered saline (PBS; pH 7.4, 8.0 g NaCl, 200 mg KCl, 1.44 g Na2HPO4, and 240 mg KH2PO4 per liter of double distilled water, ddH2O).
Table 1.
Bacterial and fungal strains used in this study.
| Genus and species | Strain | Genotype or description | Growth Medium1 | Temperature (°C) / Format2 | Source |
| Burkholderia cenocepacia | K56-2 | Environmental isolate | MSD-YC/TSA | 35/S | (38) |
| Candida tropicalis | 99916 | Clinical isolate, Foothills Hospital, Calgary, AB | TSB/TSA | 35/S | (22) |
| Escherichia coli | CFT073 | Urinary tract isolate, genome sequenced | TSB/TSA | 37/S | (39) |
| JM109 | EndA1·recA1·gyrA96·hsdR17(rk -mk -)·supE44·recA1Δ(lac-proAB); F’(traD36·proAB + ·lacIq·lacZ·M15) | LB(A) | 35/R | (40) | |
| TG1 | SupE·thi·hsd5·Δ(lac-proAB); F’(traD36·proAB+·lacIq·lacZ·M15) | LB(A), MSD | 35/R | (41) | |
| DSS640 | TG1 derivative; ΔtatABC; Knr | LB(A), MSD | 35/R | (42) | |
| Pseudomonas aeruginosa | ATCC 15442 | Standard reference strain for biocide susceptibility testing | LB(A) | 35/S | ATCC |
| ATCC 27853 | Standard reference strain for antibiotic susceptibility testing | LB(A), MSVP | 35/R | ATCC | |
| PA14 | Clinical isolate, genome sequenced | TSB/TSA | 35/S | (43) | |
| Pseudomonas chlororaphis | PcO6 | Environmental isolate | KB/KA | 20/S | (44) |
| Pseudomonas fluorescens | ATCC 13525 | Environmental isolate | LB | 27/S | ATCC |
| Staphylococcus aureus | ATCC 29213 | Standard reference strain for antibiotic susceptibility testing | LB | 35/R | ATCC |
|
1Abbreviations for growth media: KB = King’s broth; KA = King’s agar; LB = Luria-Bertani broth; LBA = Luria-Bertani agar; MSD = minimal salts dextrose; MSD-YC = minimal salts dextrose enriched with yeast extract and casamino acids; MSVP = minimal salts vitamins pyruvate; TSA = tryptic soy agar; TSB = tryptic soy broth 2Denotes the incubation temperature and CBD assay format used: R = rocking table for trough format; S = gyrorotary shaker for microtiter plate format | |||||
Biofilm cultivation
Bacterial biofilms were grown in the Calgary Biofilm Device (CBD) as previously described by Ceri et al. (15) as well as by the manufacturer (Innovotech, Edmonton, AB, Canada). This method is briefly summarized here and is illustrated in Figure 1 (A to D). To begin, the desired bacterial or fungal strain was streaked out twice on agar, and colonies were suspended into fresh broth to match the optical density of a 1.0 McFarland standard. For the bacterial strains, this corresponded to approximately 3.0 × 108 cfu ml-1; for C. tropicalis this was approximately 3.0 × 106 cfu ml-1 (as verified by viable cell counts, see below). The cultures matching the optical standard were then diluted 30-fold in the appropriate broth medium, which subsequently served as the inoculum for the CBDs.
Fig. 1. An overview for using the CBD for the purpose of microscopy and 3D visualization of microbial biofilms.
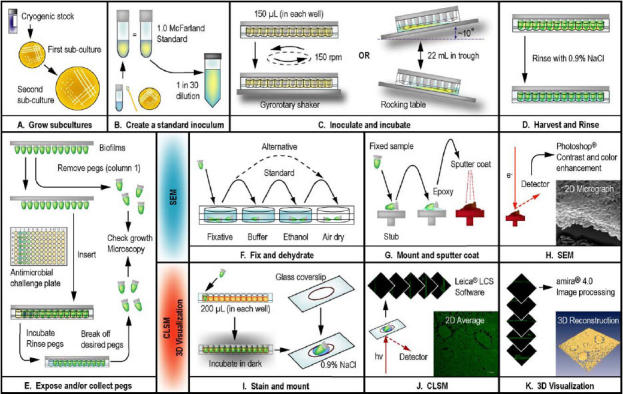
A. To begin, fresh subcultures of the microbial strain were grown on the appropriate agar medium. B. Using a cotton swab, colonies from a fresh second subculture were suspended in broth medium to match a 1.0 McFarland standard. This was diluted 30-fold in broth to create the inoculum for the CBD. C. The peg lid of the CBD was either inserted into a microtiter plate (containing 150 μL of inoculum in each well) or a corrugated trough (with 22 mL of inoculum inside). The inoculated devices were placed in a humidified incubator on a gyrorotary shaker or platform rocker, respectively. D. After cultivation, biofilms were rinsed with saline to remove loosely adherent cells. E. Pegs were removed from the CBD using pliers and the biofilms then were enumerated by viable cell counting. A second set of pegs was removed for examination by microscopy. There is an option to expose biofilms to an array of antimicrobial agents or other test conditions and then to remove a second set of pegs for microscopy. F. For SEM, pegs were first fixed and then dehydrated, which was carried out using 1 of 2 protocols. G. The fixed samples were mounted on stubs using epoxy resin, dried, and then sputter coated with gold-palladium. H. The biofilms were then examined by SEM, and the resulting images were contrast enhanced. I. For CLSM, pegs were immersed in the appropriate stain and then placed in 2 drops of 0.9% saline on a glass coverslip. J. Images of the biofilms were captured using CLSM, and the instrument software was used to generate 2D averages of image z-stacks. K. The z-stacks were imported into amira™ for advanced image processing and 3D visualization.
There were two methods used to cultivate biofilms on the polystyrene pegs of the CBD: the first method involved the use of a corrugated trough (the MBEC™-High Throughput assay) (15), the second utilized a microtiter plate (the MBEC™-Physiology and Genetics assay) (21). For the first format of this assay, 22 mL of the inoculum was transferred into the trough and the peg lid was then fitted inside of this. The assembled CBD was then placed on a rocking table (Bellco Biotechnology, Vineland, NJ, USA) at ~3.5 rocks per minute in a humidified incubator. For the second method of biofilm cultivation, 150 μL of the inoculum was added to each well of a 96-well microtiter plate. The peg lid was then fitted inside of this and the assembled device was placed on a gyrorotary shaker at ~150 revolutions per minute (rpm) in a humidified incubator. The cultivation method used for each bacterial or fungal strain is indicated in Table 1, and the method used was chosen based on which approach gave the greatest biofilm cell density, with the stipulation that the growth was statistically equivalent between the different rows of pegs (data not shown). The evaluation and choice for the method of biofilm growth on the surface of the CBD pegs using these two different assay formats has been previously described (15, 17). Following the desired period of incubation, the biofilms were rinsed by inserting the peg lids into microtiter plates with 200 μL of either 0.9% saline or PBS in each well for 2 min.
The polystyrene pegs of the CBD have a surface area of approximately 109 mm2 and bear an overall neutral electrostatic charge. The rounded “tip” of each peg extends approximately 3-4 mm into the growth medium. Corresponding to this, the “air-liquid-surface interface” occurs approximately 4-5 mm above the tip after the inoculated device is agitated on a rocking table or gyrorotatry shaker. Note that to facilitate the growth of C. tropicalis 99916 on the surface of the CBD, the pegs were coated with L-lysine as previously described (22). This was accomplished by immersing the pegs into a solution of 1.0% L-lysine for 1 h, then by drying the peg lids upside down in a laminar flow hood for 30 min prior to use.
Viable cell counting
Viable cell counts were determined after biofilms had been rinsed (as described above). Sample pegs were broken from the lid of the CBD using a pair of flamed pliers, then inserted into 200 μL of 0.9% saline in the wells of microtitre plate (Fig. 1E). Biofilms were disrupted from the peg surface using an Aquasonic 250HT ultrasonic cleaner (VWR International, Mississauga, ON, Canada) set at 60 Hz for 5 min. The disrupted biofilm cells were serially diluted in either 0.9% saline or PBS, and then plated onto the appropriate agar medium. Agar plates were incubated for up to 48 h at the temperatures summarized in Table 1 and then enumerated. Viable cell counts for planktonic cultures (ex. starting inocula) were similarly carried out by serial dilution in 0.9% saline or PBS, and then by plating onto agar as described for biofilm cells.
Scanning electron microscopy (SEM)
Pegs were broken from the lid of the CBD using pliers and then rinsed once with 0.9% saline to disrupt loosely adherent planktonic cells. Two approaches were used for fixing the biofilms. In the first approach, biofilms were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) at 4°C for 20 hours. Following this, pegs were washed with 0.1 M cacodylate buffer and then rinsed with ddH2O (for 10 min at each step). Subsequently, the pegs were dehydrated with 70% ethanol and then air dried for 72 h before mounting. An alternate approach was used to examine extracellular polymeric substance (EPS) production. In this case, the rinsed biofilms were fixed with 0.1 M cacodylate buffer (pH 7.2) at room temperature for 2 h, then air dried for 120 h before mounting. SEM was performed using a Hitachi model 450 scanning electron microscope as previously described (23). SEM images were contrast and brightness enhanced using Adobe® Photoshop® 7.0 (Adobe Systems Inc., San Jose, CA, USA). This process is summarized in Figure 1 (F to H).
Confocal laser scanning microscopy (CLSM)
Pegs were broken from the lid of the CBD using pliers (as described above) and then rinsed once with 0.9% saline to disrupt planktonic bacteria. Prior to examination by CLSM, biofilms were fluorescently stained with one of the four following treatments: 1) acridine orange (AO), 2) Syto-9 and propidium iodide (PI), 3) AO and tetramethylrhodamine isothiocyanate conjugated concanavalin A (TRITC-ConA), or 4) Syto-9 and tetramethylrhodamine isothiocyanate conjugated peanut agglutinin (TRITC-PNA). The mechanism and procedure for cell staining are briefly summarized for each of these fluorescent compounds, and the general process for staining biofilms on pegs is illustrated in Figure 1 (I to K).
AO is a membrane permeant nucleic acid stain that intercalates dsDNA and binds to ssDNA as well as to ssRNA through dye-base stacking to give broad spectrum fluorescence when excited at 476 nm (24). This compound stains all cells in a biofilm, live or dead, and may also bind to nucleic acids that are present in the extracellular matrix. To stain biofilms, pegs were immersed in 0.1% w/v acridine orange (Sigma Chemical Co., St. Louis, MO, USA) in PBS for 5 min at room temperature.
Syto-9 and PI are packaged together as part of the Live/Dead® BacLight™ Kit for bacterial cell viability staining (Molecular Probes, Burlington, ON, Canada). Syto-9 (488 nm excitation, green emission) is a freely diffusible, nucleic acid intercalator that labels all cells in the microbial population regardless of viability. The counterstain, PI (543 nm excitation, red emission), is a membrane impermeant DNA intercalator that only stains cells with compromised membrane integrity. In principle, live cells stain green and dead cells stain orange-red. This has been shown to correlate well with viable cell counts for calibrated suspensions of many bacteria as well as for C. albicans (25). Here, cell viability staining of bacteria and fungi was carried out by incubating biofilms concomitantly with Syto-9 (6.7 μM) and PI (40 μM) at 30°C for 30 min as previously described by Jin et al. (25).
To stain the extracellular polysaccharides in the matrix of P. aeruginosa and C. tropicalis biofilms, pegs were immersed in 200 μg ml-1 TRITC-ConA (Molecular Probes) and incubated at 30°C for 90 min. ConA is a lectin with high specificity for mannose sugars present in the cell walls and biofilm matrix of Candida spp. (25) as well as P. aeruginosa (26). These pegs were subsequently treated with AO as described above.
In contrast, the extracellular polysaccharide component of B. cenocepacia biofilms was labeled by immersing pegs in 50 μg ml-1 TRITC-PNA and incubating at 30°C for 60 min. Peanut lectin specifically binds to D-galactose, which occurs three times in the heptasaccharide repeating unit that makes up the exopolysaccharide cepacian (which is specific to Burkholderia cepacia complex bacteria) (27). To preserve structure and extracellular biomass in the case of B. cenocepacia, biofilms were fixed with 5% glutaraldehyde for 1 h at room temperature prior to staining. These fixed biofilms were rinsed 5 times with 0.9% saline before mounting for microscopy.
In all cases, fluorescently labelled biofilms were placed in two drops of 0.9% saline on the surface of a glass coverslip. These pegs were examined using a Leica DM IRE2 spectral confocal and multiphoton microscope with a Leica TCS SP2 acoustic optical beam splitter (AOBS) (Leica Microsystems, Richmond Hill, ON, Canada). To minimize or eliminate artefacts associated with single and/or simultaneous dual wavelength excitation, all dual labelled samples were sequentially scanned, frame-by-frame, first at 476 or 488 nm and then at 543 nm. Fluorescence emission was then sequentially collected in the green and red regions of the spectrum, respectively. Line averaging (×2) was used to capture images with reduced noise. A 63 × water immersion objective was used in all imaging experiments. Image capture, two-dimensional (2D) projections of z-stacks (see below) and 3D reconstructions were performed using Leica Confocal Software (LCS, Leica Microsystems).
Three-dimensional (3D) visualization
Three-dimensional (3D) visualization of CLSM data were created using amira™ 4.0 (Mercury Computer Systems Inc., Chelmsford, MS, USA). The principle and application of using this software to the analysis of biofilm structure are briefly summarized here. CLSM data consists of a set of two-dimensional (2D), cross-sectional images in the x-y plane that is captured along a z-axis. Collectively, a set of x-y images through the z-axis is termed a z-stack. Here, each individual x-y image was a 1024 × 1024 pixel tagged image file format (TIFF) file that corresponded to a cross-section through the biofilm. Points in 2D and 3D data sets are termed pixels and voxels, respectively. For instance, the x-y images in the z-stack are composed of pixels, whereas the same point in the 3D volume data set is a voxel. There were two different methods used to visualize the microbial biofilms.
First, the method of surface rendering was used to create biofilm 3D visualizations; in this approach 3D surfaces were created to encase the biomass by interconnecting its boundary voxels. Therefore, biofilm visualizations created in this manner were a geometric representation of a surface (termed an isosurface) from a 3D volume data set. CLSM z-stacks were processed by re-sampling and segmenting the images according to a threshold that was selected according to a fluorescence intensity histogram of the TIFF files. In this manner, segmentation partitioned the images into background and biomass voxels and this was further user verified by manually comparing segmented biomass to its 2D original.
An alternative method was to use volume rendering, whereby biomass was a direct 3D visualization of the 3D volume data set, without the use of thresholding segmentation. Briefly, this method, termed ray tracing or ray casting, was based on the amount of light (in terms of colour and opacity values) that every pixel in an image was emitting and absorbing. For every pixel in the image a ray was shot into the data volume, and at a predetermined number of locations along the ray path, the colour and opacity values were obtained by interpolation, subject to a predetermined range of light intensity. In other words, every pixel displayed in the image had a colour and opacity as displayed relative to the viewing plane of the user. In comparison to surface rendering, volume rendering was computationally intense as it required more processor time and special hardware.
In both cases, 3D visualization using amira™ allowed for dynamic display of the biofilm, such that the visualization of biomass could be examined from any viewpoint. This allowed for the detection of 3D features in these biological systems that were not perceptible from static 2D CLSM image stacks or from wireframe isosurface rendering carried out using LCS. Animations produced using amira™ were edited for screen resolution using Quicktime 7.1 Pro (Apple Computers Inc., Cupertino, CA, USA).
Statistical tests and data analysis
Mann-Whitney U-tests of viable cell counts were performed using MINITAB® Release 14 (Minitab Inc., State College, PA, USA) to analyze log10-transformed raw data. Alternate hypotheses were tested at the 95% level of confidence. Mean and standard deviation calculations were performed using Microsoft® Excel XP (Microsoft Corporation, Redmond, WA, USA).
Results and Discussion
Biofilm growth in the CBD
Within our research group, every time an assay has been performed using the CBD, 3 or 4 pegs have been removed from the lid and the number of cells growing on the surface determined by viable cell counting. We have pooled the cumulative data from all of the studies previously undertaken by our research group that have specifically used the CBD to examine the microbial strains summarized in Table 1. The mean viable cell counts and standard deviations (in units of log10 cfu peg-1) for these bacterial and fungal strains (under all of the test conditions examined in this paper) are summarized in Table 2. The studies used to compile this data are indicated (where applicable), and this meta-analysis allowed the number of viable cells growing in CBD biofilms to be quantified based on 3 to 202 replicates for each strain. The rationale for this approach was that analyzing the results from a group of studies allowed for a more accurate representation of the mean and variation of biofilm growth on the surface of the CBD pegs.
Table 2.
Meta-analysis of mean viable cell counts for microbial biofilms cultivated in the CBD.
| Genus and species | Strain | Growth medium1 | Time (h) | Viable cell count (log10 cfu peg-1) | Replicates | Reference(s) |
| B. cenocepacia | K56-2 | MSD-YC | 72 | 6.6 ± 0.4 | 4 | this study |
| C. tropicalis | 99916 | TSB | 48 | 4.3 ± 0.4 | 202 | (22), unpublished data |
| 72 | 4.3 ± 0.3 | 3 | this study | |||
| E. coli | CFT073 | TSB | 24 | 5.9 ± 0.3 | 71 | unpublished data |
| JM109 | LB | 24 | 6.2 ± 0.5 | 119 | (16, 17, 45) | |
| TG1 | LB | 24 | 7.0 ± 0.3* | 84 | (17, 20) | |
| MSD | 24 | 6.1 ± 0.6** | 40 | (20) | ||
| DSS640 | LB | 24 | 6.8 ± 0.5* | 80 | (20) | |
| MSD | 24 | 4.9 ± 0.7** | 40 | (20) | ||
| P. aeruginosa | ATCC 15442 | TSB | 24 | 6.8 ± 0.5 | 181 | unpublished data |
| ATCC 27853 | LB | 9.5 | 6.9 ± 0.8 | 55 | (16, 46) | |
| MSVP | 22 | 6.1 ± 0.4 | 133 | (46), this study | ||
| PA14 | TSB | 24 | 6.3 ± 0.2 | 12 | (21), this study | |
| P. chlororaphis | PcO6 | KB | 24 | 7.3 ± 0.2 | 4 | this study |
| P. fluorescens | ATCC 13525 | LB | 24 | 6.0 ± 0.8 | 99 | unpublished data |
| S. aureus | ATCC 29213 | LB | 24 | 6.2 ± 0.9 | 76 | (16), (47) |
|
1 Abbreviations for growth media are the same as those listed in Table 1. * These values are significantly different by means of a Mann-Whitney U-Test (p < 0.01) ** These values are significantly different by means of a Mann-Whitney U-Test (p < 0.01) | ||||||
Comparative SEM analysis of biofilm structure
SEM examination of biofilms cultivated in the CBD revealed that different bacterial strains adopted various structural conformations that were distinct from the “stalk” and “mushroom cap” biofilms formed in flow cells by P. aeruginosa. For instance, P. chlororaphis PcO6 formed thick cell layers with high cell density (Fig. 2A) whereas S. aureus ATCC 29213 adhered to the pegs in clumps of approximately 2 to 20 cells (Fig. 2B). In another example, E. coli JM109 formed uneven layers of single and multiseptate cells that were clustered into mounds (Fig. 2C). The formation of multiseptate cells, which were chains of cells that did not separate from one another as is normal during planktonic cell replication and division, is a commonly observed event for many biofilm bacteria (H. Ceri, unpublished data).
Fig. 2. SEM of bacterial biofilms grown in the CBD.

For the sake of comparison, the biofilms were grown in either rich or minimal medium (as summarized in Table 1) and then fixed using 1 of 2 different protocols. These micrographs were chosen to illustrate that medium composition has an impact on the capacity of bacteria to form biofilms, which further varies between genus, species and strains. Moreover, the choice of fixing protocols influences how well microstructures may be preserved, which may impact on the interpretation of SEM data.
As researchers have begun to dissect the molecular mechanisms of biofilm formation, the concepts of “good,” “poor,” and “hyper-” biofilm forming bacterial strains have emerged. This is particularly relevant with regards to engineered mutants from strain libraries designed to identify genes important for the biofilm lifestyle. For example, wild type P. aeruginosa is considered a good biofilm former under many growth conditions (Table 2, Fig. 2D and 2G). Strains of this microorganism bearing inactivating mutations in the two-component regulatory system GacA/GacS have been labeled poor biofilm formers (21, 28), whereas small colony variant (SCV) strains of this microorganism have been labeled hyper-biofilm formers (29). In the example here, an E. coli mutant lacking the twin-arginine translocase (tat), a strain that is considered a poor biofilm former (20, 30), was compared to its wild type parental strain using comparative SEM analysis.
When grown in rich medium and examined using a standard fixing protocol, biofilms of E. coli TG1 (wild type) and DSS640 (tatABC- ) similarly produced surface-adherent layers of cells that had little EPS, and similarly, were thickest near the air-liquid-surface interface (Fig. 2E and 2F). A previous study by our group has revealed that the mean number of viable cells in E. coli DSS640 biofilms is significantly less than that of the isogenic, wild type strain TG1 (Table 2, by means of a two-population Mann-Whitney U-test, p < 0.001) (20). It is important to note that this difference was not perceptible from examination by SEM (Fig. 2). Therefore, a first caveat to comparative studies using this technique is that viable cell counts (with statistical analysis) are required to give meaning to microscopy (Table 2).
A variable influencing biofilm formation in the CBD was the choice of growth medium. For instance, biofilms of E. coli TG1 and DSS640 had a cell density that was 8 and 79 times greater in LB than it was in MSD, respectively. To provide another cross comparison, biofilms of E. coli TG1 grown in MSD had a mean viable cell count that was 16 times greater than biofilms of E. coli DSS640 grown in MSD. This was a statistically significant difference (Table 2, by means of a Mann-Whitney U-test, p < 0.001) that was readily observable by SEM (Fig. 2H and 2I). Therefore, under conditions of nutrient restriction, E. coli DSS640 may be considered a poor biofilm former relative to the isogenic, wild type strain. It is interesting to contrast these results to those for biofilms of E. coli TG1 and DSS640 grown in rich medium, which, using comparative SEM analysis both appeared to be good biofilm formers. A caveat that emerges from this data set is that growth conditions are an important consideration when evaluating biofilm formation, as a good biofilm former in one medium may be a poor biofilm former in another.
A final feature of SEM that warrants attention is that samples must be fixed and dehydrated, which may introduce experimental artifacts that affect the observation and interpretation of biofilm structure. This was examined here by using two different protocols for fixing biofilm E. coli TG1 and DSS640 cells to the CBD pegs. The first (standard) protocol employed two rinse steps after incubation of biofilm pegs in a glutaraldehyde fixative, followed by ethanol dehydration (Fig. 2E and 2F). The alternative protocol required extended air drying after the incubation of pegs in a glutaraldehyde fixative, but did not utilize rinse or ethanol dehydration steps (Fig. 2J to 2L). The alternative protocol preserved biofilm extracellular polymers and revealed a tight organization of biofilm cells in situ. Using this latter protocol, biofilms of E. coli TG1 and DSS640 biofilms appeared similar to those we have previously reported for hyper-biofilm forming SCV strains of P. aeruginosa PA14 (fixed with the standard protocol) (21). By comparison, the standard protocol removed much of the adherent biomass and exposed a portion of the underlying cells. In other words, the method of fixing biofilm cells to pegs may introduce artifacts that affect the judgment concerning the capacity of a particular microbe to form biofilms. Thus, this experiment emphasizes the importance of a consistent experimental approach to create valid comparisons in SEM analysis. In this case, E. coli DSS640 may be considered a poor biofilm former in minimal media, a good biofilm former in rich media, or mistaken for a hyper-biofilm former when treated with an alternative fixing protocol.
Acridine orange staining of microbial biofilms
AO may be used as a fluorescent biofilm biomass indicator as it stains cells as well as the nucleic acids that are a normal component of the extracellular matrix (31). Here, this technique was used in conjunction with CLSM to illustrate the variety of structural formations that may be adopted by biofilm bacteria grown in the CBD. In the following examples, CLSM z-stacks were processed using Leica Confocal Software (LCS), which was used to generate 2D average projections as well as 3D visualizations of biofilms using an isosurface rendering algorithm. Each experiment was performed in triplicate and a representative example of each is shown here.
There are many limitations to the interpretations that may be drawn from viability staining of biofilms using Syto-9 and PI; however, here we will address the limitation of sampling. A single field of view in a microscope does not represent a random sample from the entire biofilm population on the peg. Systematic collection of fields of view is a possible (but impractical) solution to quantify population survival in the CBD, which is more simply done through viable cell counting. Therefore, Live/Dead staining of CBD biofilms (as described here) is qualitative, and the discussion of semi-quantitative image analysis using this method is beyond the scope of this manuscript.
Despite pragmatic limitations on sampling, this type of assay is highly useful for spatial localization of dead cells in CBD biofilms following many antimicrobial treatments (19, 22, 25) as well as for qualitative assays such as the one described here. We acknowledge that dead cells are also a normal component of late logarithmic and stationary phase planktonic cell suspensions cultured in the CBD (J. J. Harrison, H. Ceri and R. J. Turner, unpublished data), and thus part of this dead biomass may be incorporated into the biofilm during growth. Nonetheless, this reinforces the notion that control groups are of pivotal importance when using Syto-9 and PI to evaluate the efficacy of anti-biofilm treatments, as every growing biofilm population normally contains a portion of dead biomass.
Staining of biofilm extracellular polysaccharides using fluorescent lectins
A distinguishing feature of biofilms is an extracellular matrix that is composed of short and long chain oligonucleotides (32), species specific proteins (33) and polysaccharides (31), as well as the biochemical derivatives and monomeric units of these compounds (26, 34). A strategy that has been employed to visualize extracellular polymers relies upon fluorophore-conjugated lectins (see Materials and methods). In this study, we used TRITC-ConA and TRITC-PNA in conjunction with AO and Syto-9, respectively, to stain the biofilm extracellular matrix and surface-attached cells of C. tropicalis 99916 (Fig. 5A to 5C) and B. cenocepacia K56-2 (Fig. 5D to 5F). Each experiment was performed in triplicate using the CBD and a representative example of each is shown here.
Fig. 5. TRITC-conjugated lectin staining of C. tropicalis and B. cenocepacia biofilms.
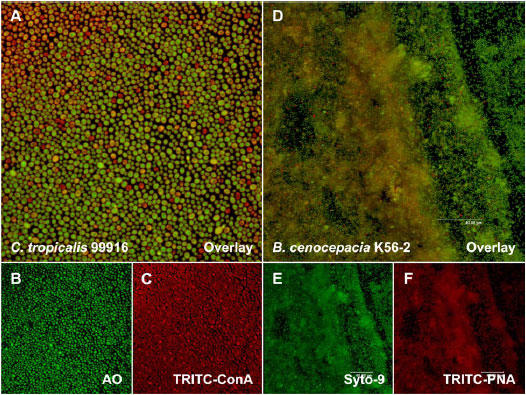
A. An overlay image of a C. tropicalis biofilm that was stained with AO (green emission) and TRITC-ConA (red emission). B. AO staining of the section illustrated in panel A. C. TRITC-ConA staining of the section illustrated in panel A. D. An overlay image of a B. cenocepacia biofilm that was stained with Syto-9 and TRITC-PNA. E. Syto-9 staining of the section illustrated in panel D. F. TRITC-PNA staining of the section illustrated in panel D. Each panel represents a square surface area of approximately 238 × 238 μm.
Fig. 3. CLSM of AO stained bacterial biofilms grown in the CBD.
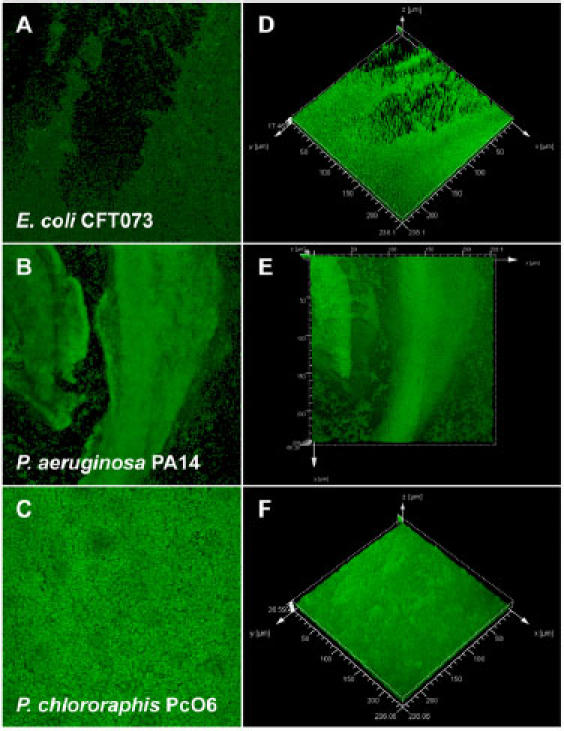
The images on the left are 2D averages of image z-stacks, whereas the images on the right are isosurface rendered 3D visualizations of the same data set (prepared using Leica® Confocal Software). These data sets illustrate that mature biofilms may adopt a number of structures that are distinct from the archetypal “stalk and mushroom” microcolony structures that are well characterized for P. aeruginosa. Each panel represents a square surface area of approximately 238 × 238 μm.
Correlative to previous reports, we observed that ConA highlighted C. tropicalis 99916 cell walls and stained EPS to a lesser extent (8, 22). An overlay of AO and ConA 2D average images revealed that yeast cells were in physical contact with their neighbours, joined either by their cell walls or a thin layer of EPS (Fig. 5A). The distribution of the extracellular biomass was also uneven, a feature that was shared in common with P. aeruginosa PA14 biofilms stained in a similar fashion (Fig. 6, discussed below with regards to 3D visualization). An overlay of Syto-9 and TRITC-PNA 2D average images additionally showed that B. cenocepacia K56-2 biofilms were encased in a heterogeneously distributed layer of EPS (Fig. 5D). Collectively, these data suggest that the production of extracellular polymers occurs non-uniformly throughout microbial biofilms.
Fig. 6. 3D visualization of microbial biofilms using amira®.
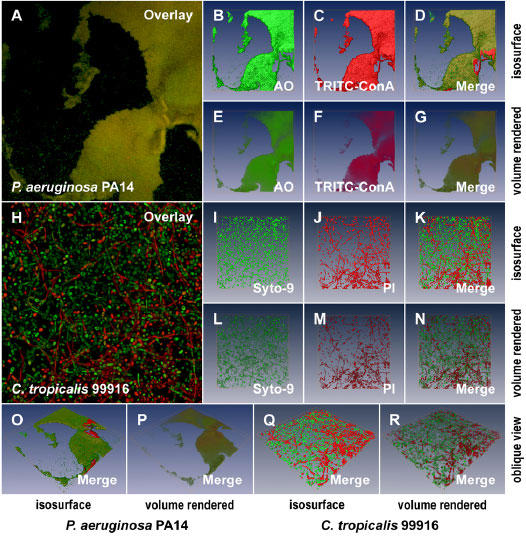
A. A 2D average of an image z-stack for a P. aeruginosa PA14 biofilm stained with AO and TRITC-ConA. B and C. Isosurface rendering of the 3D volume data sets for AO and TRITC-ConA, respectively, extrapolated from the image z-stacks used to create the image in panel A. D. The merged, isosurface rendered 3D volume data set for AO and TRITC-ConA. E to G. Volume rendering corresponding to the data sets presented in panels B to D. H. A 2D average of an image z-stack for a C. tropicalis 99916 biofilm stained with the Live/Dead® cell viability kit. I and J. Isosurface rendering of the 3D volume data sets for Syto-9 and PI respectively, extrapolated from the image z-stacks used to create the image in panel H. K. The merged, isosurface rendered 3D volume data set for Syto-9 and PI. L to M. Volume rendering corresponding to the data sets presented in panels I to K. O and P. The oblique view of the P. aeruginosa PA14 biofilm pictured in panel A visualized using isosurface and volume rendering, respectively. Q and R. The oblique view of the C. tropicalis 99916 biofilm pictured in panel H visualized using isosurface and volume rendering, respectively. Each 2D image panel or 3D model represents a square surface area of approximately 238 × 238 μm.
In the case of B. cenocepacia K56-2, biofilms were fixed using glutaraldehyde and rinsed several times following staining with TRITC-PNA. As discussed for SEM, any method used to preserve the biofilm may affect community structure and in particular, remove components of the EPS. Based on the data presented in Figure 2, it is likely that ethanol dehydration of samples for SEM is for the most part responsible for the removal of the EPS layer. The advantage of CLSM is that the samples remain in an aqueous environment and as such it is reasonable to expect that a larger portion of EPS is retained using this technique than with SEM.
3D visualization of CLSM data
Image processing and analysis methods are widely used in microbiological research to qualitatively and/or semi-quantitatively characterize microorganisms growing in biofilms. There are three open-source software packages that have been applied to the examination of multiple channel CLSM data sets from biofilms: daime (35), PHLIP (36), and COMSTAT (37). Here we describe the use of amira™, a professional software package for 3D visualization of volume data sets that utilizes hardware accelerated OpenGL 3D graphics with texture mapping. This software package was used to examine two additional CLSM data sets that were part of the proof-in-principle experiments described in this manuscript. First, P. aeruginosa PA14 biofilms were examined by staining samples with AO and TRITC-ConA (Fig. 6A to 6G). This was carried out with the specific aim of visualizing the heterogeneous distribution of extracellular polymers throughout the surface-attached community. Second, C. tropicalis 99916 biofilms (grown for 72 h on a gyrorotary shaker) were stained with Syto-9 and PI (Fig. 6H to 6N). This visualization was performed to illustrate that dead cells were a prevalent component of these biofilm communities. With regards to biofilm cultivation in the CBD and fluorescent staining, each of these experiments was performed in triplicate and a representative example of each was visualized using computer graphics (CG), as shown in the data presented here. Furthermore, two computational methods were used to examine these biofilm CLSM data sets.
The first visualization method used was isosurface rendering, whereby biofilm biomass was illustrated as a hollow shell that corresponded to the interconnected boundary voxels of the fluorescent, 3D volume data set. This method required that CLSM z-stacks were segmented, a user refined step carried out to separate background noise from biomass voxels. The advantage of this method was fast computer time and real-time user manipulability of the models without the requirement of specialized hardware (ex. expanded computer memory and specialized graphics cards). Isosurface rendering of CLSM data for P. aeruginosa PA14 and C. tropicalis 99916 is illustrated in Figure 6 (panels B to D and O for P. aeruginosa; panels I to K and Q for C. tropicalis) as well as Supplementary Videos 1 and 2, respectively.
The second method used to visualize biofilms was volume rendering, whereby 3D visualizations were a direct representation of the 3D volume data set (see Materials and methods). This method did not require the use of image segmentation to separate noise from the emitted fluorescence signals, thereby reducing user manipulation of the CLSM data sets. This method required greater processor time as well as hardware acceleration. Volume rendering of CLSM data for P. aeruginosa PA14 and C. tropicalis 99916 is illustrated in Figure 6 (panels E to 6 and P for P. aerugionsa; panels L to N and R for C. tropicalis) as well as Supplementary Videos 3 and 4, respectively.
In summary, 3D visualization was used to dynamically illustrate 3D volume data sets from CLSM of biofilms cultivated in the CBD. With regards to the underlying biology of microbial biofilms, these data illustrated two important points that were the focus of the proof-in-principle experiments carried out in this manuscript. First, biofilms cultivated in the CBD displayed heterogeneity in the production of extracellular polysaccharides. Second, CBD biofilms contained a significant proportion of dead cells amongst the population. In this example, C. tropicalis 99916 biofilms grown for 72 h had a number of viable cells equivalent to those cultivated for 48 h (Table 2). However, the aged biofilms were thicker, contained multiple cell morphotypes, as well as a much greater portion of dead cells in the population (compare Fig. 4B and 6H).
Fig. 4. CLSM of Live/Dead® (Syto-9 and PI) stained microbial biofilms grown in the CBD.
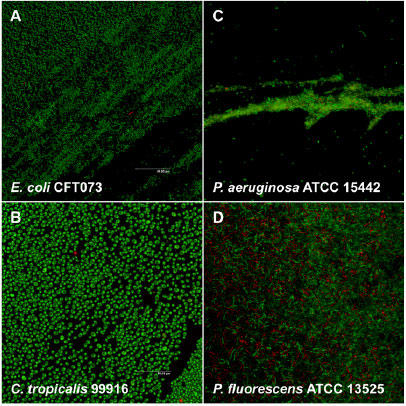
Cell death is a normal part of biofilm development (the extent of which may vary) and therefore dead biomass constitutes a portion of every biofilm. Each panel represents a square surface area of approximately 238 × 238 μm.
Conclusions
In this manuscript we have described the use of microscopy and 3D visualization methods to qualitatively evaluate the structure of microbial biofilms cultivated in the CBD. A feature of this model system for microbial cultivation was the ability to examine up to 96 biofilms in one batch culture apparatus. Therefore, biofilm structure may be evaluated under multivariate conditions and/or concomitantly with antimicrobial susceptibility testing or other biological assays (Fig. 1). For example, certain pegs may be removed from the device for microscopy, whereas others may be used for determining starting biofilm viable cell counts, and others still may be used for susceptibility determinations. In this fashion, the methods in this manuscript may be used to perform structure-function analysis of microbial biofilms under a combinatorial matrix of test conditions.
We acknowledge that an important constraint of biofilm growth in the CBD is complex fluid dynamic conditions. This must be considered when comparing data obtained from other experimental configurations (that feature controlled fluid dynamic conditions) to the data obtained from the CBD.
With regards to comparative analysis by microscopy, an experimental approach must be taken where biofilms of different microbial strains are treated in an identical fashion. Considerations for setting up microscopy analysis included the fixing protocol, choice of growth medium, viable cell counting, cross-talk between fluorophores, as well as the method of 3D visualization. Additionally, growth does not evenly cover the surface of a CBD peg. For instance, growth is typically greatest near the air-liquid-surface interface; therefore examination of pegs should be made in a systematic way to reflect this. In summary, microscopy and 3D visualization are important tools for biofilm research, and correctly designed studies using the CBD may allow for structure-function analyses on a high-throughput scale.
Supplemental Information
Supplementary videos of an AO and TRITC-ConA stained P. aeruginosa PA14 biofilm visualized using isosurface and volume rendering (Supplementary Videos 1 and 3, respectively) as well as a Live/Dead® stained C. tropicalis 99916 biofilm visualized using these two methods (Supplementary Videos 2 and 4, respectively) are available on the authors' website (http://www.acs.ucalgary.ca/~turnerr/biofilm.htm). These videos may be viewed using Quicktime 7, which is available free of charge from Apple Computers Inc. on the World Wide Web (http://www.apple.com/quicktime/win.html).
Acknowledgments
This work has been supported through discovery grants from the Natural Sciences and Engineering Research Council (NSERC) of Canada to R.J.T., H.C. and Y.H.. NSERC has also provided a Canada Graduate Scholarship (CGSD) to J.J.H., who was additionally supported by a Ph.D. Studentship from the Alberta Heritage Foundation for Medical Research (AHFMR). The Canadian Institue of Health Research (CIHR) has also provided a research grant to R.J.T.. R.M. has been supported by the Westaim Chair for Biofilm Research. J.Y. was supported by an Informatics Circle of Research Excellence (iCORE) Postgraduate Scholarship Award. SCLM was made possible through a Canadian Foundation for Innovation (CFI) Bone and Joint Disease Network grant to H.C.. The Alberta Innovation and Science Research Investments Program (ISRIP) has also funded this project through a grant to R.J.T., H.C. and R.M.. Additional thanks to Liz Middlemiss, Erin A. Badry, Kimberley M. Sproule, Pernilla Stenroos, Matthew L. Workentine and Lyriam L. R. Marques for their valuable technical assistance and expert advice. H.C. is the Director for Business Development at Innovotech, and this corporation donated the CBD plates used in this study.
Appendix
Protocols
Protocol 1 – Cultivating biofilms in the Calgary Biofilm Device
Reagents
Sterile CBD peg lid with 96-well microtiter plate or corrugated trough
Sterile 96-well microtiter plates
Sterile cotton swabs
Sterile 16 × 100 mm glass culture tubes
Sterile physiological saline solution (ex. PBS, 0.9% NaCl)
Sterile micropipette tips (2-200 μl), in racks of 96
Sterile 1 ml and 25 ml pipettes
Sterile 50 ml culture tubes
Sterile reagent reservoirs
Agar and broth growth media specific for the microorganism to be cultured
Protocol
Cultivation of biofilms in the CBD using a microtiter plate
**The following protocol describes the cultivation of biofilms in 96-well microtiter plates. An amendment to this protocol must be made for using the trough format of the CBD. This is described at the end of this section.
If using a cryogenic stock (at -70°C), streak out a first sub-culture of the desired bacterial or fungal strain on an appropriate agar plate. Incubate at the optimum growth temperature of the microorganism for a suitable period of time. For many bacterial strains, the first sub-culture may be sealed to minimize evaporation and stored at 4°C for up to 14 days.
Check the first sub-culture for purity (ie. only a single colony morphology should be present on the plate).
From the first sub-culture or from a clinical isolate, streak out a second sub-culture on an appropriate agar plate. Incubate at the optimum growth temperature of the microorganism for an appropriate period of time. The second sub-culture should be used within 24 h starting from the time it was first removed from incubation.
Verify the purity of the second sub-culture.
Open a sterile 96-well microtiter plate. For each CBD used, fill 4 ‘columns’ of the microtiter plate from ‘rows’ A to F with 180 μl of a physiological saline solution.
Put 1.5 ml (plus 1.0 ml for each additional CBD being inoculated at the same time) of the desired broth growth medium into a sterile glass test tube.
Using a sterile cotton swab, collect the bacterial colonies on the surface of the second agar sub-culture. Cover the tip of the cotton swab with a thin layer of bacteria.
Dip the cotton swab into the broth to suspend the bacteria. The goal is to create a suspension that matches a 1.0 McFarland standard (ie. 3.0 × 108 cfu ml-1). Be careful not to get clumps of bacteria in the solution.
Suspend more colonies, if necessary, to match the optical standard.
Put 29 ml of the appropriate broth growth medium (e.g. TSB) into a sterile 50 ml polypropylene or glass tube. To this, add 1.0 ml of the 1.0 McFarland standard bacterial suspension. This 30 fold dilution of the 1.0 McFarland standard (ie. 1.0 × 107 cfu ml-1) serves as the inoculum for the MBEC™ device.
Open the sterile package of CBD. Pour the inoculum into a reagent reservoir. Using the multichannel pipette, add 150 μl of the inoculum to each well of the 96-well tissue culture plate packaged with the CBD. Place the peg lid into the microtiter plate. Ensure that the orientation of the plate matches the orientation of the lid (i.e. peg A1 must be inserted into well A1 of the microtiter plate, otherwise the device will not fit together correctly). Label the device appropriately.
**The volume of inoculum used in this step has been calibrated such that the biofilm covers a surface area that is immersed, entirely, by the volume of antimicrobials used in the challenge plate set up in Step 3 (below). Using a larger volume of inoculum may lead to biofilm formation high on the peg that physically escapes exposure in this challenge step.
Place the device on the gyrorotary shaker in a humidified incubator at the appropriate temperature. The shaker should be set to between 100 and 150 revolutions per minute (rpm).
Serially dilute (ten-fold) a sample of the inoculum (do 3 or 4 replicates). These are controls used to verify the starting cell number in the inoculum.
Spot plate the serial 10 fold dilutions of the inoculum from 10-6 to 10-1 on an appropriately labeled series of agar plates. Incubate the spot plates for an appropriate period of time and score for growth.
Cultivation of biofilms in the CBD using a trough
All of the steps for biofilm cultivation are identical to those described above, except for the following two steps, which replace steps 11 and 12 from the protocol above.
Open the sterile package of the CBD. Using a sterile pipette, add 22 ml of the inoculum to the trough packaged with the device. Place the CBD peg lid onto the trough.
Place the device on the rocking table in a humidified incubator at the appropriate temperature. The table should be set to between 3 and 5 rocks per minute.
**It is critical that the angle of the rocking table is set to between 9° and 16° of inclination. This motion must be symmetrical.
Protocol 2 – Coating the CBD pegs with L-lysine
Reagents
Sterile CBD peg lid with 96-well microtiter plate or corrugated trough
Sterile 96-well microtiter plates
Sterile 1% L-lysine solution
Sterile micropipette tips (2-200 μl), in racks of 96
1% L-lysine solution
Add 1.0 g of L-lysine to 100 mL of ddH2O
Sterile filter at 0.20 μm
Protocol
Add 200 μL of 1.0% L-lysine solution to each well of a microtiter plate.
Remove the sterile CBD from its package and insert the peg lid into the microtiter plate containing the 1 % L-lysine solution. Incubate for 1 hour at room temperature.
Remove the peg lid from the microtiter plate and place upside down in a laminar flow hood for 30 min to air dry.
Use the peg lid to cultivate biofilms as described in Protocol 1.
Protocol 3 – Standard protocol for fixing biofilms onto the CBD pegs for SEM
**Compared to Protocol 4, this harsh fixing technique is more destructive to biofilms, but allows the cell structure and organization of the underlying bacteria to be examined.
Reagents
Phosphate buffered saline (PBS, pH 7.2)
70% glutaraldehyde, EM grade
cacodylate buffer (0.1 M)
2.5% or 5% glutaraldehyde in cacodylate buffer (0.1M)
0.9% saline
96-well microtiter plate
Micropipette tips (2-200 μl), in racks of 96
Cacodylate buffer (0.1 M)
16 g of sodium cacodylate in 1 L of double distilled (ddH2O)
Adjust to pH 7.2
**Wear gloves
Glutaraldehyde (2.5% or 5%)
To prepare a 2.5% solution, dissolve 2 ml of 70% glutaraldehyde in 52 ml of cacodylate buffer (0.1 M)
To prepare a 5% solution, dissolve 2 ml of 70% glutaraldehyde into 26 ml of cacodylate buffer (0.1 M)
**Wear gloves and use in a fume hood
Protocol
Break pegs from the CBD using a pair of flamed pliers.
Rinse pegs in 0.9% saline for 2 min by placing the pegs into 200 μL of 0.9% saline in the wells of a 96-well microtiter plate. This disrupts loosely adherent planktonic bacteria from the peg surface.
Fix the pegs in by placing into 2.5% glutaraldhyde in 0.1 M cacodylic acid (pH 7.2). Pegs are placed in this solution at 4°C for 16 h. Alternatively, pegs may be placed into 2.5% glutaraldehyde at room temperature for 2 h. A 5% solution of glutaraldehyde may be substituted for the 2.5% solution.
Following this fixing step, wash the pegs once in cacodylate buffer (0.1 M) for approximately 10 min.
Wash the pegs once in ddH2O for approximately 10 min.
Dehydrate the pegs in 70% ethanol for 15 to 20 minutes.
Air dry for a minimum of 24 h.
Mount specimens for SEM.
Protocol 4 - Alternative protocol for fixing biofilms onto the CBD pegs for SEM
**Compared to Protocol 3, this technique is relatively gentle and can be used to observe EPS and biofilm ultrastructure, albeit dehydrated.
Reagents
Phosphate buffered saline (PBS, pH 7.2)
70% glutaraldehyde, EM grade
cacodylate buffer (0.1 M)
2.5% glutaraldehyde in cacodylate buffer (0.1M)
0.9% saline
96-well microtiter plate
Micropipette tips (2-200 μl), in racks of 96
Protocol
Prepare solutions of cacodylate buffer (0.1 M) and glutaraldehyde (2.5%) as described in Protocol 3 above.
Break pegs from the CBD using a pair of flamed pliers.
Rinse pegs in 0.9% saline for 2 min. This disrupts loosely adherent planktonic bacteria from the peg surface.
Fix the biofilms in glutaraldhyde (2.5%). Pegs are placed in this solution at room temperature for 2 h.
Air dry for at least 120 h.
Mount specimens for SEM.
Protocol 5 - Fixing biofilms onto the CBD pegs for CLSM
**This protocol is not compatible with fluorescent staining using the Live/Dead® kit for bacterial cell viability.
Reagents
Phosphate buffered saline (PBS, pH 7.2)
5% glutaraldehyde in PBS
0.9% saline
96-well microtiter plate
Protocol
Break pegs from the CBD using a pair of flamed pliers.
Rinse pegs in 0.9% saline for 1 min. This disrupts loosely-adherent planktonic bacteria.
Fix the pegs in 5% glutaraldhyde in PBS (pH 7.2). Pegs are placed in this solution at 30°C for 30 min to 1 h.
Rinse pegs in 0.9% saline for 1 min.
Stain pegs with the appropriate fluorphores and examine using the confocal laser scanning microscope as indicated in Protocol 6.
Protocol 6 – Fluroescent staining and mounting of pegs for CSLM
Reagents
0.9% saline
96-well microtiter plate
Micropipette tips (2-200 μL), in racks of 96
Protocol
**There is an option here to fix the biofilms using glutaraldehyde. To do this, perform Protocol 5 instead of steps 1 and 2 below.
Break pegs from the CBD using a pair of flamed pliers.
Rinse the biofilms by immersing the pegs into 200 μL of 0.9% saline in the wells of a microtiter plate. Incubate for 1 min at room temperature.
Prepare the fluorescent stains as indicated in Table 3, or alternatively use the directions supplied by the manufacturer. Place 200 μL of the fluorescent stain in the required number of wells in the microtiter plate.
Using a pair of tweezers, transfer the pegs from the rinse into the fluorescent staining solution. Wrap the microtiter plate with tin foil and incubate for the required period of time.
If required as part of a second step, transfer the pegs into the required counterstain, wrap in tin foil, and then incubate for the required period of time.
Using a wax pencil, draw a circle on the surface of a 10 × 30 mm glass coverslip. With an eyedropper, put two drops of 0.9% saline in the middle of this circle.
**Do not use PBS for microscopy, as it may interfere with the fluorescence of some stains.
Use the tweezers to transfer the desired peg in to the saline on the glass coverslip.
Examine the biofilms using the confocal microscope as required by the system manufacturer.
Protocol 7 – amira™ for 3D visualisation of biofilms
**See the manufacturer’s website for minimum hardware requirements (Mercury Computer Products, http://www.tgs.com). Thirty-day trial version software is also available free of charge from the company at this website.
Protocol
Importing CLSM image data
Execute the software Amira 4.0 from a workstation.
Choose the Load command from the File menu to make the file dialog box to appear
Navigate to the location of the image files that were in the “tiff” format and select all of them by clicking the first file and shift clicking the last one.
Click Load in the file dialog box to load all the images.
The Image Read Parameter dialog appears that prompts you for entering the physical dimension of the bounding box or alternatively, the size of the voxel. Default values were used for the current study.
Click OK to continue.
An icon in green representing the loaded images appears in the object pool under the menu bar. Right click on the icon to popup a submenu. Select Compute from the submenu to open a list of commands. Choose Resample from the list to generate a new icon in the object pool called Resample in red.
Select the Resample icon and edit its ports in the working area below the object pool. For a coarser resolution, change the Resolution port from x=1024, y=1024 into x=256, y=256.
Press the Apply button to execute the resampling process.
After the resampling process is complete, a new icon in green representing the resampled images appears in the object pool.
Right click on this new icon to popup a submenu and select BoundingBox to display a bounding box.
Isosurface rendering
Right click on the icon of the resampled images to popup a submenu. Select Display from the submenu to open a list of commands. Choose IsoSurface from the list to generate a new icon in the object pool called IsoSurface in yellow.
Select the IsoSurface icon and edit its ports in the working area. Select the desired color and transparency from the Colormap port and adjust the value in the Threshold port to segment the resampled images. For semi-transparent display of IsoSurface, the Draw Style port needs to be set to transparent.
Press the Apply button to generate the 3D surfaces of the segmented images in the Viewer window.
Right click on the icon of the resampled images to popup a submenu. Select OrthoSlice from the submenu to generate a new icon in the object pool called OrthoSlice in orange and to overlay the original CLSM images to the 3D surfaces in the Viewer window.
Select the IsoSurface icon to ensure its ports visible in the working area. Adjust the value in the Threshold port and press Apply button to update the new value. Repeat the process so that the 3D surfaces of the segmented images overlay exactly to the biomass in the original CLSM images.
Repeat from step 2 to step 11 and from step 12 to step 16 to add a new stack of CLSM images for isosurface rendering.
Volume rendering
Right click on the icon of the resampled images to popup a submenu. Select Display from the submenu to open a list of commands. Choose Voltex from the list to generate a new icon in the object pool called Voltex in yellow.
Select the Voltex icon and edit its ports in the working area. Select the desired color and transparency from the Colormap port.
Press the Apply button to generate the 3D volume rendering of the resampled images in the Viewer window.
Repeat from step 2 to step 11 and from step 18 to step 20 to add a new stack of CLSM images for volume rendering.
Table 3.
Fluorescent stains for CLSM of microbial biofilms cultivated in the CBD (as described in this manuscript).
| Stain 1a | Stain 2a | Excitiation (nm) | Collected emission (nm) | Incubation time (min) | |||
| λ1 | λ2 | λ1 | λ2 | Stain 1 | Stain 2 | ||
| AO (0.1% in PBS) | n/a | 476 | n/a | 505-535 | n/a | 5 | n/a |
| TRITC-ConA (200 μg ml-1) | AO (0.1% in PBS) | 543 | 476 | 555-615 | 505-535 | 60 | 5 |
| Syto-9b (6.7 μM) | PIb (40 μM) | 488 | 543 | 510-540 | 610-670 | 30 (concomitant) | |
| TRITC-PNA (50 μg ml-1) | Syto-9b (6.7 μM) | 543 | 488 | 555-615 | 510-540 | 60 | 5 |
|
aAbbreviations for fluorescent stains: AO = acridine orange; PI = propidium iodide; TRTIC-ConA = tetramethylrhodamine isothiocyanate conjugated concanavalin A; TRITC-PNA = tetramethylrhodamine isothiocyanate conjugated peanut agglutinin. bSyto-9 and PI were diluted 500-fold from the stock solutions provided by the manufacturer (Molecular Probes). n/a denotes an item that is not applicable. | |||||||
References
- Chandra J, Zhou G, Ghannoum MA. Fungal biofilms and antimycotics. Curr Drug Targets. 2005;6:887–894. doi: 10.2174/138945005774912762. [DOI] [PubMed] [Google Scholar]
- Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: From the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- Harrison JJ, Turner RJ, Marques LLR, Ceri H. Biofilms: A new understanding of these microbial communities is driving a revolution that may transform the science of microbiology. Am Sci. 2005;93:508–515. [Google Scholar]
- Camilli A, Bassler BL. Bacterial small-molecule signaling pathways. Science. 2006;311:1113–1116. doi: 10.1126/science.1121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsek MR, Greenberg EP. Sociomicrobiology: the connections between quorum sensing and biofilms. Trends Microbiol. 2005;13:27–33. doi: 10.1016/j.tim.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Stoodley P, Sauer K, Davies DG, Costerton JW. Biofilms as complex differentiated communities. Annu Rev Microbiol. 2002;56:187–209. doi: 10.1146/annurev.micro.56.012302.160705. [DOI] [PubMed] [Google Scholar]
- Xu KD, Stewart PS, Xia F, Huang C, McFeters GA. Spatial physiological heterogeneity in Pseudomonas aeruginosa biofilm is determined by oxygen availability. Appl Environ Microbiol. 1998;64:4035–4039. doi: 10.1128/aem.64.10.4035-4039.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra J, Kuhn DM, Mukherjee PK, Hoyer LL, McCormick T, Ghannoum MJ. Biofilm formation by the fungal pathogen Candida albicans: development, architecture, and drug resistance. J Bacteriol. 2001;183:5385–5394. doi: 10.1128/JB.183.18.5385-5394.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borriello G, Werner E, Roe F, Kim AM, Ehrlich GD, Stewart PS. Oxygen limitation contributes to antibiotic tolerance of Pseudomonas aeruginosa in biofilms. Antimicrob Agents Chemother. 2004;48:2659–2664. doi: 10.1128/AAC.48.7.2659-2664.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purevdorj-Gage B, Costerton WJ, Stoodley P. Phenotypic differentiation and seeding dispersal in non-mucoid and mucoid Pseudomonas aeruginosa biofilms. Microbiology. 2005;151:1569–1576. doi: 10.1099/mic.0.27536-0. [DOI] [PubMed] [Google Scholar]
- Garcia-Medina R, Dunne WM, Singh PK, Brody SL. Pseudomonas aeruginosa acquires biofilm-like properties within airway epithelial cells. Infect Immun. 2005;73:8298–8305. doi: 10.1128/IAI.73.12.8298-8305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley NR, Lazazzera BA. Environmental signals and regulatory pathways that influence biofilm formation. Mol Microbiol. 2004;52:917–924. doi: 10.1111/j.1365-2958.2004.04036.x. [DOI] [PubMed] [Google Scholar]
- Donlan RM. Biofilms: microbial life on surfaces. Emerg Infect Dis. 2002;8:881–890. doi: 10.3201/eid0809.020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tay JH. The essential role of hydrodynamic shear force in the formation of biofilm and granular sludge. Water Res. 2002;36:1653–1665. doi: 10.1016/S0043-1354(01)00379-7. [DOI] [PubMed] [Google Scholar]
- Ceri H, Olson ME, Stremick C, Read RR, Morck DW, Buret AG. The Calgary Biofilm Device: New technology for rapid determination of antibiotic susceptibilities in bacterial biofilms. J Clin Microbiol. 1999;37:1771–1776. doi: 10.1128/jcm.37.6.1771-1776.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JJ, Ceri H, Stremick C, Turner RJ. Biofilm susceptibility to metal toxicity. Environ Microbiol. 2004;6:1220–1227. doi: 10.1111/j.1462-2920.2004.00656.x. [DOI] [PubMed] [Google Scholar]
- Harrison JJ, Turner RJ, Ceri H. High-throughput metal susceptibility testing of microbial biofilms. BMC Microbiol. 2005;5:53. doi: 10.1186/1471-2180-5-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EO, Ward MK, Raney DC. Two simple media for the demonstration of pyocyanin and fluorescein. J Lab Clin Med. 1954;44:301–307. [PubMed] [Google Scholar]
- Teitzel GM, Parsek MR. Heavy metal resistance of biofilm and planktonic Pseudomonas aeruginosa. Appl Environ Microbiol. 2003;69:2313–2320. doi: 10.1128/AEM.69.4.2313-2320.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JJ, Ceri H, Badry EA, Roper NJ, Tomlin KL, Turner RJ. Effects of the twin-arginine translocase on the structure and antimicrobial susceptibility of Escherichia coli biofilms. Can J Microbiol. 2005;51:671–683. doi: 10.1139/w05-048. [DOI] [PubMed] [Google Scholar]
- Davies JA, Harrison JJ, Marques LLR, Foglia GR, Stremick CA, Storey DG, Turner RJ, Olson ME, Ceri H. The GacS sensor kinase controls phenotypic reversion of small colony variants isolated from biofilms of Pseudomonas aeruginosa PA14. FEMS Microbiol Ecol. 2007;59:32–46. doi: 10.1111/j.1574-6941.2006.00196.x. [DOI] [PubMed] [Google Scholar]
- Harrison JJ, Rabiei M, Turner RJ, Badry EA, Sproule KM, Ceri H. Metal resistance in Candida biofilms. FEMS Microbiol Ecol. 2006;55:479–491. doi: 10.1111/j.1574-6941.2005.00045.x. [DOI] [PubMed] [Google Scholar]
- Morck DW, Lam K, McKay SG, Olson ME, Prosser B, Ellis BD, Cleeland R, Costerton JW. Comparative evaluation of fleroxacin, ampicillin, trimethoprim-sulfamethoxazole, and gentamicin as treatments of catheter associated urinary tract infections in a rabbit model. Am J Med. 1994;94:23S–30S. [Google Scholar]
- Bernas T, Asem EK, Robinson JP, Cook PR, Dobrucki JW. Confocal fluorescence imaging of photosensitized DNA denaturation in cell nuclei. Photochem Photobiol. 2005;81:960–969. doi: 10.1562/2004-11-11-RA-369. [DOI] [PubMed] [Google Scholar]
- Jin Y, Zhang T, Samaranayake YH, Fang HHP, Yip HK, Samaranayake LP. The use of probes and stains for improved assessment of cell viability and extracellular polymeric substances in Candida albicans biofilms. Mycopathologia. 2005;159:353–360. doi: 10.1007/s11046-004-6987-7. [DOI] [PubMed] [Google Scholar]
- Wozniak DJ, Wycoff TO, Starkey M, Keyser R, Azadi P, O'Toole GA, Parsek MR. Alginate is not a significant component of the extracellular polysaccharide matrix of PA14 and PAO1 Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci USA. 2003;100:7907–7912. doi: 10.1073/pnas.1231792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha MV, Sousa SA, Leitão JH, Moreira LM, Videira PA, Sá-Correia I. Studies on the involvement of the exopolysaccharide produced by cystic fibrosis isolates of the Burkholderia cepacia complex in biofilm formation and in persistence of respiratory infections. J Clin Microbiol. 2004;42:3052–3058. doi: 10.1128/JCM.42.7.3052-3058.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkins MD, Ceri H, Storey DG. Pseudomonas aeruginosa GacA, a factor in multihost virulence, is also essential for biofilm formation. Mol Microbiol. 2001;40:1215–1226. doi: 10.1046/j.1365-2958.2001.02469.x. [DOI] [PubMed] [Google Scholar]
- Kirisitis MJ, Prost L, Starkey M, Parsek M. Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl Environ Microbiol. 2005;71:4809–4821. doi: 10.1128/AEM.71.8.4809-4821.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ize B, Porcelli I, Lucchini S, Hinton JC, Berks BC, Palmer T. Novel phenotypes of Escherichia coli tat mutants revealed by global gene expression and phenotypic analysis. J Biol Chem. 2004;279:47543–47554. doi: 10.1074/jbc.M406910200. [DOI] [PubMed] [Google Scholar]
- Branda SS, Vik S, Friedman L, Kolter R. Biofilms: the matrix revisited. Trends Microbiol. 2005;13:20–26. doi: 10.1016/j.tim.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Whitchurch CB, Tolker-Neilsen T, Ragas PC, Mattick JS. Extracellular DNA required for bacterial biofilm formation. Science. 2002;295:1487. doi: 10.1126/science.295.5559.1487. [DOI] [PubMed] [Google Scholar]
- Branda SS, Chu F, Kearns DB, Losick R, Kolter R. A major protein component of the Bacillus subtilis biofilm matrix. Mol Microbiol. 2006;59:1229–1238. doi: 10.1111/j.1365-2958.2005.05020.x. [DOI] [PubMed] [Google Scholar]
- Sutherland IW. The biofilm matrix - an immobilized but dynamic microbial environment. Trends Microbiol. 2001;9(5):222–227. doi: 10.1016/S0966-842X(01)02012-1. [DOI] [PubMed] [Google Scholar]
- Daims H, Lücker S, Wagner M. daime, a novel image analysis program for microbial ecology and biofilm research. Environ Microbiol. 2006;8:200–213. doi: 10.1111/j.1462-2920.2005.00880.x. [DOI] [PubMed] [Google Scholar]
- Mueller LN, de Brouwer JFC, Almeida JS, Stal LJ, Xavier JB. Analysis of a marine phototrophic biofilm by confocal laser scanning microscopy using the new image quantification software PHLIP. BMC Ecology. 2006;6:1. doi: 10.1186/1472-6785-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydorn A, Nielsen AT, Hentzer M, Sternberg C, Givskov M, Ersboll BK, Molin S. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology. 2000;146:2395–2407. doi: 10.1099/00221287-146-10-2395. [DOI] [PubMed] [Google Scholar]
- Tomlin KL, Malott RJ, Ramage G, Storey DG, Sokol PA, Ceri H. Quorum-sensing mutations affect attachment and stability of Burkholderia cenocepacia biofilms. Appl Environ Microbiol. 2006;71:5208–5218. doi: 10.1128/AEM.71.9.5208-5218.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch RA, Burland V, Plunkett III G, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, Perna NT, Mobley HL, Donnenberg MS, Blattner FR. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci USA. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- Gibson TJ. Studies on the Epstein-Barr virus genome. Cambridge, UK: University of Cambridge; 1984.
- Sambasivarao D, Dawson HA, Zhang G, Shaw G, Hu J, Weiner JH. Investigation of Escherichia coli dimethyl sulfoxide reductase assembly and processing in strains defective for the sec-independent protein translocation system membrane targeting and translocation. J Biol Chem. 2001;276:20167–20174. doi: 10.1074/jbc.M010369200. [DOI] [PubMed] [Google Scholar]
- Rahme LG, Stevens EJ, Wolfort J, Shao J, Tompkins RG, Ausubel FM. Common virulence factors for bacterial pathogenicity in plants and animals. Science. 1995;268:1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- Radtke C, Cook WS, Anderson AJ. Factors affecting antagonism of the growth of Phanerochaete chrysosporium by bacteria isolated from soils. Appl Microbiol Biotechnol. 1994;41:274–280. doi: 10.1007/s002530050143. [DOI] [Google Scholar]
- Harrison JJ, Ceri H, Roper NJ, Badry EA, Sproule KM, Turner RJ. Persister cells mediate tolerance to metal oxyanions in Escherichia coli. Microbiology. 2005;151:3181–3195. doi: 10.1099/mic.0.27794-0. [DOI] [PubMed] [Google Scholar]
- Harrison JJ, Turner RJ, Ceri H. Persister cells, the biofilm matrix and tolerance to metal cations in biofilm and planktonic Pseudomonas aeruginosa. Environ Microbiol. 2005;7:981–994. doi: 10.1111/j.1462-2920.2005.00777.x. [DOI] [PubMed] [Google Scholar]
- Harrison JJ, Ceri H, Stremick C, Turner RJ. Differences in biofilm and planktonic cell mediated reduction of metalloid oxyanions. FEMS Microbiol Lett. 2004;235:357–362. doi: 10.1016/j.femsle.2004.05.005. [DOI] [PubMed] [Google Scholar]