

Abstract
14C-Labelled octulose phosphates were formed during photosynthetic 14CO2 fixation and were measured in spinach leaves and chloroplasts. Because mono- and bisphosphates of d-glycero-d-ido-octulose are the active 8-carbon ketosugar intermediates of the L-type pentose pathway, it was proposed that they may also be reactants in a modified Calvin–Benson–Bassham pathway reaction scheme. This investigation therefore initially focussed only on the ido-epimer of the octulose phosphates even though 14C-labelled d-glycero-d-altro-octulose mono- and bisphosphates were also identified in chloroplasts and leaves. 14CO2 predominantly labelled positions 5 and 6 of d-glycero-d-ido-octulose 1,8-P2 consistent with labelling predictions of the modified scheme. The kinetics of 14CO2 incorporation into ido-octulose was similar to its incorporation into some traditional intermediates of the path of carbon, while subsequent exposure to 12CO2 rapidly displaced the 14C isotope label from octulose with the same kinetics of label loss as some of the confirmed Calvin pathway intermediates. This is consistent with octulose phosphates having the role of cyclic intermediates rather than synthesized storage products. (Storage products don’t rapidly exchange isotopically labelled carbons with unlabelled CO2.)
A spinach chloroplast extract, designated stromal enzyme preparation (SEP), catalysed and was used to measure rates of CO2 assimilation with Calvin cycle intermediates and octulose and arabinose phosphates. Only pentose (but not arabinose) phosphates and sedoheptulose 7-phosphate supported CO2 fixation at rates in excess of 120 μmol h−1 mg−1 Chl. Rates for octulose, sedoheptulose and fructose bisphosphates, octulose, hexose and triose monophosphates were all notably less than the above rate and arabinose 5-phosphate was inactive. Altro-octulose phosphates were more active than phosphate esters of the ido-epimer. The modified scheme proposed a specific phosphotransferase and SEP unequivocally catalysed reversible phosphate transfer between sedoheptulose bisphosphate and d-glycero-d-ido-octulose 8-phosphate. It was also initially hypothesized that arabinose 5-phosphate, an L-Type pentose pathway reactant, may have a role in a modified Calvin pathway. Arabinose 5-phosphate is present in spinach chloroplasts and leaves. Radiochromatography showed that 14C-arabinose 5-phosphate with SEP, but only in the presence of an excess of unlabelled ribose 5-phosphate, lightly labelled ribulose 5-phosphate and more heavily labelled hexose and sedoheptulose mono- and bisphosphates. However, failure to demonstrate any CO2 fixation by arabinose 5-phosphate as sole substrate suggested that the above labelling may have no metabolic significance. Despite this arabinose and ribose 5-phosphates are shown to exhibit active roles as enzyme co-factors in transaldolase and aldolase exchange reactions that catalyse the epimeric interconversions of the phosphate esters of ido- and altro-octulose. Arabinose 5-phosphate is presented as playing this role in a New Reaction Scheme for the path of carbon, where it is concluded that slow reacting ido-octulose 1,8 bisphosphate has no role. The more reactive altro-octulose phosphates, which are independent of the need for phosphotransferase processing, are presented as intermediates in the new scheme. Moreover, using the estimates of phosphotransferase activity with altro-octulose monophosphate as substrate allowed calculation of the contributions of the new scheme, that ranged from 11% based on the intact chloroplast carboxylation rate to 80% using the carboxylation rate required for the support of octulose phosphate synthesis and its role in the phosphotransferase reaction.
Keywords: Arabinose 5- phosphate, Calvin Cycle, 14C-carbon dioxide, Chloroplasts, 14C-labelled novel compounds, Keto-group exchange, L-type pentose pathway, Spinach leaf, Octulose phosphates, Octulose-P epimerization, Phosphotransferase, Revised RPP in PS, Stromal enzymes
Introduction
The formulation of the Calvin–Benson–Bassham pathway, hereafter called Calvin cycle, of photosynthesis (PS) was heavily dependent on the elucidation of a reaction sequence for the pentose pathway (PP) of glucose metabolism in the biochemistry of tissues. A reaction sequence for the PP was proposed from the results of experiments in which liver, pea leaf and pea root enzyme preparations were used to catalyse the conversion of [1-14C] Ribose 5-P (Rib 5-P) to14C-labelled hexose 6-P and unlabelled glyceraldehyde 3-P (Gap), which were formed in vitro over a 17-h period (Horecker et al. 1954). For many years Williams and co-workers investigated and published results of research on PP in liver, some neoplasms, adipose tissue, heart, colonocytes and photosynthetic tissues (see Williams et al. 1987; Williams 2004 for published listings). These investigations showed that the conclusion drawn from the results of the fundamental experiments on which the PP reaction sequence is based (Horecker et al. 1954; Gibbs and Horecker 1954) was erroneous and because of its 17-h duration, the results were incapable of being interpreted due to the random scattering of 14C isotope in the glucose 6-P (Glc 6-P) product. The 14C labelling pattern of Glc 6-P was the result of a prediction labelling experiment that was used to hypothesize a reaction sequence for the PP. The unpredictable and randomized isotope scattering of 14C in Glc 6-P was finally shown to be due to the activity of the extensive 14C-exchange rates versus the slower rates of mass transfer reactions catalysed by the group transferring enzymes, aldolase (EC 4.1.2.13) (Ald), transketolase (EC 2.2.1.1) (TK) and transaldolase (EC 2.2.1.2) (TA) (Flanigan et al. 1993). Williams et al. (1978a) demonstrated that the reversible interconversions of Rib 5-P and hexose and triose phosphates by liver and the other tissues mentioned above, also involved ido- and altro-octulose mono- and bisphosphates, sedoheptulose 1,7-P2 (Seh 1,7-P2) and a low concentration of arabinose 5-P (Ara 5-P) as intermediates, together with a proposal for the inclusion of three new enzyme activities in the PP reaction sequence (see Fig. 1).
Fig. 1.
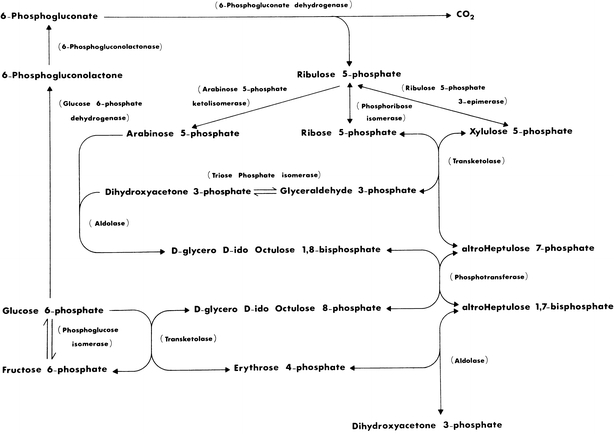
The L-Type PP of glucose metabolism. The pathway features reactions of arabinose 5-phosphate, d-glycero-d-ido-octulose and sedoheptulose mono- and bisphosphates together with new enzymes to catalyse their reactions (Williams and Clark 1971; Williams et al. 1978a; Williams 2004)
Calvin and colleagues based the reaction sequence which regenerated pentose phosphates (especially Ru 1,5-P2) from hexose and triose phosphates in PS, on a selection of the reactions of a reverse-acting classical PP (Calvin 1956), thereby providing important credibility at the time for the PP which still carried an author-imposed tentative caveat. It was later noted (Clark et al. 1974) that the omission of the above new PP intermediates and enzymes from the Calvin scheme provided further explanations of some of the early criticisms and anomalies of the Calvin pathway scheme (Clark et al. 1974; Stiller 1962; Kandler and Gibbs 1956; Beck and Hopf 1982).
The investigations described here were made in order to (a) search for evidence that d-g-d-i-oct phosphates may be formed by carbon fixation reactions in spinach during PS and (b) quantitatively measure any formation of octulose phosphates and investigate whether their proposed path of synthesis conformed with a theoretical extension of the photosynthetic reaction scheme shown in Fig. 2. This paper reports the results of studies on the formation and functions of octulose phosphates, with a particular initial focus on d-glycero-d-ido-octulose 1,8-P2 and the 8-monophosphate, together with other novel intermediates in spinach leaves and chloroplasts.
Fig. 2.
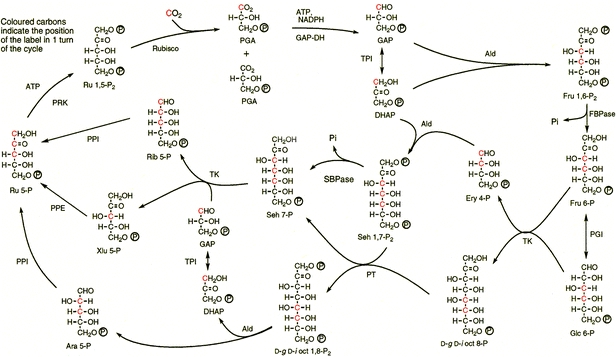
The initial hypothesis for a revised reaction scheme for the reductive PP of photosynthetic carbon reduction in C-3 plants. The hypothesis differs from the Calvin Pathway by the inclusion of new enzymes and reactions by octulose and arabinose phosphates and is tested by the results reported in this paper. The figure shows the distributions (for one cycle) of the coloured carbon of CO2 into traditional and proposed new intermediates of a modified Calvin Pathway
The initial singular attention on the ido- epimer rather than an inquiry that also included d-glycero-d-altro-octulose phosphates, rested on the exclusive roles assigned to d-g-d-i-oct phosphates in the L-type PP (Fig. 1). However, a final conclusion drawn from the study suggests that both epimeric forms of octulose phosphates occur and that both may have roles in the path of carbon in PS. The investigation used the same 14C tracer and other methodologies that were pioneered by Calvin and his team in the unravelling of a path of carbon in PS. The accompanying paper reports the results of a subsequent mass spectrometric investigation of the incorporation of 13CO2 into C4 to C8 sugar phosphates in spinach chloroplasts during light-driven PS.
Materials and methods
Growth of plant material
Spinach (Spinacia oleracea, L.,var. Yates 102) was grown by water culture in a glasshouse under natural lighting according to established methods (Anderson and Boardman 1966). Depending on the season, leaves were harvested five or eight weeks after sowing the seeds.
Chemical and chromatographic materials
Enzymes and cofactors for analysis and synthesis of sugar phosphates were obtained from either Sigma Chemical Co., St. Louis, MO 63178, USA or from Boehringer-Mannheim Corp., D-6800 Mannheim, Germany. 14C- and 32P-labelled compounds were from Amersham (Bucks, England). Other analytical reagent grade chemicals were obtained from Ajax Chemicals, Sydney, Australia, BDH Poole, BH12 4NN, UK, or E. Merck, D-6100 Darmstad, Germany. Chromatographic media were obtained from the following sources: Ion exchange resins from BioRad, Richmond, CA 94804, U.S.A. Phenylboronate agarose gel (Matrex PBA 60) from Amicon Corp., Danvers, MA 01923, USA. Thin layer chromatography plates from E.Merck, 3Mm paper from Whatman, Maidstone, UK, Sephadex G10 and Sephadex A25 from Pharmacia, Uppsala, Sweden. Chromatographic columns were from Pharmacia or LKB, Bromma, Sweden. Ultrafiltration apparatus and Diaflow PM10 membranes were obtained from Amicon, Lexington, MA, USA.
Extraction and analysis of photosynthetic tissue metabolites by gas-liquid chromatography
Young spinach leaves (6–8 weeks) were detached from plants, which had been exposed to full sunlight for 8 h, and then placed in liquid nitrogen. The tissue was ground to a fine powder using a stainless steel mortar and pestle and 6% perchloric acid was added. The temperature of this mixture was raised to 4°C and the solid was blended with the acid using a Potter-Elvejhem homogenizer. The liquid was separated from insoluble matter by centrifugation at 10,000g for 10 min. The pellet was re-extracted with perchloric acid and the two extracts combined. The solution was neutralized with 2 M KOH and stored at 2°C for several hours to complete the precipitation of potassium perchlorate which was then removed by centrifugation at 15,000g for 10 min. The supernatant fluid was treated with acid-washed activated carbon (Norite) to remove nucleotides and aromatic compounds and was lyophilized. The extract was dissolved in 5 ml of 0.2 M acetate buffer, pH 5.0 and 20 units of acid phosphatase (EC 3.1.3.2 from potato) added. Mannitol (2 μmol) and erythritol (0.5 μmol) were added as internal standards. The mixture was incubated at 30°C to achieve complete sugar phosphate hydrolysis. The free sugars were recovered by thermally denaturing the protein, followed by centrifugation and treatment of the supernatant fraction with a three-fold excess (calculated from the amount of buffer used) of mixed bed resin (Dowex 50 × 4, 100–200 mesh, H+ form; Dowex 1 × 4, 100–200 mesh, HCO−3 form). The completeness of deonization was established using a Radiometer conductivity meter. The solution was lyophilized and stored at −20°C. Lyophilized extracts were converted to their TMS ethers immediately before analysis using the procedure of Williams et al. (1984). Dimethylformamide and bis-trimethylsilyltrifluoroacetamide (50 μl of each) were added and the mixture stirred for 45 min at 55°C. The analyses were carried out using a Pye-Unicam gas chromatograph (series 204), fitted with two glass columns (180 cm × 0.4 cm) packed with 3% SP-2250 on 100/120 mesh Supelco, Bellefonte, PA; USA. FID responses were recorded on a computing integrator (Spectra-Physics, model 4100).
Photosynthesis experiments with whole spinach leaves
14C isotope tracer and pulse–chase investigations with whole spinach leaves were conducted using an exact application of the methods and apparatus described by Hatch and Slack (1966). This procedure and the method for the ethanol extraction, preparation of the soluble fraction and isolation of sugar phosphates was described by Bartlett et al. (1989). Sugar phosphates were separated by formate anion-exchange chromatography and further purified using two-dimensional TLC or paper chromatography and the GW3/GW3-phenylboronate solvents (Kapuscinski et al. 1985). The insoluble residue remaining after ethanol extraction of all sugar phosphates was extensively washed with water, ethanol and acetone. The amount of 14C radioactivity in the insoluble glucan was determined using liquid scintillation counting (Arora et al. 1987).
Preparation of whole leaf homogenate
Approximately 50 g of fresh spinach leaves were washed briefly in distilled water, de-ribbed and cut into small segments to facilitate maceration. The material was homogenized in 100 ml of 50 mM Tris–HCl buffer (pH 7.4) containing 1 mM EDTA, and strained through two layers of Miracloth (Calbiochem). After centrifugation at 10,000 × g for 15 min, the preparation was concentrated four-fold by ultrafiltration using a 10,000 MW cut-off membrane (PM10 Amicon). The concentrated solution was dialysed for 10 h against six changes of the homogenization buffer. All operations were carried out at 4°C.
Chloroplast isolation
The procedure for the isolation of intact chloroplasts is based on a method which involves a two-step gradient of the silica-sol, Percoll (Pharmacia Biotech, Sweden) (Robinson 1983). Five- to six-weeks-old spinach leaves were harvested in the early morning in order to avoid the accumulation of starch granules, which can rupture the chloroplast envelope during centrifugation. The amount of leaf material required to produce a satisfactory yield of chloroplasts was not critical but the yield depended on the quality of the leaves and increased with the amount of material used. The harvested leaves were de-ribbed and floated in basins of water. Immediately before isolating the chloroplasts, the leaves were illuminated for 30 min with incandescent light at an intensity of 800 μ E m−2 s−1. During this period, crushed ice was added to the water to maintain the temperature at 15°C. Typically, 55 g of coarsely chopped leaves were homogenized in 300 ml of medium for 1.5–2 s using a Polytron blender (setting 7) fitted with a PT35K probe. The homogenizing medium at pH 6.5 was chilled to a semi-frozen consistency before use and contained 330 mM sorbitol, 10 mM Na4P2O7·10H2O, 5 mM MgCl2, 2 mM isoascorbic acid and 0.1% bovine serum album (BSA) (Jensen and Bassham 1966). The brei was filtered through a double layer of Miracloth (Calbiochem). The filtrate was centrifuged at 1,200 × g for 1 min in a Sorvall refrigerated centrifuge (DuPont Medical Products, Newtown, CT O647-5509 USA) using either an SS34 or SA600 rotor. The supernatant fluid was discarded and the sedimented material was gently resuspended, using a fine soft brush, in 6 ml of a medium at pH 7.6 containing 330 mM sorbitol, 50 mM HEPES–KOH, 2 mM EDTA, 1 mM MgCl2 and 1 mM MnCl2 (Jensen and Bassham 1966). An aliquot (3 ml) of the suspension was then placed into each of two 40-ml Corex tubes and carefully underlaid with 4 ml of Percoll medium made to 40% with resuspension medium and centrifuged. Finally, the chloroplasts were suspended in the resuspension medium (3 ml) and stored on ice. The above procedures were carried out on ice using previously chilled equipment. All parameters (density, yield, photosynthetic activity etc.) describing chloroplast properties were related to the Chl content measured according to the following procedure. Twenty five to 65 g of leaves produced between 3 mg and 8 mg of Chl. Provided the quality of leaf material was satisfactory and the Percoll was fresh, the method unfailingly produced a maximum yield of highly active chloroplasts which were better than 95% intact. Typical examples of preparations had the following mean values and S.D.: Activity 113.7 ± 8.6 (n = 10) μmol CO2 h−1 mg−1 Chl; Intactness 95.75% ± 3.25% (n = 10).
Chlorophyll assay
The Chl content of isolated chloroplast suspensions was estimated by the method of Arnon (1949). In whole leaf experiments Chl was estimated using the method of Vernon (1960).
Measurements of chloroplast polarographic activity and intactness
The activity of the isolated chloroplast suspensions was determined polarographically using a Clark oxygen electrode. The reaction mixture (2 ml) contained, 0.33 mM sorbitol, 2 mM EDTA, 1 mM MgCl2, 1 mM MnCl2, 50 mM HEPES, 0.5 mM Pi, 5 mM pyrophosphate, 500 units of catalase (EC 1.11.1.6), 10 mM NaHCO3 and chloroplast suspension equivalent to 40 μg of Chl. The CO2-dependent O2 evolution of isolated chloroplast suspensions illuminated in an assay medium at 20°C was recorded using a Goerz Metrawatt SE120 chart recorder and the rate of oxygen evolution was calculated from the linear portion of the trace. The chloroplasts were illuminated at an intensity of 1,200 μE m−2 s−1 using the quartz-halogen light from a slide projector. In the experiments described here, the range of acceptable activity of chloroplast preparations is 100–120 μmol h−1 mg−1 Chl. Light measurements were made using a Hansatech Quantum Sensor 3/2897 (Hansatech Instruments, King’s Lynn, Norfolk PE321, JL.UK).
The intactness of chloroplasts was measured by the ferricyanide reduction method (Lilley et al. 1975).
Isotopic tracer studies with isolated chloroplasts
Experiments were carried out in the oxygen electrode (unless stated otherwise) by the procedure described for the assay of chloroplast activity. NaH14CO3 (100 μCi) was introduced when the rate of CO2-dependent oxygen evolution reached a linear rate. The reaction was terminated by rapid transfer of the mixture to boiling ethanol (80% w/v). The insoluble material was removed by centrifugation and washed once with a small volume of water. The ethanol was evaporated from the combined supernatant solutions in a rotary evaporator. After passage through a cation-exchange column (Bartlett et al. 1989), the sugar phosphates were separated and isolated by anion-exchange column chromatography and further purified by two-dimensional TLC or paper chromatography (Kapuscinski et al. 1985).
The isolation of stromal metabolites used the method of Heldt (1980) which relied on rapid separation of intact chloroplasts from a reaction medium using the following silicon oil method. Fifty μl of 10 M formic acid and 50 μl of silicone oil mixture (AR100 and AR150 mixed in a ratio of 3:1) were placed in a 400-μl microfuge tube followed by 200 μl of a chloroplast suspension that had been preincubated with 32P-orthophosphate (specific radioactivity 50 mCi mmol−1). The extracts of the pellets containing the aqueous fractions that were pooled from four tubes were lyophilized and the sugar phosphates therein were isolated by anion-exchange column chromatography as described in Methods. Sugar phosphate peaks isolated in the column eluates were identified and quantitatively measured using the specific enzymatic and colourimetric procedures of Williams et al. (1978b) and Kapuscinski et al. (1985).
Preparation of stromal enzyme extracts (SEP) from spinach chloroplasts
The soluble (stromal) enzyme fraction was prepared essentially as described by Furbank and Lilley (1981) for pea chloroplasts. Intact spinach chloroplasts were isolated with assayed rates of CO2-dependent O2 evolution in excess of 100 μmol h−1 mg−1 Chl. Typically a chloroplast suspension equivalent to 2 mg of Chl was pelleted by centrifugation at 1,000 × g for 1 min in an SS34 Sorvall rotor at 4°C. The supernatant solution was removed and 10 ml of chilled 10 mM HEPES buffer, pH 8.0 containing 3 mM DTT was added. The pellet was resuspended by gentle shaking. The centrifuge tube was kept on ice for approximately 10 min after which it was centrifuged for 15 min at 9,000 × g in a refrigerated Sorvall centrifuge RC-5 to separate the soluble proteins from the membrane fraction. The clear supernatant fluid, representing the stromal fraction of the chloroplasts, was concentrated to 4 ml in an Amicon ultrafiltration cell, using a PM10 membrane (nominal MW cut-off 10,000). Finally, dialysis was carried out against 3 × 500 ml of dialysis buffer containing 10 mM HEPES–KOH (pH 8.0), 3 mM DTT, 15 mM MgCl2 and 10 mM KCl. This last step was completed in 4 h. The pelleted membranes were retained for Chl analysis. The activity of the extracts was stable for 4–6 h from completion of the dialysis step. Some experiments used an alternative stromal isolation procedure based on the method of Kaiser and Bassham (1979). The chloroplast suspension was pelleted as described above but lysis of organelles was achieved with 25 mM HEPES–NaOH, 2 mM MgCl2, 1 mM EDTA (pH 7.6) and the same buffer was used for dialysis.
Assay of the reductive PP using stromal enzyme preparation (SEP)
The activity of the reductive PP in the stromal extracts was determined by the method of Furbank and Lilley (1981). The progress of the reaction was followed spectrophotometrically using 0.2-cm light path cuvettes. The assay mixture contained 40 mM HEPES–KOH buffer (pH 8.0), 20 mM MgCl2, 8 mM KCl, 1 mM NADH, 0.8 mM EDTA, 10 mM DTT, 4 mM ATP, 10 mM creatine phosphate, 10 mM NaHCO3, 2 units of creatine kinase (EC 2.7.3.2), 1.25 units of glyceraldehyde-phosphate dehydrogenase (EC 1.2.1.12), 1.7 units of phosphoglycerate kinase (EC 2.7.2.3). SEP equivalent to 5–50 μg of Chl was added to make a final volume of 500 μl. SEP was preincubated for 5 min at 25°C in 20 mM MgCl2, 10 mM DTT, 10 mM NaHCO3 before addition of the test sugar phosphate substrate. Usually the following test substrates were individually used at 2 mM concentration in the reaction mixture: Rib 5-P, dihydroxyacetonephosphate (DHAP), or Seh 1,7-P2.
Maximum catalytic capacity of the non-oxidative segment of the PP
This was determined by following the conversion of ribose 5-P to hexose 6-P in a coupled enzyme assay as described by Williams et al. (1984). The reaction mixture contained 80 mM TEA–HCl buffer, pH 7.4, 0.4 mM NADP+, 10 mM MgCl2, 1.5 units of phosphoglucose isomerase (EC 5.3.1.9) (PGI), 2 units of glucose 6-phosphate dehydrogenase (EC 1.1.1.49) (Glc 6-PDH), 0.4 mM ribose 5-P and stromal extract equivalent to 50 μg of Chl in a total volume of 1.0 ml.
Synthesis of isotopically labelled compounds
[U-14C] Arabinose 5-phosphate, [8-14C] d-g-d-a-oct 8-P and [8-14C] d-g-d-i-oct 8-P were prepared by the method of Arora et al. (1987). [1-32P]-Sedoheptulose 1,7-bisphosphate was prepared by phosphorylation of sedoheptulose 7-P (altro-heptulose) with [γ-32P] ATP using rabbit muscle phosphofructokinase (EC 2.7.1.11) (PFK). The reaction mixture (0.50 ml) contained: 20 μmol Tris–HCl buffer (pH 7.6), 2.5 μmol of MgCl2, 0.2 μmol of [γ-32P] ATP (100 μCi), 2.5 μmol of Seh 7-P and 6 units of PFK. The mixture was incubated at 25°C and was 95% complete in 1 h. The title compound was purified using formate-anion exchange chromatography and TLC as described above and was shown by enzymatic analysis to be free of Fru 1,6-P2. Unlabelled d-glycero-d-ido- and d-glycero-d-altro-octulose 8-P and 1,8-P2 esters were prepared as described in Kapuscinski et al. (1985).
Measurements of phosphotransferase activity
Arora et al. (1985) published three assay procedures for the minimum estimate of the activity of d-glycero-d-ido-octulose 1,8-P2 or d-glycero-d-altro-octulose 1,8-P2: d-altro-heptulose 7-phosphotransferase (PT). Two of these assays measured the reaction for the non-oxidative PP (Fig. 1) in the direction of octulose 8-P formation. The third procedure, and the one detailed here, measured the activity in the flux direction of the RPP (see Fig. 2 and Eqs. (1) and (2)).
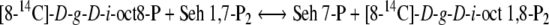 |
1 |
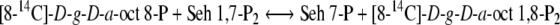 |
2 |
Only the third method was effective when SEP was the crude source of the putative enzyme activity. It is a radiochromatographic discontinuous-stop assay and is applied as follows. The reaction mixture in a volume of 0.50 ml contained 1 μmol of [8-14C]-octulose 8-P (0.8 μCi μmol−1), 3 μmol Seh 1,7-P2, 20 μmol of TEA–HCl buffer (pH 7.4) and 16 μmol of KCl. The mixture was incubated for 30 min at 30°C and was initiated by the addition of SEP equivalent to 50 μg of Chl. Aliquots (0.1 ml) were sampled at 5.0 min intervals during the time-course of the reaction and injected into an equal volume of 6% perchloric acid. Denatured protein was removed by centrifugation, the pH of the supernatant fluid adjusted to 6.8 with KOH and precipitated KClO4 separated by centrifugation. The activity of the enzyme was estimated by separating the labelled octulose mono- and bis-phosphates by ion-exchange chromatography as follows: A precisely known volume of the above supernatant fraction was applied onto a column (5 mm × 5 mm) of ion-exchange resin (AG 1 X8, 200–400 mesh, formate form) followed by 2 ml of deionized water. The sugar phosphates were eluted by successive washes with 2 M HCOOH (approx. 12 ml), which quantitatively removed monophosphate esters, and 5 ml of 4 M HCOOH–1 M HCOONH4 solution which eluted sugar bisphosphates. Fractions (2 ml) of the eluted sugar phosphates at each time point were analysed for radioactivity and the amount of octulose bisphosphate formed was calculated from the integrated peaks of radioactivity. Confirmation of the identity and amount of radioactivity in the 14C-labelled octulose phosphates used two-dimensional radiochromatography of each of the above fractions and the GW3/GW3-phenylboronate solvents (Kapuscinski et al. 1985). Control experiments using only a single substrate accompanied each activity measurement.
Arabinose 5-P activity with SEP
The reaction mixture (2.0 ml) containing 80 μmol TEA–HCl buffer (pH 7.4), 3.6 μmol Rib 5-P, 0.4 μmol of [U-14C]-Ara 5-P (0.2 μCi) and SEP equivalent to 200 μg of Chl was incubated for 1 h at 30°C. The reaction was terminated by heating and all sugar phosphates were isolated with greater than 90% recovery of 14C isotope. An identical control reaction using boiled SEP accompanied all incubations. Processed samples were chromatographed on paper using either the GW3 or GW3–2% PBA solvents and the chromatograms exposed to X-ray film (Kodak X-O mat RP), (Bleakley et al. 1984).
Positional isotopic analysis of sugar phosphates
Following PS experiments with leaves or chloroplasts, 14C-labelled Glc 6-P, Rib 5-P, d-g-d-i-oct 1,8-P2 and PGA were isolated, resolved by ion-exchange chromatography and purified by two-dimensional TLC or paper chromatography. The fraction containing Glc 6-P was concentrated and chromatographed twice on paper (3MM, Whatman) using the GW3 solvent of Wood (1968) followed by solvent A of Bandurski and Axelrod (1951). The 14C-labelled Glc 6-P was located by autoradiography and comparison with a stained standard marker compound (Rosenberg 1959) and was then eluted from the paper with deionized water. The above regime ensured that the isolated 14C-labelled Glc 6-P was free of any other hexose phosphates. The phosphate group of Glc 6-P was removed by enzymatic hydrolysis as follows: Solid Glc 6-P was dissolved in 5 ml of 50 mM glycine–NaOH buffer, pH 10.4 and 5 units of Alkaline phosphatase (EC 3.1.3.1) from Escherichia coli was added and the mixture incubated at 37°C. The progress of the reaction was monitored by assaying the release of inorganic phosphate using the method of Tashima and Yoshimura (1975).
When the reaction was completed the mixture was poured into a suspension of mixed-bed resin (1 meq of Dowex 50W, H+ form and 1 meq of Bio AG1 x 4, HCO−3 form) which was stirred for approx. 60 min. The solution was filtered through a sintered glass funnel (porosity 3) and the residual cake of resin thoroughly washed with deionized water. The 14C-labelled glucose filtrate was freeze-dried and stored at 4°C until used. The percentage distribution of 14C isotope in the individual carbons of glucose was determined as 14CO2 using a combination of microbiological and chemical methods (Williams et al. 1971).
14C-Labelled octulose phosphates from spinach leaves or chloroplast extracts were isolated and purified as above and degraded using the methods of Williams et al. (1985). Following isolation by anion-exchange and TLC chromatography, 14C-labelled Rib- and Ara 5-phosphates were dephosphorylated (Williams et al. 1984) and Ara and Rib were chemically degraded using the periodate method (Genovese et al. 1970). [14C]-3-Phosphoglyceric acid derived from DHAP was chemically degraded by the procedure of Andrews et al. (1965).
Recovery of 14CO2 as BaCO3 and measurement of radioactivity
Discs (2 cm) of Whatman No. 42 filter paper were used to collect barium carbonate precipitates. The discs were washed with water, absolute ethanol and diethyl ether, dried and weighed before use. BaCO3 was deposited on the papers using gentle suction, then washed with CO2-free water, alcohol and ether. The discs were dried under vacuum over silica and equilibrated at room temperature before weighing on a semi-micro balance. Approx. 20 mg of dried BaCO3 was exactly weighed, placed into a vial with 10 ml of scintillation cocktail and counted for radioactivity as a suspension in the “Cab-O-Sil” (Godfrey L. Cabot Inc.) scintillant of Cluley (1962).
Replication and statistical analysis
Data presented are the mean values (±S.D.) from results of five or more experiments or mean values (±P.E.) (Probable Error) for all experiments conducted on different days.
Results and discussion
GLC analysis of perchloric acid extracts of spinach leaves
Principal aims of the experiments were an investigation of the occurrence, identity and quantitative levels of the ido- and altro-octuloses in fresh photosynthetic tissue and the assessment of their hypothesized functions (especially that of d-g-d-i-oct 1,8-P2) in PS. It was noted that Charlson and Richtmyer (1959) and Begbie and Richtmyer (1966) had isolated and characterized octulose from avocado and Primula officinalis, respectively. In a remarkable finding Howarth et al. (1996) showed that d-glycero-d-ido-octulose represented 90% of the total carbohydrates in fully hydrated leaves and 50% of dried leaf matter of the resurrection plant Craterostigma plantagineum. Heath (1984) also showed evidence for the L-type, but not the F-type, PP in Chlorella sorokiniana. Since the mono- and bisphosphate esters of d-g-d-i-oct are a unique feature of the L-type PP (Fig. 1), the need to identify and measure these esters in fresh spinach leaf and chloroplasts is obvious. Different methods were used to establish whether d-glycero-d-ido-octulose 1,8-P2 occurred in spinach leaves. The first approach used the simple and sensitive GLC method. A gas chromatogram of the pertrimethylsilyl (TMSi) derivatives of the dephosphorylated sugar phosphates extracted from spinach leaves is shown in Fig. 3.
Fig. 3.
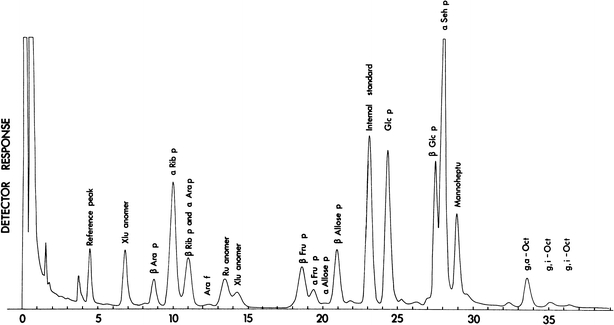
Gas liquid chromatogram of dephosphorylated TMSi-derivatized sugars extracted from young spinach leaves during light-driven carbon fixation. Octuloses and arabinose feature in the chromatogram. For details see Materials and methods section
The high molecular weight region was well resolved and allowed tentative identification (by retention times and co-chromatography with authentic compounds) of d-g-d-i-octulose and its 5′-altro-epimer, d-g-d-a-oct. Estimation of the amounts of these compounds from the known response factors of the gas chromatograph gave values of 11.8 ± 0.98 nmol mg−1 Chl for ido-octulose and 18.2 ± 0.78 nmol mg−1 Chl for altro-octulose. These values are tentative because other compounds may co-elute with the octuloses, e.g. the TMS ethers of heptitols have similar retention times to those of eight carbon sugars. In addition, the GLC method used in this study cannot differentiate between free and phosphorylated sugars. Different approaches were therefore adopted to determine the levels of specifically phosphorylated octuloses present in photosynthesizing tissue.
14C-d-g-d-i-oct 1,8-P2 isolation from spinach leaves: 14CO2 fixation and pulse–chase studies
The second approach used the combination of formate ion-exchange chromatography followed by phenylboronic acid and two-dimensional TLC, to achieve isolation, identification and complete separation of d-g-d-i-oct 1,8-P2 from all other sugar phosphates (see Methods). This methodology both confirmed and extended the GLC findings and unequivocally demonstrated that d-g-d-i-oct 1,8-P2 occurred in spinach leaf extracts.
The conditions adopted for the study of the time-course of 14CO2 fixation into the products of PS involved an exact application of the method of Hatch and Slack (1966). This provided steady-state conditions for PS in intact spinach leaves at a CO2 concentration of 470 ppm that followed from a 50 ml injection of 0.80 mCi of 14CO2 and light intensity of 50% direct sunlight (1,200 μE m−2 s−1). The rate of 14CO2 incorporation into the soluble leaf fraction (an index of the total 14CO2 fixed) was approximately linear (1.9 × 106 cpm s−1 mg−1 Chl) during a 120-s interval (Fig. 4, panel A). The 14C labelling of PGA was marginally more rapid and extensive than that of Glc 6-P which reached 14C isotope saturation in the interval 75–120 s.
Fig. 4.
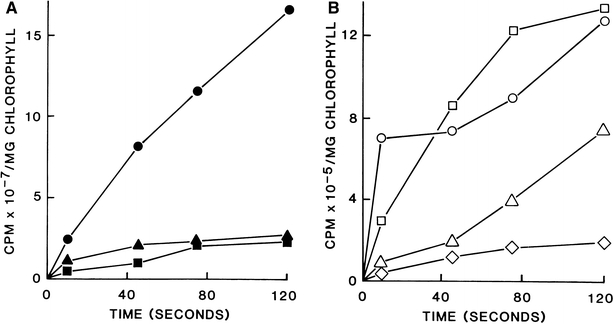
Shows the kinetics of 14C isotope incorporation from 14CO2 into a selection of sugar phosphates in whole spinach leaves during light-driven PS for 120 s. The time course of the total 14C fixed in the soluble fraction of the leaf extract is also shown. Labelled sugar phosphates were resolved by anion-exchange chromatography and were then individually isolated and further purified by two-dimensional paper or TLC chromatography as described in the Materials and methods Section. The 14C-radioactivity incorporated in glucose 6-P and PGA (panel A) was an order of magnitude greater than that incorporated in the sugar phosphates shown in panel B. Panel A: Total 14C isotope fixed, •; radioactivity incorporated in Glc 6-P, ■ and PGA, ▴. Panel B. Rib 5-P, □; other pentose 5-P, ○; Seh 1,7-P2, Δ and d-g-d-i-oct 1,8-P2, ◊
Glc 6-P was labelled at an approx rate of 2.8 × 105 cpm s−1 mg−1 Chl (Fig. 4, Panel A). The radioactivity in PGA and Glc 6-P was an order of magnitude greater than that fixed in all other sugar phosphates investigated (see Fig. 4, panel B). The fast rate of 14C incorporation into Rib 5-P was initially inclined to linear for 75 s (2.0 × 104 cpm −1 mg−1 Chl). The bisphosphates Seh 1,7-P2 and d-g-d-i-oct 1,8-P2 were more slowly labelled at 7.3 × 103 and 2.4 × 103 cpm s−1 mg−1 Chl, respectively. The kinetics and relative degree of 14C isotope incorporation into individual PS intermediates shown at Fig. 4 are very similar to the results for 14CO2 incorporation in Scenedesmus (Benson et al. 1952). Moreover, d-g-d-i-oct 1,8-P2 was labelled early and was saturated with 14C isotope at 75 s, at which point its concentration in the leaf was 1.6 nmol mg−1 Chl and the rate of label incorporation was 0.13% of that fixed in the soluble fraction. When the above experiments were conducted in the dark the distribution of 14C in the leaf soluble fraction was uneven and at 120 s was only 0.01% of that measured in the light. None of the sugar phosphates shown at the A and B panels of Fig. 4 were dark labelled.
Hatch and Slack (1966) described an apparatus for measurements of the kinetics of 14CO2 fixation and 12CO2 exchange by pulse–chase in bulk samples of intact leaves undergoing PS. Their method was able to delineate Calvin Cycle intermediates, which rapidly exchanged 14C for 12C during and following a short pulse with 12CO2, thereby identifying rapidly cycling compounds. These are clearly distinguished by their delabelling kinetics (Figs. 5 and 6) from the cytoplasmic storage compounds sucrose, starch and cationic compounds that retained or increased their levels of 14CO2 following the pulse phase (Fig. 5).
Fig. 5.
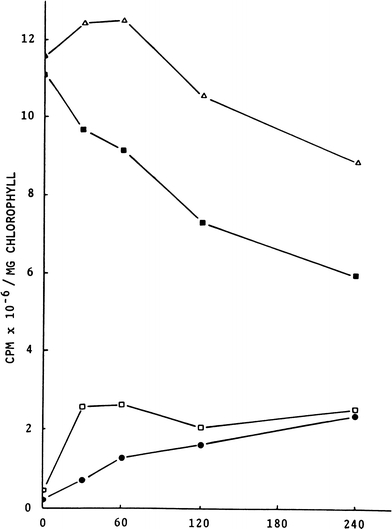
Shows the results of pulse–chase experiments where spinach leaves were confined in 14CO2 for 20 s (the pulse) and were then exposed to non-radioactive air (the chase). See methods. The figure shows the time-course of the partition of 14C isotope into various fractional groups of compounds in spinach leaves following their exposure to the ‘chase’ with unlabelled CO2 for 240 s and illuminated to the equivalent of 50% direct sunlight. It is of note that starch and cationic compounds retained and continued to show increasing 14C radioactivity while the radioactivity in the total soluble fraction and the pool of anionic compounds declined. The time-course of changes in 14C radioactivity are shown for starch, •; neutral and anionic compounds, ■; the cationic fraction, □ and total soluble radioactivity, Δ
Fig. 6.
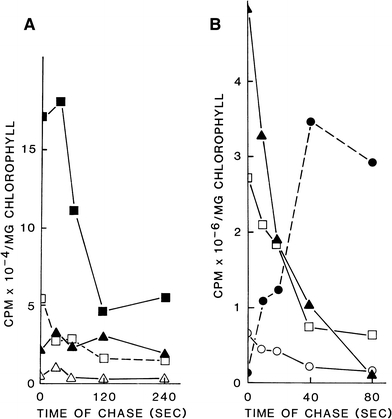
Shows the change in the 14C-radioactivity of individual sugar phosphates and sucrose in spinach leaves during the time-course of the ‘chase’ with unlabelled CO2. Sugar phosphates from leaves that had been subjected to the ‘pulse-chase’ experiments (See Fig. 4b results) were separated by anion-exchange chromatography and individually purified by two dimensional TLC or paper chromatography. See Materials and methods for details. Panel A shows the kinetics over 240 s of the decline in radioactivity of Rib 5-P, ■; Fru 1,6-P2, ▴; DHAP, □; and d-g-d-i-oct 1,8-P2, •. Panel B shows the time-course over 80 s of the decline in radioactivity of PGA, ▴; Glc 6-P, □; Fru 6-P, ○; and the initial increase in radioactivity in sucrose, •
Taken together the results of Figs. 4 and 6 showed that d-g-d-i-oct 1,8-P2 was rapidly labelled and saturated with isotope during 14CO2 fixation. It was delabelled on exposure to 12CO2 during a comparative study which showed that it exhibited the same kinetics of 14C loss as other cycling bisphosphate intermediates of the Calvin Pathway. It was therefore unambiguously distinguished from slow 14C-release carbon storage and other cytoplasmic end products of PS by the above kinetics.
d-glycero-d-ido-octulose 1,8-P2 in spinach chloroplasts
The above results suggested that d-g-d-i-oct 1,8-P2 may have a chloroplast origin. In order to test this proposal, PS experiments were carried out with isolated spinach chloroplasts. If d-g-d-i-oct 1,8-P2 is synthesized by enzymes of the reductive PP, it should become rapidly labelled following the metabolism of 32P-inorganic phosphate (Pi) or 14C-bicarbonate. It was noted above that d-g-d-i-oct 1,8-P2 incorporated 14C isotope during PS by isolated chloroplasts in the presence of NaH14CO3 (see Methods). These observations were further tested using 32P-orthophosphate as the isotopic marker (see Methods section) in order to investigate whether d-g-d-i-oct 1,8-P2 was exclusively present in the organelles and was not the result of external reactions catalysed by enzymes released from any ruptured chloroplasts. Intact chloroplasts were rapidly filtered through a layer of silicone oil (Heldt 1980) and the 32P-labelled metabolites were separated by ion-exchange chromatography (see Methods section). The unambiguous identification of the [32P]-d-g-d-i-oct 1,8 P2 was made by rechromatographing the recovered octulose phosphate fractions on paper using the GW3 and GW3/PBA solvents. From the known specific radioactivity of inorganic phosphate in the reaction mixture and the absolute amount of 32P-isotope in the purified d-g-d-i-oct 1,8-P2, the concentration of the sugar phosphate in the stroma was estimated.
Using the value of the sorbitol impermeable space of 26 μl mg−1 Chl (Schafer et al. 1977), the concentration of d-g-d-i-oct 1,8-P2 was estimated to be 1.3 nmol mg−1 Chl ±0.50 (50 μM, three determinations). This value is less than that measured for ido-oct in the whole leaf PS study, but is in the same concentration range as that of some other Calvin Cycle metabolites in spinach chloroplasts (Portis et al. 1977; Heldt et al. 1978, 1980; Stitt et al. 1980; Petterson and Ryde-Petterson 1988). Measurement of the amount of the altro-epimer of the octulose bisphosphate in chloroplasts was not made in this study (see the accompanying paper for the results of a mass spectrometric study of 13C labelled sugar phosphates formed in spinach chloroplasts during PS). A carry-through of the external medium may affect the estimation of the stromal volume as a sorbitol impermeable space and consequently the determination of the molar concentration of metabolites contained in the stroma. However, the amount of d-g-d-i-oct 1,8-P2 in the stroma is quoted per mg of Chl and is thus independent of the volume of external medium that may be carried through silicon oil with the organelles. The results of control experiments, using 14CO2, showed that following centrifugation, the external medium contained less than 10% of the 14C-labelled d-g-d-i-oct 1,8-P2 found in the pelleted chloroplast fraction. Thus it is concluded that d-g-d-i-oct 1,8-P2 is probably synthesized and may react exclusively in the stromal compartment of spinach chloroplasts. Its concentration is estimated to be 50 μM using the above data and the following equation:
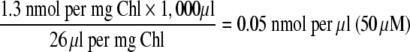 |
Origin of d-g-d-i-oct 1,8-P2 and distributions of 14C isotope in a selection of sugar phosphates isolated from spinach leaves and chloroplasts assimilating 14CO2 in photosynthesis
The most likely biosynthetic routes leading to d-g-d-i-oct phosphate formation involve catalysis by the carbon–carbon cleavage and group-transferring enzymes of sugar phosphate metabolism, namely transketolase (Eq. (3)), aldolase (Eq. (4)) and transaldolase (Eq. (5)).
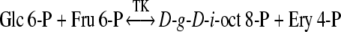 |
3 |
 |
4 |
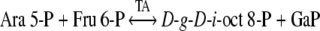 |
5 |
The short-term PS experiments described in the Methods and Results sections were used to identify which of the above three enzymes may have operated for the synthesis of d-g-d-i-oct phosphate in spinach leaves and chloroplasts. However, it is of note that the initial hypothesis (Fig. 2) for the modified reductive PP (Clark et al. 1974; Williams et al. 1987), proposed that d-g-d-i-oct-8-P was formed by the reaction of (Eq. (3)) and d-g-d-i-oct-1,8-P2 by the PT reaction of (Eq. (6)) (Arora et al. 1985).
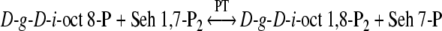 |
6 |
Figure 2 shows that in one cycle of 14CO2 fixation, d-g-d-i-oct 1,8-P2 is labelled in carbons 5 and 6. In order to test this aspect of the mechanism of the pathway, the distributions of 14C isotope into the individual carbons of Glc-6P, phosphoglyceric acid (PGA), Rib-5-P and d-g-d-i-oct 1,8-P2 were determined in samples isolated from spinach leaves and chloroplasts during light-driven PS in 14CO2. The procedures used (see Methods section) allowed each of the carbons of Glc 6-P and PGA to be isolated for the determination of the degree of positional carbon isotopic labelling. However, only carbons 1, 2, 3, 4 and 8 of d-g-d-i-oct 1,8-P2 were able to be individually isolated for 14C measurement and carbons 5, 6 and 7 were collectively measured as a group (Williams et al. 1985). Only the 14C isotope levels of carbons 1 and 5 of Rib-5-P were uniquely determined by the methods applied in this study, with carbons 2, 3 and 4 being estimated as a group 14C-measurement. The data of Table 1 show the results of these experiments using spinach leaves photosynthetically assimilating 14CO2 for 10, 45, 75 and 120 s, respectively (see Methods section). The data of Table 2 show similar 14C isotope distributions in PGA, Glc 6-P and d-g-d-i-oct 1,8-P2 isolated from intact spinach chloroplasts after 60 s of PS in 14CO2.
Table 1.
Distribution of 14C isotope in sugar phosphates isolated from spinach leaves following specified intervals of PS in 14COa2
| Carbon | 10 s of 14CO2 fixation | 45 s | 75 s | 120 s | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glc 6-P | d-g-d-i-oct 1,8-P2 | Rib 5-P | PGA | Glc 6-P | d-g-d-i-oct 1,8-P2 | Glc 6-P | d-g-d-i-oct 1,8-P2 | Glc 6-P | |||||||||||
| Found | Predicted | ||||||||||||||||||
| 1 | 3.2 | 35.6 | (33.3) | 23.4 | 84.9 | 12.7 | 20.4 | (38.7) | 15.1 | 29.9 | (37.3) | 18.5 | |||||||
| 2 | 3.2 | 25.3 | (33.3) | } | 7.2 | 7.4 | 21.1 | (22.6) | 10.3 | 30.6 | (25.4) | 11.8 | |||||||
| 3 | 37.2 | 39.1 | (33.3) | 71.6 | 7.9 | 24.6 | 58.5 | (38.7) | 22.0 | 39.5 | (37.3) | 17.2 | |||||||
| 4 | 50.5 | 21.2 | (3.1) | 38.1 | 13.1 | (8.5) | 33.4 | 13.3 | (12.1) | 25.0 | |||||||||
| 5 | 2.5 | } | } | 5.0 | 7.0 | } | } | 9.1 | } | } | 10.7 | ||||||||
| 6 | 3.4 | (60.7) | (93.2) | 10.2 | 79.2 | (79.8) | 10.1 | 63.7 | (76.0) | 17.1 | |||||||||
| 7 | |||||||||||||||||||
| 8 | 18.1 | (3.5) | 7.7 | (11.7) | 23.0 | (11.9) | |||||||||||||
| % recovery of 14C-isotope | 88 | 91b | 76c | 86 | 101 | 106 | 82b | 75c | 93 | 85b | 89c | 90 | |||||||
aThe conduct of the experiments is described in the Methods section. Sugar phosphates were isolated and purified by ion-exchange and paper chromatographic procedures. Results are mean values of duplicate degradations except for all of the compounds isolated after 10 s of 14CO2 fixation, which were degraded in triplicate. b, c% 14C recovered in the top 3 and bottom 5 carbons of d-g-d-i-oct 1,8-P2
Table 2.
Distribution of 14C isotope in sugar phosphates isolated from intact spinach chloroplasts following carbon fixation by light-driven PSa
| Carbon | Percent specific radioactivity | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 40 s | 60 s | ||||||||
| PGA | DHAP | Glc 6-P | PGA | Glc 6-P | d-g-d-i-oct 1,8-P2 | ||||
| Found | (Theory) | ||||||||
| 1 | 55.1 | 59.4 | 12.5 | 46.6 | 13.6 | 56.7 | (32.5) | ||
| 2 | 18.4 | 17.2 | 7.3 | 29.2 | 14.4 | 28.4 | (35.0) | ||
| 3 | 26.5 | 23.4 | 23.0 | 24.2 | 18.3 | 14.9 | (32.5) | ||
| 4 | 39.1 | 33.6 | 18.4 | (17.0) | |||||
| 5 | 8.5 | 9.2 | } | } | |||||
| 6 | 9.6 | 10.9 | 64.0 | (70.0) | |||||
| 7 | |||||||||
| 8 | 17.6 | (13.0) | |||||||
| % 14C recovered | 98 | 101 | 89 | 97 | 95 | 88b, 94c | |||
a The 14C-labelling experiments were carried in an oxygen electrode apparatus as described in the Methods section. Illuminated chloroplast suspensions (100 μg Chl ml−1) were incubated with 10 mM NaHCO3 until a linear rate of CO2-dependent O2 evolution was attained. One hundred μCi of NaH14CO3 was added to adjust the specific radioactivity of the substrate to 5 μCi μmol−1. The subsequent steps are detailed in the Methods section. The specific radioactivity of individual carbons of sugar phosphates are the mean value of triplicate determinations. b,c The percent recovery of 14C isotope from the top 3 and bottom 5 carbon atoms of d-g-d-i-oct 1,8-P2, respectively. The method for calculating the theoritical bracketed values in Tables 1 and 2 is given in the Appendix
Degradative data for PGA, DHAP and Glc 6-P following 40 s of PS are also listed. Both sets of results implicate a TK-catalysed reaction (Eq. (3)) as the most likely route for the biosynthesis of d-g-d-i-oct 8-P leading to the bisphosphate by the reaction of Eq. (6).
In order to compare the above TK path of synthesis with the 14C distribution pattern that may be imposed by the reactions of Eqs. (4) and (5), catalysed by either Aldolase (ALD) or TA, theoretical distributions of 14C isotope in d-g-d-i-oct 1,8-P2 are also presented as bracketed values in Tables 1 and 2. The Appendix shows the method for calculating the theoretical values. These distributions were calculated using the experimentally measured isotope levels in Glc 6-P (Table 1) and an assumption that Fru 6-P formed in the reductive pathway is in chemical and isotopic equilibrium with Glc 6-P formed by the action of PGI. It is also assumed that the 14C distributions in the mono- and bisphosphates of the d-g-d-i-octuloses of Eqs. (3) and (6) are the same.
Equation (3a) shows detail of the spread of 14C in each carbon of d-g-d-i-oct 8-P at 10 s (see data Table 1) using the mean values of triplicate estimations of 14C isotope in each of the six carbons of Glc 6-P and the assumptions described above. The individual distributions measured in the top three carbons of the d-g-d-i-oct 1,8-P2 sampled at all times support the proposal that d-g-d-i-oct 8-P in spinach leaves (Table 1) and chloroplasts (Table 2) may be formed by the TK reaction of Eq. (3). However, the leaf concentrations of 14C in C-4 and C-8 of d-g-d-i-oct 1,8-P2 in the 45-s and 75-s samples and the 60-s chloroplast sample are greater than expected from contributions of C-1 and C-6 of hexose phosphates to these carbons of oct 8-P formed by the reaction of Eq. (3). The level of 14C in position 8 of some ido-octulose 1,8-P2 samples was significant and may be the result of a second cycle of 14C labelling or action by a phenomenon that was first noted by Van Sumere and Shu (1957). Their results showed an inversion of 14C-labelling of [1-14C] Rib and production of [5-14C]-Xlu via the intermediacy of 14C-d-arabinitol formed by d-arabinitol: NAD+ 4-oxidoreductase (EC 1.1.1.11). Rephosphorylation and isomerization to form the isotopically inverted aldopentose 5-P and its Aldx catalysed exchange with d-g-d-i-oct 1,8-P2 will specifically label position 8. Contamination, isolation, recovery and analytical errors may be ruled out since authentically labelled d-g-d-i-oct 1,8-P2 samples subject to the above procedures showed that the error in 14C analysis of C-8 was < 8%. However, it is important to note that neither the results of the Calvin group, the reactions of the Calvin pathway, the scheme of Fig. 2, nor the results of Table 1 show pentose phosphates labelled at positions other than carbons 1, 2 and 3. The scrambling of 14C into carbons 2 and 3 of 3-PGA (see Table 2 for 60-s chloroplast data) by recycling in the path of carbon in PS is predicted to lead to some enrichment of 14C isotope into positions 5 and 6 of d-g-d-i-oct 1,8-P2 (via the prior labelling of Fru 6-P from PGA). However, only heavily labelled [1-14C] PGA was found after 10 s PS in spinach leaves (Table 1). After 120 s of 14CO2 exposure there was an almost uniform 14C distribution in all carbons of Glc 6-P consistent with extensive recycling of the isotope in the carbon path of PS.
The results of the partial degradations of d-g-d-i-oct 1,8-P2 samples do not support exclusive paths of synthesis by the reactions of Eqs. (4) and (5), since both of these reactions will very heavily label carbon 3 of octulose phosphate. Because [1-14C]-PGA is the earliest and most heavily labelled intermediate detected during 14C fixation in C-3 plant PS and is the precursor of [1-14C]-DHAP, then it follows that the aldolase reaction of Eq. (4) forms a predominantly labelled [3-14C]-d-g-d-i-oct 1,8-P2. The TA reaction of Eq. (5) uses Fru-6-P which will be most heavily, but unequally, labelled (Gibbs Effect) in positions 3 and 4 in short time-course experiments (Tables 1 and 2). Thus the transfer of the [3-14C]-DHA-TA moiety from [3,4-14C]-Fru 6-P to Ara 5-P in the reaction of Eq. (5) forms d-g-d-i-oct 8-P which would have C-3 notably labelled. These predictions cannot be made in longer term experiments (much beyond 30 s in leaves) because of the PS-induced spread of 14C label in PGA and hexose 6-P (Tables 1 and 2). The most supportive evidence for the TK reaction in the synthesis of d-g-d-i-oct phosphate is best shown in the 10 s fixation in spinach leaves and 60 s in spinach chloroplasts. It is of note that the inability of the degradative procedures (see Methods section) to provide quantitative individual 14C distributions for all eight carbons of the octulose phosphates was addressed using the mass spectrometric method (MacLeod et al. 2001), the results of which are reported in the accompanying paper. The above quantitative 14C- labelling of positions 5 and 6 of d-g-d-i-oct 1,8-P2 was qualitatively confirmed by Bartlett et al. (1989), who used two-dimensional NMR Spectrometry and 60 s of 13CO2 fixation by PS with intact spinach leaves (Hatch and Slack 1966) to demonstrate formation of [5,6-13C]-d-g-d-i-oct 1,8-P2. Their results also included the first demonstration of Aldx catalysis by showing that the [5,6-13C]-d-g-d-i-oct 1,8-P2 and [1,2,3-13C]-Rib 5-P, both products of PS, interconverted with [4,5,6-13C]-d-g-d-a-oct 1,8-P2 and [2,3-13C]-Ara 5-P.
While the above results and the scheme of Fig. 2 support the proposal that the TK reaction leading to Oct 8-P involved Glc and Fru 6-phosphates, it should be noted that TK can accept various other substrates (Williams et al. 1987). Using the theoretical analysis method dealt with earlier in this section, it can be shown that Seh 7-P serving as a two-carbon fragment donor will give the same labelling result as that imposed by Fru 6-P. On the other hand, reaction between Glc 6-P and Xlu 5-P results in a different pattern, with C-1 and C-2 of the resulting d-g-d-i-oct 8-P having much heavier labelling than C-3 and thus this reaction possibility is excluded. Finally, another potential TK donor substrate is β-hydroxypyruvate. Daley and Bidwell (1977) showed that in its phosphorylated form, 3-phosphohydroxypyruvate together with phosphoserine, accounted for a substantial portion (35%) of the 14C fixed by Phaseolus vulgaris leaves during the first minute of exposure to 14CO2 in PS. If P-hydroxypyruvate is synthesized by a carboxylation process, as the above authors imply, and is converted to hydroxypyruvate, then reaction of the latter with Glc 6-P would produce d-g-d-i-oct 8-P labelled in the same fashion as that derived from Glc 6-P and Fru 6-P, since the distribution of 14C in hydroxypyruvate resembles that in carbons 1–3 in hexose monophosphate. However, lack of data prevents our further speculation on the possible role of hydroxypyruvate as a path-of-carbon intermediate in the synthesis of octulose phosphates in spinach during PS.
This section on octulose phosphate synthesis and reactivity in PS would be incomplete without mention of the 5′-epimerization of octulose phosphates (Williams et al. 1978a). In such a mechanism d-g-d-a-oct 8-P or the corresponding bisphosphate may be reversibly generated from the ido-epimeric phosphate esters. The manner of this epimerization and its significance in the proposed modification of the path of carbon in PS is treated in the last section of this Discussion.
Stromal enzyme preparation and photosynthesis
In 1981, Furbank and Lilley showed that reaction sequences of a complete photosynthetic carbon reduction pathway could be demonstrated using a SEP (see Methods section) from the chloroplasts of peas. By supplying SEP with ATP, NAD(P)H, Mg2+ and dithiothreitol (DTT) and operating at pH 8.0, the need for light was eliminated. When SEP was provided with a small priming level of 3-PGA, there was an immediate and rapid oxidation of NAD(P)H which was spectrophotometrically monitored at 340 nm. The amount of NAD(P)H oxidized was equated with the number of moles of 3-PGA added. During the period of rapid NAD(P)H oxidation it was shown, in parallel experiments using 14CO2, that no carbon fixation took place. However, after this initial rapid period there followed an interval of further NAD(P)H oxidation at a slower rate, which was linear for at least 15 min and which was accompanied by CO2 fixation. It was assumed that CO2 was fixed by Ru 1,5-P2 production for the ribulosebisphosphate carboxylase (EC 4.1.1.39) reaction which was supported by the test sugar-P added to the SEP reaction mixture (Table 3). Furbank and Lilley hypothesized that, in spite of a 500-fold dilution of the intact enzyme complement of chloroplasts and the loss of any attendant structural regulation, the pathway catalysed by SEP is mechanistically similar to the overall carbon fixation rate that occurs in intact organelles, although we suggest that it may not be quantitatively equivalent for all intermediates. Thus it was of interest to prepare SEP from spinach chloroplasts and investigate its comparative ability to react octulose and arabinose phosphates, and adopt its use for measurement of catalytic activity of the phosphotransferase enzyme, and the rates of the oxidative and reductive PPs (see Methods).
Table 3.
Rates of reductive PP sequences, measured by NADH oxidation, in reactions catalysed by spinach SEP with a variety of sugar phosphates as substrates
| Substrate | Concentration (mM) | Rate of NADH oxidation (μmol h−1 mg−1 Chl) | |
|---|---|---|---|
| Spinacha | Peab | ||
| Ru 1,5-P2 | 2.0 | 592 ± 79 (9) | 546 |
| Rib 5-P | 2.0 | 743 ± 92 (10) | 453 |
| Rib 5-P | 0.2 | 366 ± 39 (10) | – |
| Seh 7-P | 2.0 | 183 ± 32 (12) | 184 |
| Seh 1,7-P2 | 2.0 | 76 ± 14 (10) | 112 |
| Fru 1,6-P2 | 2.0 | 10.1 ± 2.7 (10) | 74.1 |
| Fru 1,6-P2 | 1.0 | 18.2 ± 2.9 (11) | – |
| Fru 6-P | 2.0 | 1.0c ± 0.3 (13) | 77.6 |
| Fru 6-P + DHAP | 2.0/0.20 | 1.8 ± 0.4 (10) | 142 |
| Fru 6-P + PGA | 2.0/0.20 | 2.9 ± 0.4 (8) | – |
| DHAP | 2.0 | 13.1 ± 3.1 (10) | 53 |
| DHAP | 0.20 | 5.0 ± 1.4 (11) | 22.6 |
| Ara 5-P | 2.0 | 1.1 (9)d | – |
| d-g-d-i-oct 1,8-P2 | 2.0 | 3.7 ± 1.7 (5) | – |
| d-g-d-a-oct 1,8-P2 | 2.0 | 9.3 ± 1.5 (6) | – |
| d-g-d-i-oct 8-P | 2.0 | 6.3 ± 2.4 (5) | – |
| d-g-d-a-oct 8-P | 2.0 | 8.2 ± 3.0 (5) | – |
a Shows the results of this study. Results are mean values ± standard deviation. The number of determinations using different batches of SEP are shown in brackets
b Shows the data of Furbank and Lilley (1981) for peas
c When preparations were made using sonicated spinach chloroplast suspensions and Fru 6-P as substrate, varying (non-reproducible) rates up to 5 μmol h−1 mg−1 Chl were recorded
d Results were variable with different SEP batches, three of the SEP preparations with Ara 5-P did not support any NADH oxidation
Determination of CO2 fixation using SEP and activity of the reductive PP with various sugar phosphate substrates
The relative capacity of SEP to fix CO2 (see Methods) depended primarily on the nature of the sugar-P primer in the incubation. In Table 3, the rates of spinach SEP-catalysed NADH oxidation in the presence of various sugar phosphates are compared with the data reported by Furbank and Lilley (1981) using SEP from peas.
In general, metabolites which immediately precede the carboxylation reaction in the scheme of the reductive PP, namely Rib 5-P, Ru 5-P, Ru 1,5-P2, Seh 7-P, Seh 1,7-P2, gave very high rates of CO2 fixation as indicated by the oxidation rate of NADH, consistent with their role in autocatalysis (Walker and Lilley 1974). However, triose, arabinose, fructose, ido- and altro-octulose phosphates exhibited low rates that were notably below the maximum rate of CO2 fixation in intact chloroplasts. Triose-P and hexose-P rates using pea SEP were also below the physiological PS rate. In particular Fru 6-P and Glc 6-P (results not shown) were very inefficient substrates for spinach SEP giving much lower values than those reported by Furbank and Lilley (Table 3) for pea preparations, results which we were able to confirm.
Lilley and Walker (1979) also reported that substrate concentrations of Fru 6-P and a reconstituted chloroplast system yielded very low rates for the enablement or support of Calvin Cycle regenerative activity by this substrate.
A number of modifications of the original procedure were introduced in an attempt to obtain higher rates of CO2 fixation with several sugar phosphates, Fru 6-P in particular. These modifications included variations in the composition of the low osmolality buffer used for the disruption of the organelles, freeze–thawing and sonication of chloroplasts, the inclusion of various components such as ThPP or glycerol in the dialysis medium and the omission of selected steps from the procedure. Notably better reproducibility, in rates of NADH oxidation, were obtained using 10 mM HEPES buffer (pH 8.0) with 3 mM DTT. Consequently, this method of SEP preparation was used for all reported results. The low rates obtained with hexose and the octulose phosphates were initially thought to be due to depressed activity of a key group-transferring enzyme, particularly transketolase. This was shown to be unlikely since the maximum catalytic activity of TK, as well as that of several other enzymes of the reductive PP in SEP, was sufficient to support rates of CO2 fixation in excess of 100 μmol h−1 mg−1 Chl (Table 4). The enzyme activities were measured between 4 and 8 h after chloroplasts were disrupted and are representative of their activities in SEP during progressive or serial assays of NADH oxidation.
Table 4.
Maximum catalytic activity of selected enzymes of the reductive PP in Spinach SEP
| Enzyme | Catalytic activity (μmol h−1 mg−1 Chl) | |
|---|---|---|
| This study | Data of Latzko and Gibbs (1969) for Spinach leaf extract | |
| Phosphoglycerate kinasea | 1990 | 2423 |
| Glyceraldehyde 3-phosphate dehydrogenase (NADP+)b | 207 | 269 |
| Transketolasec | 199i; 103j | 194 |
| Fructose-bisphosphate aldolased | 113 | 102 |
| Fructose-bisphosphatasee | 53 | 46 |
| Sedoheptulose-bisphosphatasef | 35k | 3.8 |
| Phosphotransferaseg | 2.7l; 13.6m | – |
| Transaldolaseh | 2.4 | 7.7 |
| Phosphotransferaseg in Pea leaf extract | 25.3n | |
| Phosphotransferaseg in Chlorella fusca extract | 20.9n | |
| Phosphotransferaseg in Spinach leaf extract | NDn; 19.3m | |
Enzymes were assayed using the methods of: a, b Latzko and Gibbs (1969); c Brin (1974); d Bergmeyer and Bernt (1974); e Latzko and Gibbs (1974); f Woodrow and Walker (1982); g Arora et al. (1985); h Brand (1974).
i Assayed using Ery 4-P and Xlu 5-P as substrates
j Assayed using Rib 5-P and Xlu 5-P as substrate
k Assayed using the Pi liberation method
l Assayed using [8-14C]-d-g-d-i-oct 8-P and Seh 1,7-P2 as substrates
m Assayed using [8-14C]-d-g-d-a-oct 8-P and Seh 1,7-P2 as substrates
n Assayed using d-g-d-i-oct 1,8-P2 and Seh 7-P as substrates
ND: not detected
Both the mono- and bisphosphates of d-g-d-a-oct were marginally more active substrates for the support of CO2 fixation (Table 3) than the ido-oct phosphate esters. d-g-d-i-oct 1,8-P2 was least effective and has not been assigned a substrate role in a new reaction sequence for PS presented as Scheme 1 and is a conclusion of this paper. This is a curious finality to this study of the octuloses in PS since d-g-d-i-oct 1,8-P2, because of its identification in the L-type PP, was the initial substrate of choice to test the hypothesis of this study (see Introduction). Although d-g-d-i-oct 1,8-P2 has a degree of 14C isotopic equilibrium with all the octulose phosphates listed in Table 3, we find no other evidence or reason to now support its inclusion in the new PS scheme. d-g-d-a-oct 1,8-P2 is the most reactive octulose substrate in both reductive CO2 fixation and in its formation from the monophosphate by PT catalysis in the reductive direction of PS (see later discussion on PT). However, d-g-d-a-oct 1,8-P2 forms Rib 5-P if it is cleaved by the ALD activity of SEP, so the higher CO2 fixation by d-g-d-a-oct 1,8-P2 (Table 3) may in part be attributed to some Rib 5-P release. d-g-d-i-oct 8-P is also more effective in the support of CO2 fixation (Table 3) than its bisphosphate and it is also independent, in the new PS scheme, of any need to transact the PT step at reaction XII of the new scheme (see later section). It is also noted that the Furbank and Lilley (1981) assay for CO2 fixation identified slow reacting intermediates in the reduction assay (Table 3). These intermediates may accumulate and thus be confined to a linear, non-autocatalytic (Walker and Lilley 1974) expression of PS in vitro with resultant slow synthesis of Ru 1,5-P2.
Determination of the activity of the non-oxidative segment of the PP
The capacity to convert Rib 5-P to hexose 6-P is the measure of the activity of the enzymes comprising the non-oxidative segment of the PP (Williams et al. 1978b, 1987; Fig. 1). When the products of this conversion are removed by NADP+ reduction, using an excess of PGI and Glc 6-PDH, the reduction rate of the pyridine nucleotide is the measure of the maximum catalytic capacity of the preparation (see Methods). The average max rate of Rib 5-P to hexose 6-P conversion by SEP was 2.29 μmol h−1 mg−1 Chl (S.D. ± 0.41, n = 17). The ability of SEP from spinach chloroplasts (Furbank and Lilley 1981) to catalyse the above process (half-life 24 h) was greater than its stability in measurements of the reductive PP (half-life 6 h). When SEP was prepared by the method of Kaiser and Bassham (1979), it catalysed the non-oxidative segment reactions of the PP at the rate of 3.22 μmol h−1 mg−1 Chl and this was enhanced, at all test times, by 30% over a 48-h stability time course by the inclusion of ThPP (0.1 mM). This suggests that TK may have been susceptible to inactivation by the assay system (Murphy and Walker 1982). However, comparison of these data with the above results for the reductive PP show that the maximum catalytic capacity of the non-oxidative segment of PP in SEP, using Rib 5-P as substrate, is only 0.6% of Rib 5-P carboxylating activity in the reductive PP (Table 3). The above finding thus discounts the possibility that the complete network of the PP (Fig. 1) in SEP can make any significant overlapping contribution to the flux of the reductive PP of PS.
Phosphotransferase activity
The proposed alternative mechanism for the regenerative phase of the path of carbon in PS (Fig. 2) presents a number of features in common with the L-type PP in animal tissues (Fig. 1) (Williams et al. 1978a, 1987; Williams 1980). In particular, both pathways require the participation of a specific phosphotransferase, PT (see Methods). The presence and an excess of PT activity to support the maximum flux of Rib 5-P conversion to hexose and triose-P in vitro was reported for a number of animal and plant tissues (Arora et al. 1985). PT catalysed the reaction of Eq. (6), using the same rat liver enzyme catalyst that generated results used for the initial hypothesis of the classical (F-type) PP (Horecker et al. 1954; Horecker 2002; Williams et al. 1987). PT was also detected in French Press extracts of green algae (Chlorella fusca) and enzyme extracts of pea leaf (Arora et al. 1985) (see Table 4). The PT enzyme has not previously been investigated for any putative role in the metabolism of plants.
The detection of 13C and 14C-labelled d-g-d-i-oct 1,8-P2 in spinach leaves (Bartlett et al. 1989) and isolated chloroplasts following short periods of 14CO2 assimilation (this paper), raised the question of the origin of this bisphosphate compound. The reaction of Eq. (3) shows the formation of d-g-d-i-oct 8-P and the data reported here examine the activity and mechanism of phosphate donor and transfer for the formation of d-g-d-i-oct 1,8-P2. In particular the phosphotransferase proposed by Williams and Clark (1971), Clark et al. (1974) and Arora et al. (1985) is featured. The L-type PP (Fig. 1) and the hypothesized path of carbon in PS (Fig. 2) proposed a phosphotransferase which catalysed a reversible phosphate transfer between octulose and sedoheptulose phosphates (Eq. (6)). The investigations reported on this reaction were made using spinach leaf extracts and SEP preparations from spinach chloroplasts. PT activity was not detected using either spinach leaf extract or SEP to catalyse the reaction of Eq. (6) when the measurements were performed in the oxidative direction of d-g-d-i-oct 8-P formation (Fig. 2). A study of SEP-catalysed phosphate transfer from Seh 1,7-P2 to [8-14C]-d-g-d-i-oct 8-P (Eq. (1) in the reductive direction of the PS pathway), using the radiochromatographic stop assay procedure (see Methods), showed PT activity was 2.7 μmol h−1 mg−1 Chl (S.D. ± 0.80; n = 7) (Table 4). This value exceeded the maximum rate of SEP-catalysed conversion of Rib 5-P to hexose phosphates but was only 16% of the chloroplast CO2 fixation rate required by the PT reaction step (See New Scheme) and 15% of the degree of carboxylation required for the ab initio synthesis of octulose (120/8) by PS. 14C-labelled d-g-d-a-oct 8-P was a more effective substrate in this PT assay with activity of 13.6 μmol h−1 mg−1 Chl (P.E. ± 3.0; n = 4; see Table 4), which is 80% of the required chloroplast CO2 fixation rate at the PT step of the revised RPP scheme and 91% of the activity necessary for octulose formation. Significant PT activity was found in spinach leaf extract (19.3 μmol h−1 mg−1 Chl, see Table 4) using the above radiochromatographic method and the [8-14C]-d-g-d-a-oct 8-P substrate (Eq. (2)). No activity was detected in the leaf extract using d-g-d-i-oct 1,8-P2 and oxidative direction assays (see Table 4). However, it is not concluded that positive PT measurement in the reductive PS mode is evidence for an exclusive unidirectional flux mechanism. Rather the failure in the application of the oxidative-mode methods (Arora et al. 1985) is attributed inter alia to subversively high levels of Mg+ and DTT in SEP.
A test of the absolute specificity of the PT reaction was made using [1-32P]-Seh 1,7-P2 substrate and d-g-d-i-oct 8-P. The reaction was carried out using SEP and exactly the same conditions as the above radiochromatographic stop assay. All 32P-labelled products of the reaction were resolved using formate ion-exchange chromatography (see Methods) and their radioactivity measured. 32P-d-g-d-i-oct 1,8-P2, which coincided with the elution peak of the authentic compound, was recovered and its identity further confirmed by rechromatography with DEAE-Sephadex A25 (borate form) (Kapuscinski et al. 1985). Aldolase cleavage of the purified Oct 1,8-P2 and isolation of 32P-DHAP using short column (0.9 × 15 cm2) formate ion-exchange chromatography, showed that >90% of the incorporated 32P was in this compound. The results of this experiment indicated that qualitatively SEP has enzymatic activity capable of specifically phosphorylating d-g-d-i-oct 8-P at position 1 using Seh 1,7-P2 as the phosphate donor. There was also an extensive 12% conversion of the [1-32P]-Seh 1,7-P2 to 32Pi and 30% formation of [32P]-Fru 1,6-P2. Only 25% of the [1-32P]-Seh 1,7-P2 substrate remained after 30 min of reaction time. When 10 mM Mg2+ and 10 mM DTT were included in the above reaction mixture, 93% of the 32P label of [1-32P]-Seh 1,7-P2 was converted to 32Pi and only 0.08% to the isotopically labelled d-g-d-i-oct 1,8-P2. It is suggested that high concentrations of Mg2+ in SEP may have activated chloroplastic bisphosphatases and account in part or completely for an inability to demonstrate significantly higher activities of PT. Inclusion of DTT and the endogenous levels of aldolase in SEP and spinach leaf extracts possibly acted to further diminish the full expression of the activity of the PT enzyme. Hence the minimum qualification for the measurements of the crude activity of PT.
During the conduct of the above investigations and measurements of PT activity, the various SEP preparations were also used to test the possibility that other intermediary compounds and enzymes may act to phosphorylate d-g-d-i-oct 8-P. Among the test substrates were [γ-32P]-ATP, 1,3-diphosphoglycerate, inorganic pyrophosphate, Fru 1,6-P2 and Ru 1,5-P2. The following purified enzymes and enzyme preparations were also investigated; PFK from rabbit muscle, phosphoribulokinase (EC 2.7.1.19) from spinach, the endogenous kinases of SEP using [γ-32P]-ATP as substrate and sedoheptulose 1,7-bisphosphatase. In summary, none of these enzymes or substrates was effective except for muscle PFK which catalysed the formation of [1-32P]-d-g-d-i-oct 1,8-P2 from the reaction of [γ-32P]-ATP and d-g-d-i-oct 8-P. Notwithstanding this, when SEP was used to catalyse the same reaction only a miniscule rate of labelled d-g-d-i-oct 1,8-P2 formation was measured (0.3 μmol h−1 mg−1 Chl). PFK accepts Fru 6-P and Seh 7-P as substrates (Karadsheh et al. 1973) and on the basis of the above finding d-g-d-i-oct 8-P may be added to the list. However, the maximum catalytic activity of PFK in spinach chloroplasts is only 2.5 μmol h−1 mg−1 Chl (Kelly and Latzko 1977) and it is therefore unlikely to be a significant contributor to d-g-d-i-oct 1,8-P2 formation in RPP reactions in PS. The results of this section (Table 4) showed that minimum estimates of PT were compromised when SEP is the source of activity. The results also showed that d-g-d-a-oct phosphate was the most active substrate and that PT may be the rate-limiting enzyme for octulose involvement in path of carbon reactions. This limitation in minimum estimates of PT activity in SEP (Table 3) may account for altro-octuloses being restricted to the support of only 11% of the rate of CO2 fixation by intact spinach chloroplasts and/or 80% of the maximum CO2 rate required at the PT reaction step (See later discussion of the new reactions in Scheme 1).
Scheme 1.

Arabinose 5-phosphate reactions with spinach stromal enzyme preparation
In the L-type PP (Fig. 1), the prior formation of Ara 5-P, and its use by aldolase is the key step for the formation of d-g-d-i-oct 1,8-P2. In contrast, the hypothetical mode of d-g-d-i-oct 8-P synthesis and reaction in the PS scheme of Fig. 2 results in the later formation of Ara 5-P and its contribution to Rib 5-P (via Ru 5-P). Thus it was anticipated that Ara 5-P by its initial assignment of an intermediary role in the flux of the pathway of Fig. 2, should support CO2 fixation in the SEP assay system. The result (Table 3) of the test of this proposition was negative. Arabinose 5-P, as a single test substrate, had negligible ability to support CO2 fixation in SEP-catalysed reductive PP reactions in PS. A second question raised by the hypothetical scheme of Fig. 2 is whether SEP is able to catalyse Ara 5-P dissimilation and interconvert it into other intermediates of either the Calvin Pathway or octulose phosphates. SEP catalysed the incorporation of radioactivity from [U-14C]-Ara 5-P into the carbons of the following sugar phosphate intermediates of the reductive PP; triose-P, ketopentose 5-phosphates, hexose and heptulose mono- and bisphosphates (Fig. 7).
Fig. 7.
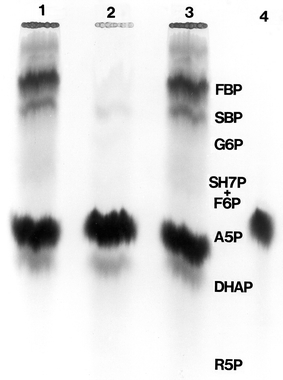
Radioautogram showing the spinach SEP-catalysed conversion of [ U-14C]-Ara 5-P, in the presence of a nine-fold excess of unlabelled Rib 5-P, to other radioactively labelled sugar phosphates. [ U-14C]-Ara 5-P and Rib 5-P were incubated for 1 h with spinach SEP as detailed in Materials and methods. The products of the reaction were separated by descending paper chromatography using the GW3-PBA solvent. Lanes 1 and 3 of the chromatogram show the mixture of 14C-labelled sugar phosphates formed from Ara 5-P dissimilation. Lane 2 shows the radioautograph of products in the absence of Rib 5-P. Lane 4 shows an absence of product formation when boiled SEP was used. The identity, by visualization, of the 14C-labelled sugar phosphates at Lanes 1 and 3 was established by comparison with the positions (not shown in Fig. 7) of the following sugar phosphate marker standards that accompanied the four lanes of the chromatogram, Fru 1, 6-P2 (FBP), Seh 1,7-P2, Seh-7-P, Glc 6-P, Fru 6-P, Ara 5-P, DHAP, Ru 5-P, Xlu 5-P. The one-dimensional chromatography system used does not permit the identification of octulose phosphates (see Kapuscinski et al. 1985 for a list of Rp-i values for some of the above sugar phosphates resolved by the GW3-PBA solvent). The visualization and tentative identification of the labelled products was distinct except for the band running just ahead of Ara 5-P which may have involved ketopentose 5-phosphates and DHAP
However, 14C-incorporation into these products only occurred in the presence of an excess (nine-fold) of unlabelled Rib 5-P. The process only faintly labelled Rib 5-P. When 2 mM [U-14C]-Ara 5-P was the sole substrate, its conversion to other identified 14C-labelled sugar phosphates was only 2% and there was clear incorporation into an unresolved band of phosphorylated compounds with an Rf value coincident with the ketopentose phosphates and also close to DHAP (Fig. 7). It was imperative to ensure that 14C-incorporation into the sugar phosphates was not the result of non-enzymatic conversions occurring during sample processing. This possibility was eliminated on the basis of several control experiments which included different ways of terminating the reaction (boiling and perchloric acid treatment) and incubations with SEP boiled before the addition of [U-14C]-Ara 5-P.
When the incubations were carried out with SEP at pH 8.0 in the presence of 10 mM MgCl2 and 10 mM DTT (conditions favouring activation of some key regulatory enzymes of the reductive PP), there was no incorporation of 14C isotope from [U-14C]-Ara 5-P (and excess Rib 5-P) into any other metabolites. Since the experiments used SEP that had provided approx 10% 14C incorporation from [U-14C]-Ara 5-P (in the presence of a nine-fold excess of Rib 5-P) into hexose 6-P and by visual judgement of the chromatogram, incorporations into Triose-P, Ru 5-P, Fru- and Seh-bisphosphates, it was concluded that Mg2+ or the reductant may be inhibitory. These latter observations were not pursued further. The above is the first report of Ara 5-P reaction in a chloroplast preparation.
Arabinose 5-P is a competitive inhibitor of TA-catalysed reactions, with a Ki of 70 μM (Williams et al. 1978a). d-manno-Heptulose 7-P, a product of TK catalysis using a ketosugar-P donor and Ara 5-P, is a competitive inhibitor of TK-catalysed reactions involving Ery 4-P (Arora 1984). Moreover, in all animal and neoplastic tissues investigated (Williams et al. 1987), Ara 5-P acted as a powerful competitive inhibitor (Ki: 40 μM, Arora 1984) when the tissue enzyme preparations converted Rib 5-P to hexose and triose phosphates using standard assay conditions for the non-oxidative PP (Methods section). Thus it was reasoned that enzyme inhibition may be responsible for the failure to find uncontentious evidence that Ara 5-P is a utilized substrate in reactions catalysed by spinach SEP. This proposition was tested with SEP using [Ara 5-P]:[Rib 5-P] ratios ranging from 0.25 to 1.0. Some inhibition was found but it was notably less than that uniformly encountered with animal and neoplastic tissue enzyme preparations. Even a 1:1 mixture of Ara 5-P and Rib 5-P with SEP exhibited 75% of the control rate measured with Rib 5-P alone. This small degree of inhibition is therefore not an explanation for the inability of SEP to catalyse Ara 5-P as a substrate in carboxylation reactions. In experiments that involved incubation of SEP with unlabelled Ara 5-P and NaH14CO3, slow 14C fixation occurred (1 μmol h−1 mg−1 Chl.).
Replacement of Ara 5-P with Rib 5-P resulted in a rapid rate of formation of acid stable 14C-labelled product consistent with the peak activity of Rib 5-P in reductive PP reactions, as shown by the data of Table 3. Moreover, using the polarographic method for measuring CO2-dependent O2 evolution (Methods section) and intact spinach chloroplasts, it was found that the inclusion of 0.2 mM Rib 5-P stimulated the rate of O2 evolution by 27% and the same concentration of Ara 5-P stimulated the rate by 7.5%. Schafer et al. (1977) found that free Ara and Rib were transported into intact chloroplasts at equal rates and the above data suggest that spinach chloroplasts may be differentially permeable to the phosphates, with Rib 5-P being 3.6 times more accessible and effective than Ara 5-P. There is also controversy about the substrate role of d-Ara 5-P in higher organisms (a brief statement of its intermediary metabolic history is given in Williams et al. 1987). Interconversion of Ara 5-P with other pentose phosphates (via isomerization to Ru 5-P) by microorganisms has been known for a considerable time (Volk 1960) but the direct evidence for such a process in rat liver was first demonstrated by Bleakley et al. (1984). The enzyme equilibrating Ara 5-P with other pentose phosphates was originally proposed to be of the epimerase type (Williams and Clark 1971), however, work with pig liver suggested that utilization and conversion of Ara 5-P to other metabolites may proceed via Ru 5-P (Williams unpublished results). The essence of the controversy concerning the metabolism of Ara 5-P is not so much its formation and presence in plant and other tissues but its comparatively low rate of substrate utilization by tissue enzyme preparations in vitro, usually only at 2–10% of the rate of Rib 5-P dissimilation. This is the case reported here where SEP catalysed utilization of Ara 5-P was extremely low. This controversy was settled when Bartlett et al. (1989) and Flanagan et al. (1993) demonstrated the role that catalytic quantities of Ara 5-P and Rib 5-P were able to play in TA and ALD exchange reactions. These exchange reactions affected 5′-epimerase interconversions of d-g-d-i-oct 8-P and d-g-d-a-oct 8-P and their bisphosphates. It was shown that Ara 5-P was reversibly exchanged into positions 4–8 of the octulose phosphates with exchange rates that were greater than the mass transfer flux rates catalysed by the maximum catalytic capacities of each of the group transferring enzymes TA and ALD acting in the reactions of the following equations.
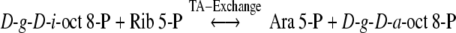 |
7 |
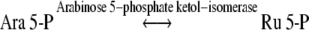 |
8 |
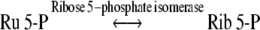 |
9 |
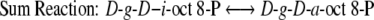 |
10 |
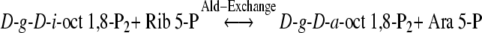 |
11 |
The roles of Eqs. (7) and (8) are shown at reactions IX and X in the following new reaction sequence (Scheme 1) for the path of carbon in PS. Taken together these reactions catalyse the epimerization of the ido- and altro-octulose monophosphates. Equation (11) shows the interconversion of ido- and altro-octulose bisphosphates by the aldolase exchange reaction (Bartlett et al. 1989; Flanagan et al. 1993). It is proposed that the reaction of 11 assists in the prevention of an accumulation of the much slower reacting d-g-d-i-oct 1,8-P2 by rapidly channelling its carbon into the more reactive 5′-epimeric form, d-g-d-a-oct 1,8-P2 (see XII and XIII in Scheme I involving octulose phosphates in PS). The experimental results of this paper and the reactions of IX, XII and XIII in the reaction scheme suggest that d-g-d-a-oct mono- and bisphosphates become the reactive forms of ido-octulose phosphates by the reactions of (7) and (11) using Rib 5-P and Ara 5-P as cycling catalytic co-factors in reactions catalysed by TAx and Aldx. We now discount any role for d-glycero-d-ido-octulose 1,8-P2 as was initially proposed for the Fig. 2 hypothesis. The revised scheme only differs significantly from Fig. 2 at reaction IX, where 5′-epimerization of d-g-d-i-oct 8-P to d-g-d-a-oct 8-P occurs. From that point ido-octulose phosphates are omitted from any further involvement in the completion of the cycle. Using PT activity data with d-g-d-a-oct 8-P as substrate (Table 4), it is calculated that the revised sequence may make a contribution that ranges from 11% (13.6/120), based on the rate of CO2 fixation (120 μmol h−1 mg−1 Chl) in intact chloroplasts, to a maximum of 80% (13.6/17) based on the required CO2 fixation rate (17 μmol h−1 mg−1 Chl) at the PT step (reaction XIII) of Scheme 1 for the RPP.
Finally, it is concluded that metabolic function has directed a distinct and confining intermediary metabolism on ido- and altro-octulose phosphates. In the L-type PP (Fig. 1) selection of d-glycero-d-ido-octulose phosphates realised the formation of the metabolically active products Fru 6-P and Glc 6-P. d-glycero-d-altro-Octulose phosphates in the pathway of Fig. 1 will only lead to the formation of Fru 6-P and the dead-end product d-allose 6-P. In Scheme 1 formulated here, the formation of d-g-d-a-oct 1,8-P2 leads to the production of Rib 5-P, the most active intermediate for the support of CO2 fixation (Table 3). Formation and further utilization of d-g-d-i-oct 1,8-P2 by the reaction scheme would produce stoichiometric amounts of Ara 5-P which, as a metabolic product, has no ability to fix CO2 (Table3).
Acknowledgements
The work was supported by grants to J.F.W. from the Australian Research Grants Committee and The Faculties Research Fund of The Australian National University. Technical assistance by K.K. Arora, I.L. Flanigan, M.K. Kapuscinski and Elisabeth Owen is acknowledged.
Abbreviations
- Ald
aldolase
- Aldx
aldolase exchange
- Ara 5-P
arabinose 5-phosphate
- Ara 5-P I
arabinosephosphate isomerase
- Chl
chlorophyll
- DHAP
dihydroxyacetone phosphate
- Ery 4-P
erythrose 4-phosphate
- FBP-ase
fructose 1,6-bisphosphatase
- Fru 6-P
fructose 6-phosphate
- Gap
glyceraldehyde 3-phosphate
- Gap-DH
glyceraldehyde 3-phosphate dehydrogenase
- Glc 6-P
glucose 6-phosphate
- Glc 6-PDH
glucose 6-phosphate dehydrogenase
- Oct
octulose
- d-g-d-i-oct 1,8-P2
d-glycero-d-ido-2-octulose 1,8-bisphosphate
- d-g-d-a-oct 1,8-P2
d-glycero-d-altro-2-octulose 1,8-bisphosphate
- PBA
phenylboronic acid
- PFK
phosphofructokinase
- PGA
3-phosphoglyceric acid
- PGI
phosphoglucoseisomerase
- PP
pentose (phosphate) pathway
- PPE
phosphoketopentose epimerase
- PRI
phosphoribose isomerase
- PRK
phosphoribulokinase
- PS
photosynthesis
- PT
phosphotransferase: d-glycero-d-ido -& d-glycero-d-altro-octulose 1,8- bisphosphate :d-altro-heptulose 7-phosphotransferase
- Rib 5-P
ribose 5-phosphate
- Ru 1,5-P2
ribulose 1,5-bisphosphate
- RPP
reductive pentose pathway (PS path of CO2 assimilation in C3 plants)
- Rubisco
ribulose 1,5 bisphosphate carboxylase/oxygenase
- Seh
sedoheptulose (altro-2-ketoheptulose)
- SBP-ase
sedoheptulose 1,7- bisphosphatase
- SEP
stromal enzyme preparation
- TA
transaldolase
- Tax
transaldolase exchange
- TK
transketolase
- TKx
transketolase exchange
- TPI
triosephosphate isomerase
- ThPP
thiamine pyrophosphate
- Xlu 5-P
xylulose 5-phosphate
Appendix
The method used to calculate the predicted labelling patterns of d-g-d-i-oct phosphates formed by the reactions of Eqs. (3) and (6) is shown. The synthesis of d-g-d-i-oct 8-P is proposed to proceed via the TK-mediated reaction of Glc 6-P and Fru 6-P (Eq. (3a)). It is assumed that the two hexose 6-phosphates are in chemical and isotopic equilibrium and therefore have the same distributions of 14C isotope. As an example, the method illustrates the application of the analytical data for the 10-s 14C estimates in the carbon atoms of Glc 6-P shown in Table 1, to quantitatively predict the 14C labelling of various carbon atoms of the octulose phosphates formed by Eqs. (3) and (6).
Octulose was isolated as the bisphosphate which is assumed to have the same labelling pattern as d-g-d-i-oct 8-P (see Results and discussion and the reaction scheme). d-g-d-i-oct 1,8-P2 was degraded by Ald cleavage to DHAP and Ara 5-P followed by selective chemical oxidation of the 3- and 5-carbon fragments (see Methods section). The predicted degree of positional carbon isotopic labelling in d-g-d-i-oct 1,8-P2 is calculated as follows. Reaction of Fru 6-P and Glc 6-P (Eq. (1)) will cause positions 1, 2 and 3 of Oct 8-P to contain hexose 6-P carbons 1, 2 and 1, respectively. By summing the activities of carbons 1, 2 and 1 of Glc 6-P and expressing the sum as 100%, one can calculate the relative activity of the top three carbons of octulose (3.2 + 3.2 + 3.2 = 9.6) then (3.2/9.6)100 = 33.3% as shown in the bracketed values in the 10-s ‘Predicted’ column of Table 1. Similarly the bottom 5 carbon atoms of d-g-d-i-oct 1,8-P2 have relative radioactivities identical to carbons 2–6 of Glc 6-P (3.2, 37.2, 50.5, 2.5 and 3.4, respectively). When their sum (96.8) is expressed as 100, the relative activity for C-4 of octulose is (3.2/96.8)100 = 3.3%, for C-5 + C-6 + C-7 ((37.2 + 50.5 + 2.5)/96.8)100 = 93.2% and for C-8, (3.4/96.8)100 = 3.5%. All of the predicted values for octulose-P data in Tables 1 and 2 are calculated in the above manner.
References
- Anderson JM, Boardman NK (1966) Fractionation of the photochemical systems of photosynthesis. I. Chlorophyll contents and photochemical activities of particles isolated from spinach chloroplasts. Biochem Biophys Acta 112:403–421 [DOI] [PubMed]
- Andrews P, Hough L, Picken JM (1965) The biosynthesis of polysaccharides. Incorporation of D-[1-14 C] glucose and D-[6-14 C] glucose into plum leaf polysaccharides. Biochem J 94:75–80 [DOI] [PMC free article] [PubMed]
- Arnon DI (1949) Copper enzymes in isolated chloroplasts. Polyphenol oxidase in Beta vulgaris. Plant Physiol 24:1–15 [DOI] [PMC free article] [PubMed]
- Arora KK (1984) Studies on carbohydrate metabolism in cancer. Ph D thesis. Australian National University, Canberra, Australia
- Arora KK, Cortis P, Bleakley PA, Williams JF (1985) Identification and measurement of d-glycero-d-ido octulose 1,8-bisphosphate: d-altro-heptulose 7-phosphotransferase enzyme in tissues with L-type pentose phosphate pathway activity. Int J Biochem 17:1329–1337 [DOI] [PubMed]
- Arora KK, MacLeod JK, Williams JF (1987) High yield synthesis of 14C labelled intermediates of the L-type pentose pathway: octulose mono and bisphosphates, sedoheptulose 1,7- bisphosphate and d-arabinose 5-phosphate. J Label Compd Radiopharmaceut 24:205–218 [DOI]
- Bandurski RS, Axelrod B (1951) The chromatographic identification of some biologically important phosphate esters. J Biol Chem 193:405–410 [PubMed]
- Bartlett MRE, Collins JG, Flanigan IL, MacLeod JK, Williams JF (1989) CO2 labelling of octulose bisphosphates during photosynthesis. An NMR study using intact spinach leaves. Biochem Int 18:35–46
- Beck E, Hopf H (1982) Carbohydrate metabolism. Prog Bot 44:132–153
- Begbie R, Richtmyer NK (1966) The isolation of some heptoses, heptuloses, octuloses and nonuloses from Primula offiicinalis. J Carbohydr Res 2:272–288 [DOI]
- Benson AA, Kawauchi S, Hays P, Calvin M (1952) The path of carbon in photosynthesis XVI Kinetic relationships of the intermediates in steady state photosynthesis. J Am Chem Soc 74:4474–4482 [DOI]
- Bergmeyer HU, Bernt E (1974) Fructose-1,6-diphosphate aldolase; UV assay, Manuel method. In: Bergmeyer HU (ed) Methods of enzymatic analysis, vol 2. Academic Press, New York, pp 1100–1105
- Bleakley PA, Arora KK, Williams JF (1984) Evidence that aldolase and arabinose 5-phosphate are components of pentose pathway reactions in liver in vitro. Biochem Int 8:491–500 [PubMed]
- Brand K (1974) Transaldolase. In: Bergmeyer HU (ed) Methods of enzymatic analysis, vol 2. Academic Press, New York, pp 710–714
- Brin K (1974) Transketolase. In: Bergmeyer HU (ed) Methods of enzymatic analysis, vol 2. Academic Press, New York, pp 703–709
- Charlson AJ, Richtmyer NK (1959) Isolation of D-glycero-D-manno-octulose from the avocado. J Am Chem Soc 81:1512–1513 [DOI]
- Calvin M (1956) The photosynthetic carbon reduction cycle. J Chem Soc (London):1895–1915
- Clark MG, Williams JF, Blackmore PF (1974) Exchange reactions in metabolism. Catal Rev 9:35–77
- Cluley HJ (1962) Suspension scintillation counting of carbon-14 barium carbonate. The Analyst 81:170–177 [DOI]
- Daley LS, Bidwell RGS (1977) Phosphoserine and phosphohydroxypyruvic acid. Evidence for their role as early intermediates in photosynthesis. Plant Physiol 60:109–114 [DOI] [PMC free article] [PubMed]
- Flanagan IL, Collins JG, Arora KK, MacLeod JK, Williams JF (1993) Exchange reactions catalyzed by group-transferring enzymes oppose the quantitation and unravelling of the identity of the pentose pathway. Eur J Biochem 213:477–485 [DOI] [PubMed]
- Furbank RT, Lilley RMcC (1981) Reductive pentose phosphate cycle and oxidative carbohydrate metabolic activities in pea chloroplast stroma extracts. Plant Physiol 67:1036–1041 [DOI] [PMC free article] [PubMed]
- Genovese J, Schmidt K, Katz J (1970) Enzymic degradation of isotopically labeled compounds I. Degradation of 14C-labelled glycerol. Anal Biochem 34:161–169 [DOI] [PubMed]
- Gibbs M, Horecker BL (1954) The mechanism of pentose phosphate conversion to hexose monophosphate II. With pea leaf and pea root preparations. J Biol Chem 208:813–820 [PubMed]
- Hatch MD, Slack CR (1966) Photosynthesis by sugar cane leaves. A new carboxylation reaction and the pathway of sugar formation. Biochem J 101:103–111 [DOI] [PMC free article] [PubMed]
- Heath RL (1984) A new type of hexose monophosphate shunt in Chlorella sorokiniana. Plant Physiol 75:964–967 [DOI] [PMC free article] [PubMed]
- Heldt HW (1980) Measurement of metabolite movement across the envelope and of the pH in the stroma and the thylakoid space in intact chloroplasts. In: Pierro S (ed) Methods in enzymology, vol 69C. Academic Press, New York, pp 604–613
- Heldt HW, Chong CJ, Lorimer GH (1978) Phosphate requirement for the light activation of ribulose 1,5 bisphosphate carboxylase in intact spinach chloroplasts. FEBS Lett 92:234–240 [DOI]
- Heldt HW, Portis AR, Lilly RMcC, Mosbach A, Chon CJ (1980) Assay of nucleotides and other phosphate-containing compounds in isolated chloroplasts by ion-exchange chromatography. Anal Biochem 101:278–287 [DOI] [PubMed]
- Horecker BL (2002) The pentose phosphate pathway. J Biol Chem 277:47965–47971 [DOI] [PubMed]
- Horecker BL, Gibbs M, Klenow H, Smyrniotis PZ (1954) The mechanism of pentose phosphate conversion to hexose monophosphate I. With liver enzyme preparation. J Biol Chem 207:393–403 [PubMed]
- Howarth OW, Pozzi N, Vlahov G, Bartels D (1996) NMR structural analysis of a tri-O-isopropylidene derivative of d-glycero-d-ido-octulose, the major sugar found in resurrection plant Craterostigma plantagineum. Carbohydr Res 289:137–142 [DOI]
- Jensen RG, Bassham JA (1966) Photosynthesis by isolated chloroplasts. Proc Natl Acad Sci 56:1095–1101 [DOI] [PMC free article] [PubMed]
- Kaiser WM, Bassham JA (1979) Carbon metabolism of chloroplasts in the dark: oxidative pentose cycle versus glycolytic pathway. Planta 144:193–200 [DOI] [PubMed]
- Kandler O, Gibbs M (1956) Asymmetric distribution of C14 in the glucose phosphates formed during photosynthesis. Plant Physiol 31:411–412 [DOI] [PMC free article] [PubMed]
- Kapuscinski MK, Franke FP, Flanigan IL, MacLeod JK, Williams JF (1985) Improved methods for the enzymic preparation and chromatography of octulose phosphates. Carbohydr Res 140:69–79 [DOI]
- Karadsheh NS, Tejwani GA, Ramaiah A (1973) Sedoheptulose-7-phosphate kinase activity of phosphofructokinase from different tissues of rabbit. Biochim Biophys Acta 327:66–81 [DOI] [PubMed]
- Kelly GJ, Latzko E (1977) Chloroplast phosphofructokinase. I. Proof of phosphofructokinase activity in chloroplasts. Plant Physiol 60:290–294 [DOI] [PMC free article] [PubMed]
- Latzko E, Gibbs M (1969) Enzyme activities of the carbon reduction cycle in some photosynthetic organisms. Plant Physiol 44:295–300 [DOI] [PMC free article] [PubMed]
- Latzko E, Gibbs M (1974) Alkaline C1-fructose-1,6-diphosphatase. In: Bergmeyer HU (ed) Methods of enzymatic analysis, vol 2. Academic Press, New York, pp 881–884
- Lilley RMcC, Walker DA (1979) Studies with the reconstituted chloroplast system. In: Gibbs M, Latzko E (eds) Encyclopedia of plant physiology new series, vol 6. Springer Verlag, Berlin, pp 41–53
- Lilley RMcC, Fitzgerald MP, Rienits KG, Walker DA (1975) Criteria of intactness and the photosynthetic activity of spinach chloroplast preparations. New Phytol 75:1–10 [DOI]
- MacLeod JK, Flanigan IL, Williams JF, Collins JG (2001) Mass spectrometric studies of the path of carbon in photosynthesis: positional isotopic analysis of 13C-labelled C4 to C7 sugar phosphates. J Mass Spectrom 36:500–508 [DOI] [PubMed]
- Murphy DJ, Walker DA (1982) The properties of transketolase from photosynthetic tissue. Planta 155:316–320 [DOI] [PubMed]
- Pettersson G, Ryde-Pettersson U (1988) A mathematical model of the Calvin photosynthesis cycle. Eur J Biochem 175:661–672 [DOI] [PubMed]
- Portis AR, Chon CJ, Mosbach A, Heldt HW (1977) Fructose and sedoheptulosebisphosphatase. The sites of a possible control of CO2 fixation by light-dependent changes in stromal Mg2+ concentration. Biochim Biophys Acta 461:313–325 [DOI] [PubMed]
- Robinson SP (1983) Isolation of intact chloroplasts with high CO2 fixation capacity from sugarbeet leaves containing calcium oxalate. Photosynth Res 4:281–287 [DOI] [PubMed]
- Robinson SP, Walker DA (1981) The photosynthetic carbon reduction cycle. In: Hatch MD, Boardman NK (eds) The biochemistry of plants: a comprehensive treatise, vol. 8. Academic Press, New York, pp 194–236
- Rosenberg H (1959) The detection of phosphates on chromatograms. J Chromatogr 2:487–489 [DOI]
- Schafer G, Heber U, Heldt HW (1977) Glucose transport into spinach chloroplasts. Plant Physiol 60:286–289 [DOI] [PMC free article] [PubMed]
- Stiller M (1962) The path of carbon in photosynthesis. Annu Rev Plant Physiol 13:151–170 [DOI]
- Stitt M, Wirtz W, Heldt HW (1980) Metabolite levels during induction in the chloroplast and extrachloroplast compartments of spinach protoplasts. Biochim Biophys Acta 593:85–102 [DOI] [PubMed]
- Tashima Y, Yoshimura N (1975) Control of rabbit liver fructose-1,6-diphosphatase activity by magnesium ions. J Biochem (Tokyo) 78:1161–1169 [DOI] [PubMed]
- Van Sumere CF, Shu P (1957) Mechanism of biosynthesis of mycelial glucosan from pentoses by Aspergillus niger. Can J Biochem Physiol 35:445–448 [PubMed]
- Vernon LP (1960) Spectrophotometric determination of chlorophylls and pheophytins in plant extracts. Anal Biochem 32 :1144–1150 [DOI] [PubMed]
- Volk WA (1960) Purification and properties of phospharabinoseisomerase from Propionibacterium pentosaceum. J Biol Chem 235:1550–1553
- Walker DA, Lilley RMcC (1974) Autocatalysis in a reconstructed chloroplast system. Plant Physiol 54:950–952 [DOI] [PMC free article] [PubMed]
- Williams JF (1980) A critical examination of the evidence for the reactions of the pentose pathway in animal tissues. Trends Biochem Sci 5:315–320 [DOI]
- Williams JF (2004) Pentose phosphate pathway, History of. In: Lennarz WJ, Lane M Daniel (eds) Encyclopedia of biological chemistry, vol 3. Elsevier Inc, Oxford, pp 216–225
- Williams JF, Clark MG (1971) An error in metabolism: the pentose phosphate cycle. Search 2:80–88
- Williams JF, Rienits KG, Schofield PJ, Clark MG (1971) The pentose phosphate pathway in rabbit liver. Studies on the metabolic sequence and quantitative role of the pentose phosphate cycle by using a system in situ. Biochem J 123:923–943 [DOI] [PMC free article] [PubMed]
- Williams JF, Blackmore PF, Clark MG (1978a) New reaction sequences for the non-oxidative pentose phosphate pathway. Biochem J 176:257–282 [DOI] [PMC free article] [PubMed]
- Williams JF, Clark MG, Blackmore PF (1978b) The fate of 14C in glucose 6-phosphate synthesised from [1-14C] ribose 5-phosphate by enzymes of rat liver. Biochem J 176:241–256 [DOI] [PMC free article] [PubMed]
- Williams JF, Clark MG, Arora KK, Reichstein IC (1984) Glucose 6-phosphate formation by L-type pentose pathway reactions of rat liver in vitro: further evidence. Hoppe Seyler’s Z Physiol Chem 365:1425–1434 [DOI] [PubMed]
- Williams JF, Clark MG, Arora KK (1985) 14C labelling of octulose bisphosphates by L-type pentose pathway reactions in liver in situ and in vitro. Biochem Int 11:97–106 [PubMed]
- Williams JF, Arora KK, Longnecker JP (1987) The pentose pathway: a random harvest. Impediments which oppose acceptance of the classical (F-type) pentose cycle for liver, some neoplasms and photosynthetic tissue The case for the L-type pentose pathway. Int J Biochem 19:749–817 [DOI] [PubMed]
- Wood T (1968) The detection and identification of intermediates of the pentose phosphate cycle and related compounds. J Chromatogr 35:352–361 [DOI] [PubMed]
- Woodrow IE, Walker DA (1982) Activation of wheat chloroplast sedoheptulose bisphosphatase: a continuous spectrophotometric assay. Arch Biochem Biophys 216:416–422 [DOI] [PubMed]