

Summary
HS1, the leukocyte-specific homolog of cortactin, regulates F-actin in vitro and is phosphorylated in response to TCR ligation, but its role in lymphocyte activation has not been addressed. We demonstrate that HS1-deficient T cells fail to accumulate F-actin at the immune synapse (IS) and, upon TCR ligation, form actin-rich structures that are disordered and unstable. Early TCR activation events are intact in these cells, but Ca2+ influx and IL-2 gene transcription are defective. Importantly, HS1 tyrosine phosphorylation is required for its targeting to the IS and for its function in regulating actin dynamics and IL-2 promoter activity. Phosphorylation also links HS1 to multiple signaling proteins, including Lck, PLCγ1, and Vav1, and is essential for the stable recruitment of Vav1 to the IS. Taken together, our studies show that HS1 is indispensable for signaling events leading to actin assembly and IL-2 production during T cell activation.
Introduction
In response to interaction with APCs, T cells undergo dramatic shape changes to form a flattened contact site enriched in F-actin and signaling molecules, termed the immune synapse (IS) (Bromley et al., 2001; Kupfer and Kupfer, 2003). Actin polymerization at the IS stabilizes conjugate formation and facilitates T cell activation (Fuller et al., 2003). Several proteins have been implicated in regulating actin dynamics at the IS, including the proximal tyrosine kinases Lck and ZAP-70 and the adaptors LAT and SLP-76 (Bubeck Wardenburg et al., 1998; Bunnell et al., 2001; Morgan et al., 2001; Zeng et al., 2003). In addition, the Rho family guanine nucleotide exchange factor Vav1 regulates actin responses through the small GTPases Cdc42 and Rac1 (Turner and Billadeau, 2002; Zeng et al., 2003). Finally, we recently showed that the Tec kinase Itk and the large GTPase Dynamin 2 collaborate with Vav1 to control actin responses at the IS (Dombroski et al., 2005; Gomez et al., 2005; Labno et al., 2003). While much has been learned about upstream signaling pathways, the immediate effectors controlling actin dynamics are still poorly understood. The two Rho family proteins regulated by Vav1, Rac1 and Cdc42, both play pivotal roles. Rac1 activates the WAVE2 complex, which we recently showed is required for actin-dependent signaling at the IS (Nolz et al., 2006), while Cdc42 regulates Wiskott-Aldrich Syndrome protein (WASp) (Cannon et al., 2001). Although WASp activation is required for T cell activation (Badour et al., 2004), recent experiments indicate that WASp-deficient T cells can retain the ability to polymerize actin at the IS (Cannon and Burkhardt, 2004). Thus, other mechanisms must contribute to the regulation of actin dynamics at the IS.
Hematopoietic lineage cell-specific protein 1 (HS1, HCLS1, LckBP1) is a major substrate for tyrosine phosphorylation during T and B cell activation (Ruzzene et al., 1996; Takemoto et al., 1996; Yamanashi et al., 1993). HS1, which is expressed only in hematopoietic cells, is related to cortactin (EMS1), a widely expressed oncogene that was recently identified as an actin regulatory protein (Daly, 2004). HS1 contains an Arp2/3 complex binding domain followed by a region of 37 amino acid tandem repeats and a coil-coiled region, both of which bind F-actin (Hao et al., 2005), a proline-rich domain that binds to Lck (Takemoto et al., 1996), and a C-terminal SH3 domain. In vitro studies show that HS1 and cortactin can activate Arp2/3-dependent actin polymerization and prolong the half-life of branched actin structures (Uruno et al., 2003b; Weaver et al., 2001, 2002). HS1−/− mice display defects in antigen-induced clonal expansion and lymphocyte deletion (Taniuchi et al., 1995), but the role of HS1 in T cell signaling has not been tested. Here we demonstrate that HS1 is required for sustained actin responses at the IS and Ca2+ signaling events leading to IL-2 gene expression.
Results
HS1 Regulates the Accumulation of F-Actin at the Immune Synapse
We began our analysis by examining HS1 distribution in T cells responding to APCs. HS1 colocalized with F-actin at the IS in conjugates formed with Jurkat T cells, primary human CD4+ T cells, and murine DO11.10 TCR Tg T cells (Figure 1A). Recruitment of HS1 to the cell-cell contact site was antigen dependent, as was the accumulation of F-actin (data not shown and Figure 1E). Thus, T cell activation induces recruitment of HS1 to the IS, where it colocalizes closely with F-actin.
Figure 1.
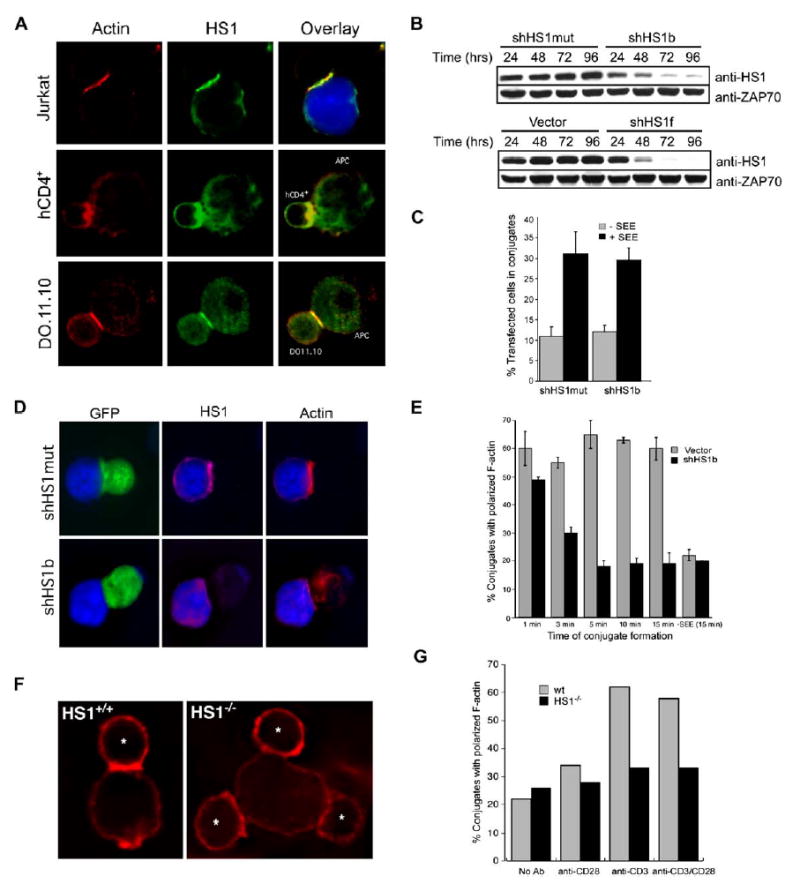
HS1 Regulates the Accumulation of F-Actin at the IS
(A) Conjugates were formed between Jurkat T cells and SEE-pulsed NALM6 B cells, between primary human CD4+ T cells and sAg-pulsed Raji B cells, or between 2° DO11.10 Tg T cells and ova-pulsed A20 B cells. Conjugates were labeled with anti-HS1 and phalloidin to label F-actin. B cells were labeled with CMAC (blue and not shown).
(B) Short hairpin RNA (shRNA) suppression time course for HS1. Jurkat cells were transiently transfected with RNA-targeting vectors (shHS1b or shHS1f), a mutated form of shHS1b (shHS1mut), or vector control. Lysates were prepared at the indicated times posttransfection and immunoblotted for HS1 and ZAP70.
(C) To assess conjugation efficiency, Jurkat cells were transfected with shHS1b or shHS1mut control vectors containing a separate GFP cassette. T cells were incubated with SEE-pulsed, PKH26-stained EBV-B cells, and the % transfected T cells in conjugates was determined by flow cytometry.
(D) To assess actin responses at the IS, Jurkat cells transfected as in (C) (GFP) were incubated with SEE-pulsed NALM6 B cells (blue). Fixed conjugates were labeled with anti-HS1 and with phalloidin to visualize F-actin.
(E) Conjugates formed as in (D) were fixed at the indicated times after initial contact and labeled with phalloidin. Conjugates containing GFP+ T cells were scored for F-actin at the IS.
(F) Wild-type and HS1−/− T cells (*) were conjugated to P815 cells bearing anti-CD3 antibodies and stained with phalloidin (red).
(G) Conjugates were formed between HS1+/+ or HS1−/− T cells and P815 cells bound with the indicated stimulating antibodies and scored for F-actin at the IS. Data are from one representative experiment. Bars in (C) and (E) represent mean ±SD from three experiments.
To test HS1 function, we suppressed HS1 expression in Jurkat T cells by shRNA-mediated gene silencing. Two different HS1 targeting vectors (shHS1b and shHS1f), but not empty vector or a scrambled version of shHS1b (shHS1mut), suppressed HS1 protein to 10% of control or less (Figure 1B). All studies were performed with both shHS1b and shHS1f, with similar results. HS1-suppressed T cells formed conjugates efficiently with SEE-pulsed B cells (Figure 1C). To assess actin responses, conjugates were fixed at 15 min and analyzed by microscopy. As expected, control transfectants showed accumulation of HS1 and F-actin at the IS (Figure 1D). HS1-suppressed cells no longer stained for HS1, verifying that HS1 was efficiently depleted. Although these cells flattened relatively normally against the APCs, F-actin labeling at the IS was absent or greatly reduced. Time-course analysis revealed that HS1-suppressed cells initially polymerized actin efficiently (Figure 1E) but failed to maintain F-actin at the IS, such that by 5 min, the response was reduced to background levels (defined by control cells without SEE).
To validate these findings, we analyzed primary lymph node T cells from HS1−/− mice responding to P815 cells decorated with anti-TCR antibodies. Wild-type T cells formed a well-defined F-actin-rich IS in this assay (Figure 1F). In contrast, although T cells from HS1−/− mice formed conjugates with P815 cells efficiently, they failed to exhibit clear F-actin polymerization at the IS. After 30 min, the frequency of HS1−/− T cells showing F-actin labeling at the IS was at baseline levels (Figure 1G). Moreover, this defect was not restored by the addition of costimulatory antibody. Thus, we conclude that HS1 is required for the stable accumulation of F-actin at the IS.
HS1-Deficient Cells Fail to Stabilize Actin after TCR Engagement
In nonhematopoietic cells, the HS1 homolog cortactin is thought to stabilize F-actin (Daly, 2004). We confirmed that neither Jurkat cells nor primary mouse T cells expressed detectable levels of cortactin mRNA (see Figure S1 in the Supplemental Data available with this article online), and we observed no compensatory up-regulation of cortactin in HS1−/− T cells or HS1-suppressed Jurkat cells. To ask whether HS1 functions by stabilizing F-actin, we analyzed the spreading of Jurkat cells stably expressing GFP-actin on anti-TCR-coated coverslips (Bunnell et al., 2001). Control cells spread in a highly ordered fashion, forming a round lamellipodial structure with uniform width (Figure 2A and Movies S1–S3). Spreading was typically maximal by 2 min, with retraction after about 5 min. DIC images (not shown) showed ruffling at the cell periphery and radial retrograde flow within the lamellar region; this process is also evident in the GFP-actin videos. In contrast, HS1-suppressed cells spread erratically (Movies S4–S7). These cells continuously sent out actin-rich protrusions, but these were asymmetric and quickly retracted, and they frequently failed to maintain contact with the coverslip (Figure 2B and Movies S4–S7). Quantitation of the coverslip contact area versus time revealed that HS1-suppressed cells spread poorly and failed to undergo the sharp increase in contact area observed in control cells during the first 90 s (Figure 2C). To measure the irregular shape of HS1-suppressed cells, the variance in radial length was calculated. Control cells became round (low variance) by 120 s, while HS1-suppressed cells were irregularly shaped at all times (Figure 2D). These data show that TCR-stimulated actin polymerization, ruffling, and lamellipodial protrusion can occur in the absence of HS1, but HS1 is required for organizing and maintaining these structures.
Figure 2.
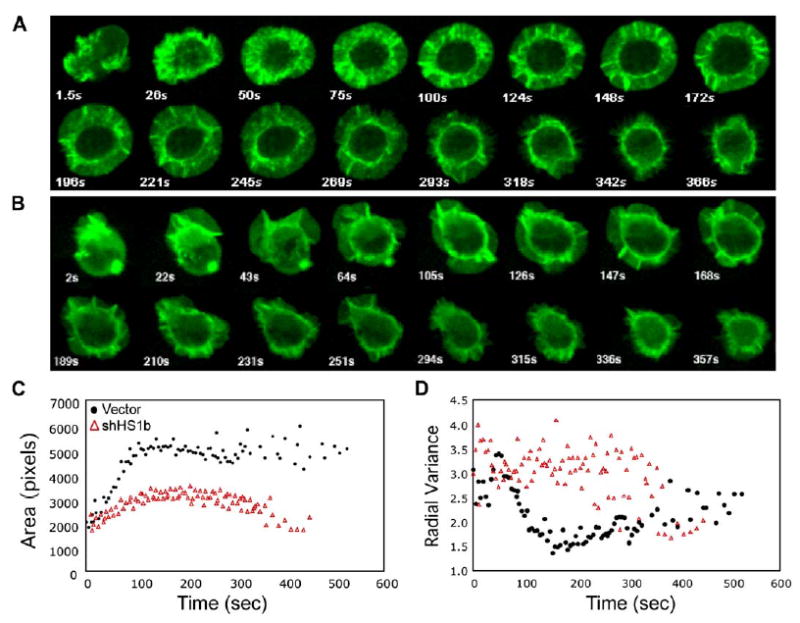
HS1-Deficient Cells Exhibit Abnormal Actin Dynamics in Response to TCR Engagement
(A and B) Jurkat cells stably expressing GFP-actin were transfected with empty vector (A) or shHS1b (B). Actin dynamics were monitored by video confocal microscopy as the cells spread on anti-TCR-coated coverslips. Selected time-lapse image projections acquired at the indicated times after contact with the coverslip are shown; see also Movies S1–S7.
(C) The contact area of each cell at each time point was determined and the average calculated for each cell population at each 5 s time point (17 cells for each condition). The difference in area was statistically significant (p = 0.0002 at 135 s).
(D) Irregularity of cell shape was assessed by measuring the radial variance of the cell outline for each cell at each time point and calculating the average values for each time point. (C) and (D), black circles, control transfectants; red triangles, HS1-suppressed cells.
Tyrosine Phosphorylation of HS1 Is Required for Actin Remodeling
As reported previously (Hutchcroft et al., 1998; Takemoto et al., 1996; Yamanashi et al., 1993), TCR and CD28 ligation induced transient tyrosine phosphorylation of HS1 (Figure 3A). Stimulation with APCs had a similar effect, although phosphorylation was more sustained (Figure 3B). Phosphorylation requires both Lck and ZAP-70, since cells lacking these kinases failed to phosphorylate HS1 (Figure 3A).
Figure 3.
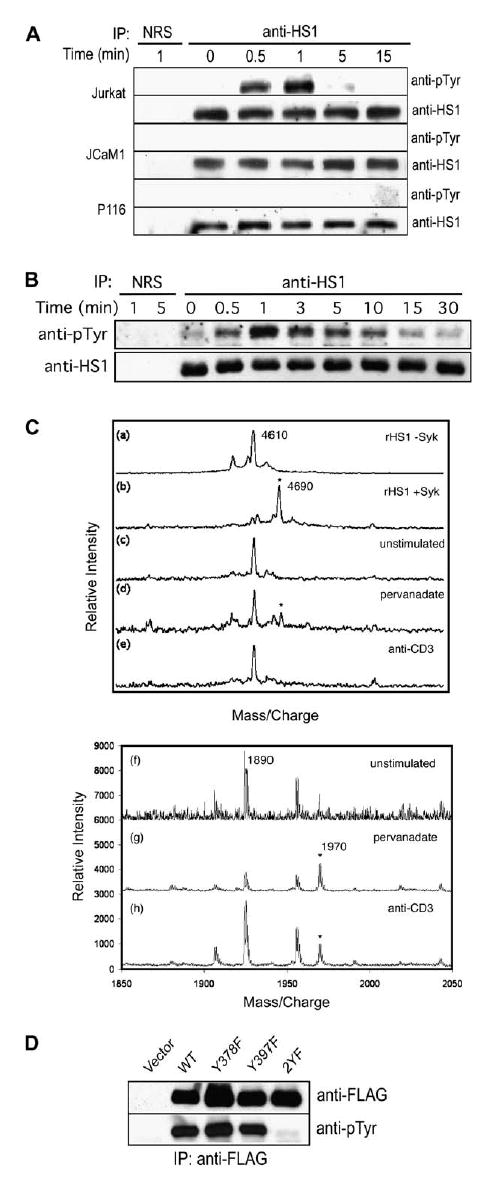
HS1 Is Phosphorylated at Tyrosines 378 and 397 in Stimulated T Lymphocytes (A) Jurkat T cells or the Jurkat-derived cell lines JCaM1 (Lck-deficient) and P116 (ZAP-70-deficient) were stimulated with anti-TCR/anti-CD28. HS1 immunoprecipitates were immunoblotted for phosphotyrosine.
(B) Jurkat T cells were unstimulated (time 0) or stimulated with fixed Raji B cells pulsed with SEE, and HS1 immunoprecipitates were immunoblotted for phosphotyrosine. In (A) and (B), rabbit IgG (NRS) was used as an immunoprecipitation control.
(C) Recombinant HS1 (rHS1, a and b) was incubated ± Syk kinase. Alternatively, endogenousHS1 was immunoprecipitated from unstimulated (c and f), pervanadate-treated (d and g), or anti-CD3-stimulated (e and h) Jurkat cells. After proteolysis, peptides were analyzed by mass spectrometry. Top, tryptic peptides containing pY378 (asterisks), detected by MALDI-TOFF. Bottom, Glu-C peptides containing pY397 (asterisks) detected by v-MALDI ion trap.
(D) Jurkat T cells were transfected with FLAG-tagged wild-type HS1 or the Y378F, Y397F, and 2YF mutants. Cells were pervanadate treated, and the FLAG-tagged proteins were immunoprecipitated and immunoblotted with anti-phosphotyrosine.
To map the sites of HS1 phosphorylation, we conducted phosphopeptide mapping by mass spectrometry. We first tested recombinant HS1, which is phosphorylated in vitro by Syk at Y378 and Y397 (Brunati et al., 1999). HS1 yields a tryptic peptide of 4610 Da, which is shifted by the 80 Da mass of a single phosphate after phosphorylation by Syk (Figure 3C). This peptide includes both Y378 and Y360, but other peptides containing Y360 showed no evidence of phosphorylation (data not shown), indicating that phosphorylation occurs at Y378. This peptide, and another that includes Y397 (not shown), were confirmed as the major peptides phosphorylated by Syk in vitro. The phosphopeptide containing Y378 was also evident in HS1 immunoprecipitated from pervanadate-treated T cells, though it could not be identified with confidence in lysates from CD3-stimulated cells. A phosphopeptide containing only Y397 was clearly visible in material isolated from both pervanadate-treated and anti-CD3-activated cells. In vitro phosphorylation of HS1 at Y222 has been documented (Brunati et al., 1999; Ruzzene et al., 1996; Takemoto et al., 1996), and there is a report of Y198 phosphorylation in T cells (Salomon et al., 2003); however, we did not detect phosphorylation at these sites. Thus, we conclude that Y378 and Y397 are the principal sites for HS1 tyrosine phosphorylation in response to TCR stimulation. Consistent with this, mutation of either individual residue (Y378F or Y397F) permitted tyrosine phosphorylation of the protein, whereas mutation of both sites (2YF) abrogated phosphorylation (Figure 3D).
To test the function of HS1 phosphorylation, T cells were transfected with YFP-tagged HS1 or HS1-2YF, and targeting to the IS was assessed. As shown in Figures 4A and 4B, YFP-HS1 colocalized with F-actin at the IS, but the 2YF mutant remained diffusely distributed. To express the HS1 mutant in cells lacking endogenous protein, we used a ‘‘suppression-re-expression’’ vector that encodes the HS1 shRNA targeting sequence together with shRNA-resistant wild-type HS1 or HS1-2YF cDNA. At 48 hr, this vector suppresses the majority of endogenous HS1 and allows re-expression at near-endogenous levels (Figure 4C, right). Wild-type HS1 restored actin polymerization at the IS, but the 2YF mutant did not (Figure 4C), indicating that phosphorylation of HS1 at Y378/Y397 is required for actin regulation at the IS.
Figure 4.
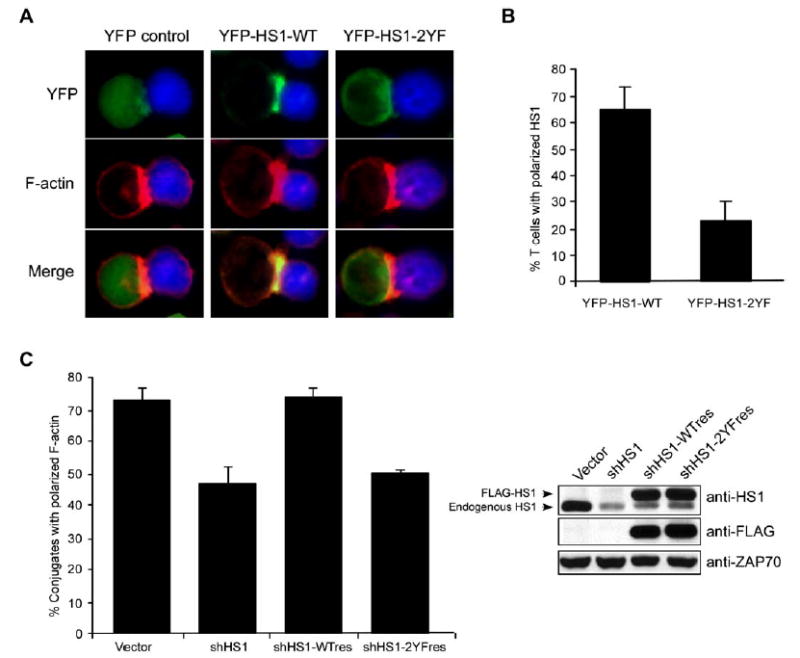
Phosphorylation at Y378/Y397 Is Required for HS1 Localization and Actin Accumulation at the IS
(A) Jurkat cells were transfected with YFP or YFP-tagged HS1 (wt or 2YF), conjugated to SEE-pulsed Raji B cells (blue), and fixed conjugates were labeled with phalloidin.
(B) Conjugates formed as in (A) were scored for YFP-HS1 localization to the IS.
(C) Jurkat T cells were transfected with the indicated constructs to allow suppression and re-expression of HS1 in the same cell. After 48 hr, cells were conjugated to SEE-pulsed NALM6 B cells and labeled with phalloidin, and conjugates containing GFP+ T cells were scored for actin polarization. Immunoblot analysis shows HS1 suppression and re-expression. ZAP-70 serves as a loading control. All error bars represent mean ± SD from three independent experiments.
Tyrosine Phosphorylation of HS1 Mediates Binding to Key Signaling Molecules
SH2 domains from several proteins known to participate in T cell actin-regulatory pathways could bind phospho-HS1 in GST pull-down assays (Figure 5A). In addition to Lck and Fyn, which are known to bind HS1 (Takemoto et al., 1996), both SH2 domains from the p85 subunit of PI3K, the second SH2 domain of PLCγ1, and the SH2 domains from all three Vav isoforms also interacted with phospho-HS1. To confirm these interactions in vivo, coimmunoprecipitation analysis was performed. Both PLCγ1 and Vav1 coimmunoprecipitated with HS1, with kinetics that mirrored HS1 tyrosine phosphorylation (Figure 5B). In contrast, Lck bound constitutively to HS1, with enhanced binding upon activation. This is consistent with previous work showing that HS1 binds both the SH3 and SH2 domains of Lck (Takemoto et al., 1996).
Figure 5.
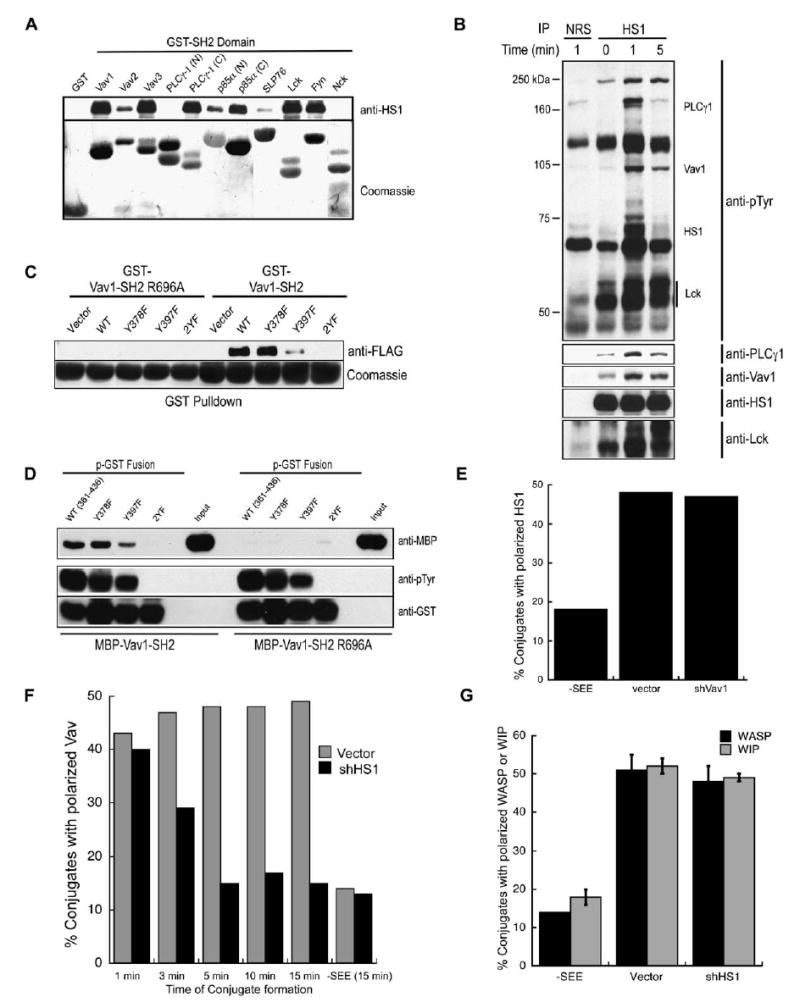
HS1 Interacts with T Cell Signaling Intermediates and Functions to Maintain Vav1 at the IS
(A) Lysates from pervanadate-treated Jurkat cells were incubated with GSH-agarose bound GST fusion proteins of various SH2 domains, and bound HS1 was detected by immunoblotting. Loading of fusion proteins was visualized with Coomassie.
(B) HS1 was immunoprecipitated from Jurkat cells after TCR/CD28 ligation, and interacting proteins were detected by immunoblotting. Rabbit IgG (NRS) was used as an immunoprecipitation control.
(C) Jurkat T cells were transfected with the indicated versions of FLAG-tagged HS1 and treated with pervanadate as in Figure 3D. Lysates were incubated with GSH-agarose bound GST-Vav1-SH2, and the bound HS1 proteins were detected by immunoblotting with anti-FLAG. Loading of fusion proteins was visualized with Coomassie.
(D) GST fusion peptides encompassing AA361–436 of HS1 were created in a phosphorylated form (p-GST) as described in Experimental Procedures. HS1 p-GST fusion proteins corresponding to wt, Y378F, Y397F, and 2YF were bound to GSH-agarose and tested for their ability to bind MBP-Vav1-SH2 in vitro. Phosphorylation of the p-GST fusions was visualized with anti-pTyr, and bound MBP-Vav1 SH2 was detected with anti-MBP. ‘‘Input’’ indicates the total amount of MBP-Vav-SH2 and Vav-SH2 R696A used per tube. In (C) and (D), Vav1-SH2 domain containing an inactivating mutation (R696A) serves as control.
(E) Jurkat T cells were transfected with control or shVav1-GFP vectors, incubated for 15 min with NALM6 B cells ± SEE, fixed, and labeled with anti-HS1. The frequency of conjugates containing GFP+ T cells exhibiting HS1 localization to the IS was scored.
(F) Jurkat cells were transfected with control or shHS1b-GFP vectors incubated with NALM6 B cells ± SEE, fixed at the indicated times, and labeled with anti-Vav1. Conjugates containing GFP+ T cells were scored for Vav1 localization to the IS.
(G) Jurkat T cells were treated as in (F), fixed at 15 min, and labeled with anti-WASp or anti-WIP. Conjugates containing GFP+ T cells were scored for WASp or WIP localization to the IS. Data represent mean ± SD from three experiments. Data in (E) and (F) are from one representative experiment.
HS1 and Vav1 Interact Specifically and Directly
Given the role of Vav1 in controlling actin responses, we focused on this interaction. As shown in Figure 5C, the GST-Vav1-SH2 fusion protein did not interact with over-expressed HS1-2YF from lysates of pervanadate-treated Jurkat cells; it bound weakly to the Y397F and more strongly to Y378F HS1. An inactivating mutant of the GST-Vav1-SH2 (R696A) failed to bind HS1, confirming the specificity of binding. We conclude that the Vav1 SH2 domain can bind to both Y378 and Y397, but binds preferentially to Y397. To ask whether HS1 binds Vav1 directly, phosphorylated GST fusion peptides (p-GST) encompassing HS1 amino acids 361–436 were used in an in vitro pull-down of MBP-Vav1-SH2. The Vav1-SH2 bound directly to the wt, Y378F, and Y397F p-GST peptides, but not to the 2YF mutant (Figure 5D), mirroring the results from pervanadate-treated cells (Figure 5C). Direct interaction was also verified in a far-Western blot (data not shown). These data show that the Vav1 SH2 domain binds directly to tyrosine-phosphorylated HS1.
HS1 Is Required for Maintaining Vav1 at the IS
Since Y378 and Y397 are required for HS1 targeting to the IS and for binding to Vav1, we reasoned that Vav1 might recruit HS1 to the IS. However, we found that HS1 recruitment was normal in T cells suppressed for Vav1 expression (Figure 5E). The converse experiment revealed a requirement for HS1 in localizing Vav1 to the IS (Figure 5F); HS1-suppressed cells initially recruit Vav1 to the IS, but Vav1 recruitment is abnormally short-lived. The kinetics of Vav1 loss closely resembles the kinetics of F-actin loss (Figure 1E). Importantly, this effect is specific to Vav1; WASp and WIP were recruited normally in HS1-suppressed cells (Figure 5G, and Figure S2). These data show that one important function of HS1 is to stabilize Vav1 at the IS. As discussed below, this is likely to involve both direct and indirect interactions. Interestingly, constitutively targeting Vav1 to the membrane via a CAAX motif (Billadeau et al., 2000b) rescued the actin defect in HS1-suppressed cells (Figure S3). This finding suggests that Vav1 plays a role in HS1-dependent actin regulation, though it is also possible that the observed response is a nonspecific consequence of constitutive Vav1 activity at the membrane. Given the other actin-regulatory functions of HS1, it seems likely that HS1 and Vav1 function together to coordinate actin dynamics at the IS.
HS1-Mediated Actin Stabilization Is Required for Sustained TCR Signaling
Since T cells from HS1−/− mice have proliferative defects (Taniuchi et al., 1995), we asked whether they exhibit defects in IL-2 production. T cells from wild-type and HS1−/− mice were cultured for 24 hr in the presence of SEB and wild-type T-depleted splenocytes, and IL-2 levels were measured. HS1−/− T cells produced significantly reduced levels of IL-2 at all doses of SEB (Figure 6A). Analysis of IL-2 reporter activity showed that HS1 suppression strongly inhibited activation of the IL-2 promoter, demonstrating that IL-2 defects occur at the transcriptional level (Figure 6B).
Figure 6.
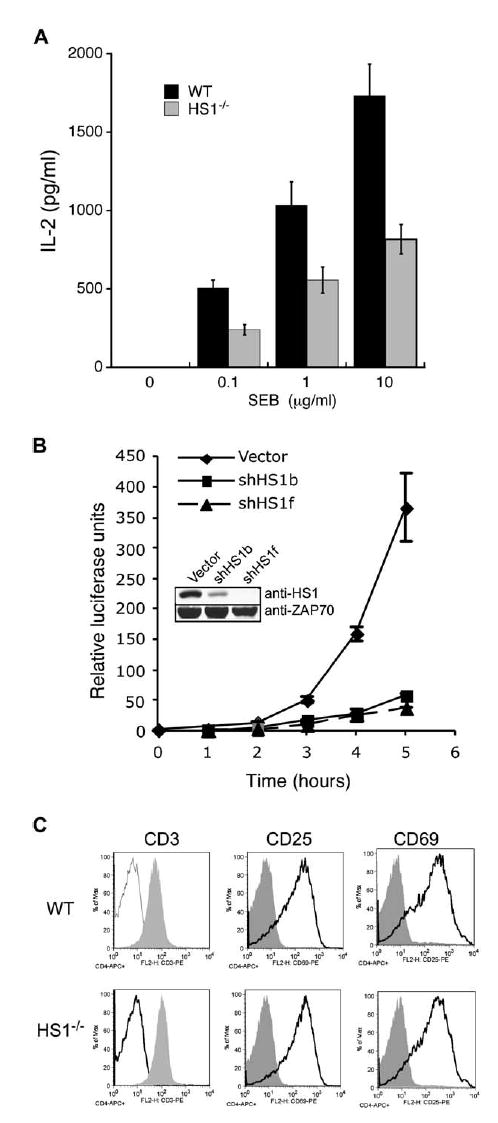
HS1 Is Required for IL-2 Production
(A) T cells isolated from wt or HS1−/− mice were cultured for 24 hr in the presence of wt T-depleted splenocytes and the indicated doses of SEB. Total IL-2 levels were measured by ELISA. Data are mean ± SD from replicate wells of one representative experiment.
(B) Jurkat T cells were cotransfected with IL2p-luc reporter and either vector control, shHS1b, or shHS1f plasmid. Cells were unstimulated or stimulated with SEE-pulsed NALM6 B cells for 5 hr, and luciferase activity was measured hourly. Data represent mean ± SD from triplicate samples.
(C) T cells isolated from wt and HS1−/− mice were unstimulated (shaded profiles) or stimulated for 24 hr with anti-CD3 and wt T-depleted splenocytes (open profiles). CD4+ T cells were analyzed for surface expression of CD3, CD25, and CD69 by flow cytometry.
The defects in HS1−/− T cells are not attributable to alterations in basal TCR expression levels or in activation-induced downregulation of CD3 or upregulation of the activation markers CD25 and CD69 (Figure 6C). As shown in Figure S4, HS1-suppressed cells showed normal APC-induced phosphorylation of ZAP-70 and coimmunoprecipitated TCRζ, indicating that Lck activation was unaffected. However, single-cell analysis by ratio-metric imaging showed a significant decrease in TCR-crosslinking-induced Ca2+ influx in HS1-suppressed cells and T cells from HS1−/− mice (Figures 7A and 7B). Because Ca2+ signaling is required for NFAT translocation, we assessed NFAT-reporter activation. As shown in Figure 7C, NFAT activity was significantly reduced in HS1-suppressed cells. Parallel analysis of NFκB also showed defects in this pathway (Figure 7D). Taken together, these results show that HS1 is dispensable for early TCR signaling events, but required for sustained signaling events leading to gene activation.
Figure 7.
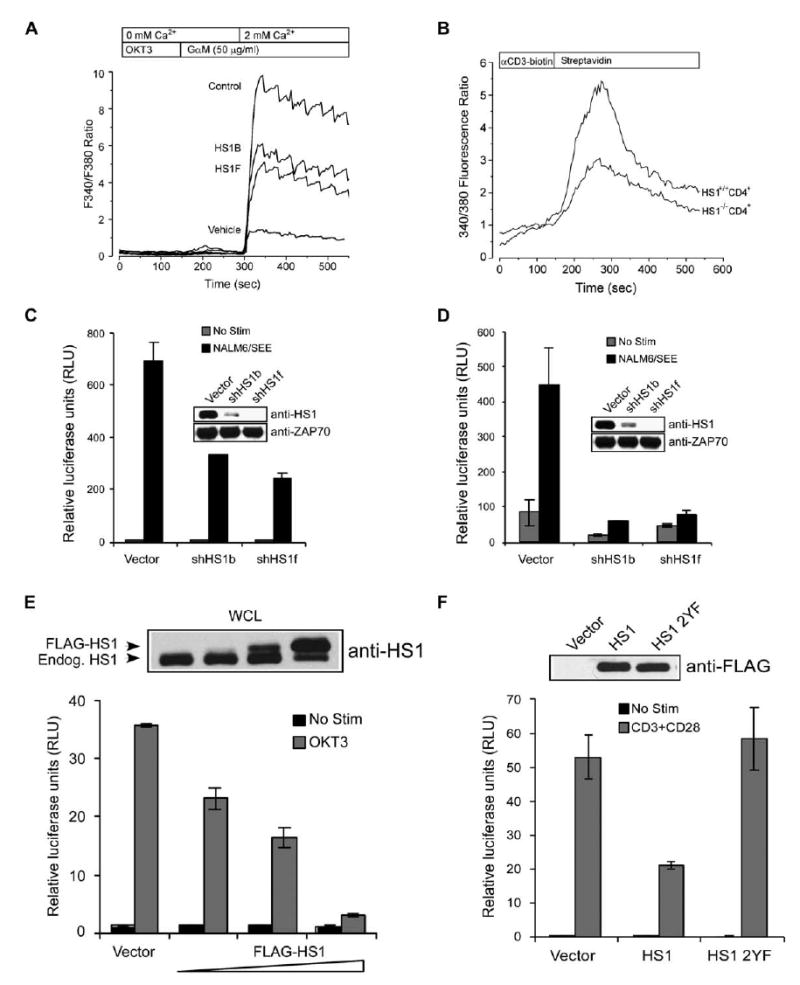
HS1 Is Required for Sustained TCR-Mediated Signaling Events Leading to Gene Activation
(A) Jurkat cells were transfected with control or shHS1 vectors. After 72 hr, TCR-induced Ca2+ signaling was examined with Fura-2 imaging. In Ca2+-free bath, increases in Fura-2 ratio reflect Ca2+ release from intracellular stores. Extracellular Ca2+ entry via activated CRAC channels was then assessed by addition of extracellular Ca2+. Each trace represents the average response of at least 100 cells.
(B) CD4+ LN T cells from wt or HS1−/− mice were loaded with Fura-2, incubated with biotinylated anti-CD3, and activated by crosslinking with streptavidin. Analysis was as in (A).
(C and D) Jurkat T cells were cotransfected with an NFAT-luc (C) or NFκB-luc (D) reporter construct along with vector control, shHS1b, or shHS1f. Cells were unstimulated or stimulated with SEE-pulsed NALM6 B cells, and luciferase activity was measured.
(E and F) Jurkat cells were transfected with IL-2p-luc along with (E) vector control (40 μg) or an increasing concentration of FLAG-HS1 expression vector (10, 20, 40 μg) or with (F) vector control, FLAG-HS1, or FLAG-HS1 2YF (20 μg each). Cells were unstimulated or stimulated and luciferase activity was measured. The accompanying immunoblots show endogenous HS1 and FLAG-HS1 expression in whole-cell lysates from each cell population. All bars represent mean ± SD from triplicate samples.
In the course of asking whether HS1’s role in controlling gene activation requires tyrosine phosphorylation of Y378/397, we found that IL-2 reporter activity is exquisitely sensitive to the expression level of HS1. Overexpression of FLAG-tagged (or untagged) HS1 inhibited IL-2 promoter activity in a dose-dependent fashion (Figure 7E and data not shown). Similar effects have been observed with other adaptors, presumably because superstoichiometric expression leads to disruption of signaling complexes. In keeping with the idea that HS1 function involves tightly controlled protein levels, high-level overexpression of HS1 led to diminished levels of endogenous protein (Figure 7E, top). Although this overexpression effect made it impossible for us to conduct transcriptional analysis based upon re-expression of HS1 mutants in shRNA-suppressed cells, we found that the 2YF mutant lacks the inhibitory effects of the wild-type molecule on IL-2 promoter activity (Figure 7F). Thus, phosphorylation of HS1 is also important for activation of IL-2 promoter activity.
Discussion
In this study, we show that HS1 is required for the stabilization of F-actin filaments after TCR engagement, for maximal Ca2+ influx, and for NFAT and NFκB-mediated gene transcription. In addition, we find that tyrosine phosphorylation is necessary for HS1 recruitment to the IS and regulates its interaction with Lck, PLCγ1, and Vav1. These findings identify HS1 as an actin-regulatory adaptor protein essential for T cell activation.
HS1-deficient T cells show unique defects in actin polymerization at the IS; actin-rich structures are formed initially but are unstable and erratic. This phenotype is distinct from that of cells lacking WASp or WAVE2, both of which also bind Arp2/3 complex. Cells lacking WASp spread essentially normally, while cells lacking WAVE2 fail to spread altogether (Nolz et al., 2006). The phenotype of HS1-deficient T cells resembles that of cortactin-deficient fibroblasts, which display disorganized lamellipod formation (Bryce et al., 2005; Kempiak et al., 2005). These phenotypes are consistent with the idea that by binding F-actin as well as Arp2/3 complex, these proteins inhibit debranching and stabilize cortical actin (Uruno et al., 2003b). An unresolved question is whether HS1 also acts via direct interactions with WASp and WIP, as reported for cortactin (Kempiak et al., 2005; Kinley et al., 2003; Uruno et al., 2003a; Weaver et al., 2001, 2002). Though we can readily detect binding of HS1 to the WIP/WASp complex in vitro, we have so far failed to verify binding in T cell lysates. We show that HS1 is not required for recruitment of WASp or WIP to the IS. Indeed, their continued presence likely contributes to the residual actin dynamics in HS1-deficient cells.
We mapped the major sites of HS1 tyrosine phosphorylation in activated T cells to amino acids 378 and 397, and our data suggest that ZAP-70 is responsible for phosphorylating these sites. Y378 and Y397 are critical for several aspects of HS1 function. Mutation of these residues leads to defective HS1 targeting to the IS, abrogates binding to Vav1, and perturbs actin responses. Finally, these residues are required for overexpression-induced inhibition of IL-2 promoter activation. Phosphorylation-dependent membrane targeting may be a general feature of HS1 function; in B cells, tyrosine phosphorylation of HS1 mediates its recruitment to lipid rafts (Hao et al., 2004). The protein(s) directly responsible for recruitment of HS1 remain to be identified. Our data rule out Vav1, because Vav1-deficient cells recruit HS1 efficiently. While it is possible that the functional defects observed in T cells expressing HS1 2YF are solely attributable to the aberrant localization of this mutant, it seems more likely that phosphorylation regulates multiple aspects of HS1 function. In addition to mediating binding to other signaling molecules (discussed below), phosphorylation may induce conformational changes that influence HS1 function, as already shown for cortactin (Huang et al., 1997; Martinez-Quiles et al., 2004).
One clear role of HS1 phosphorylation is to mediate binding to SH2 domain-containing proteins, including Lck, Vav1, and PLCγ1. The functional significance of Vav1 binding is underscored by the findings that HS1-suppressed T cells progressively lose Vav1 from the IS and that expression of a membrane-targeted Vav1 rescues the actin defect in HS1-suppressed cells. Interestingly, the loss of Vav1 from the IS in HS1-suppressed cells parallels the loss of F-actin at this site. This, and finding that the kinetics of Vav1/F-actin loss differ from those of HS1 phosphorylation, suggest that HS1 stabilizes Vav1 at the IS via a complex mechanism. Since Vav1 recruitment to the IS is dependent on interactions with SLP-76 and Itk (Dombroski et al., 2005; Zeng et al., 2003), we propose that SLP-76, Itk, and HS1 coordinately recruit Vav1 to the IS at early time points. Once this complex is localized to the IS, Vav1 initiates actin polymerization through activation of Cdc42 and Rac. HS1 participates in forming and/or stabilizing F-actin at later times, and this feeds back to stabilize Vav1 interactions with the SLP-76 complex. Stabilization could occur through direct interactions or via the F-actin scaffold. The fact that HS1 binds directly to Vav1 suggests that these important actin regulatory proteins function coordinately. It will be interesting to ask whether this interaction modifies the activity of either protein.
Phosphorylation also mediates HS1 interaction with PLCγ1, and studies are underway to probe this interaction further. Defects in signaling through PLCγ1 could account for the observed defects in Ca2+ influx in HS1-deficient T cells. However, phosphorylation of PLCγ1 at Y783 is unimpaired in HS1-suppressed cells (data not shown). Regulated interactions with PLCγ1 and the p85α subunit of PI-3K may also play a role in HS1-mediated actin regulation via effects on inositol phospholipids.
Initial analysis of HS1−/− mice demonstrated defects in T cell proliferation and negative selection (Taniuchi et al., 1995) but did not address the molecular basis of these defects. We show that HS1−/− T cells have defective actin and Ca2+ responses, as well as defective IL-2 production associated with defects in activation of NFAT and NFκB transcriptional elements. It remains to be determined how alterations in HS1 function result in the observed changes in IL-2 promoter activation. Perturbations in signaling through Vav1 and/or PLCγ1 may be involved, since both proteins are required for activation of NFAT and NFκB (Cao et al., 2002; Costello et al., 1999; Dolmetsch et al., 1998; Irvin et al., 2000). Alternatively, as we recently reported for WAVE2 (Nolz et al., 2006), HS1-dependent actin polymerization may be required to regulate CRAC activity. Studies aimed at distinguishing between these possibilities and additional analysis of HS1 function in TCR transgenic models are underway.
Experimental Procedures
Reagents and Plasmids
All reagents are from Sigma unless otherwise specified. Antibodies against PLCγ1, Vav1, Lck, and ZAP-70 have been previously described (Billadeau et al., 2000a; Karnitz et al., 1992; Ting et al., 1992; Williams et al., 1998). Anti-HS1 and anti-WIP were obtained by immunization of rabbits with a GST-fusion protein containing AA330–407 of human HS1 or a KLH-conjugated synthetic peptide corresponding to AA468–494 of human WIP (Cocalico Biologicals). Rabbit anti-WASp and anti-phosphotyrosine (4G10) were from Upstate Biotechnology. Anti-GST and anti-actin were from Santa Cruz. Anti-MBP was from Immunology Consultants Laboratory. The anti-human CD3 (OKT3) was from the Mayo Pharmacy; anti-TCR (C305) was a gift from Dr. G. Koretzky (Univ. Pennsylvania). Antibodies against murine CD3 (2C11), CD28 (PV-1), Thy 1 (AT83.A), and CD24 (J11D) were gifts from Dr. A. Sperling (Univ. Chicago). Anti-CD3PE and anti-CD4-APC were from Biolegend, and anti-CD28 (CD28.2), anti-CD25PE, and anti-CD69PE were from BD Biosciences.
Human HS1 was amplified from a cDNA library and mutated to generate the Y397F, Y378/397F, and shRNA-resistant mutants via the Quik Change kit from Stratagene (see Table S1 for primers). The IL-2p.luc, NFκB.luc, and NFAT.luc reporter constructs have been described (Cao et al., 2002).
The shRNA vectors pFRT.H1p and pCMS3.eGFP.H1p have been described (Gomez et al., 2005; Trushin et al., 2003). pCMS4.eGFP. H1p, derived from pCMS3.eGFP.H1p, contains an additional CMV promoter for driving the expression of shRNA-resistant cDNAs for reconstitution studies. shHS1b and shHS1f targeting sequences are in Table S1 along with the control shHS1mut sequence. The shRNA vector against Vav1 has been described (Zakaria et al., 2004).
Cell Culture, Mice, Transfection, and Stimulation
Jurkat-E6, JCaM-1, P116, and primary human peripheral blood CD4+ T cells and NALM6, Raji, and EBV B cells were grown in RPMI-1640 supplemented with 5% FBS, 5% FCS, and 4 mM glutamine. A Jurkat-E6 T cell line stably expressing GFP-actin was generated with the pEGFP-actin vector (Clontech). Quantitative immuno-blot analysis (Figure S5) shows that these cells express GFP-actin at ~37% of endogenous levels. P815 cells were cultured in Glutamax DMEM (GIBCO), with 5% FBS, nonessential amino acids, β-mercaptoethanol, penicillin, and streptomycin. DO11.10 2° T cells were cultured as described (McKean et al., 2001).
HS1−/− mice on the C57Bl/6 background (Taniuchi et al., 1995) were a generous gift from Dr. T. Watanabe (Univ. Tokyo). C57Bl/6 mice were from Jackson Laboratories. Primary T cells were prepared from lymph nodes of age- and sex-matched HS1−/− or wt C57Bl/6 mice by complement enrichment with anti-CD24 (J11D) hybridoma supernatant and rabbit complement H2 (Pel-Freez). Live cells were isolated by means of a Ficoll gradient, yielding 90%–95% CD3+ T cells. Primary APCs were prepared from wild-type splenocytes by complement enrichment with anti-Thy1 (AT83.A), yielding less than 1% CD3+ cells.
Transient expression and/or suppression and luciferase reporter assays in Jurkat were done as previously described (Cao et al., 2002; Nolz et al., 2006). For cell-cell stimulation in reporter assays, live NALM6 B cells (1 × 106) and 0.5 μg/ml Staphylococcal Entero-toxin E (SEE, Toxin Technologies) were added to each well. For cell-cell stimulation time courses, Jurkat cells were activated with fixed, SEE-pulsed NALM6 or Raji B cells as described (Gomez et al., 2005).
ELISAs and Flow Cytometry
Murine lymph node T cells (200,000/well) were plated in a 96-well round-bottom TC plate (Costar) with 50,000 T-depleted splenocytes and SEB (Toxin Technologies). After incubation at 37°C for 24 hr, plates were frozen at −80°C. IL-2 ELISAs were performed with eBio-sciences mouse IL-2 ELISA kit. For surface marker analysis, cells were stimulated as above, except that 1 μg/ml of anti-CD3 (2C11) was added in lieu of SEB. At 24 hr, cells were stained for CD4 and the indicated markers and analyzed with a BD FacsCalibur flow cytometer and FlowJo (Treestar).
GST Fusion Protein Coprecipitation, Immunoprecipitation, and Immunoblot Analysis
GST pull-downs, immunoprecipitations, SDS-PAGE, and immunoblotting were done as described (Gomez et al., 2005). Phosphorylated GST-HS1 fusion peptides (AA361–436) were made in the TKB1 E. coli (Stratagene). For in vitro binding assays, 30 μg of p-GST fusion protein was bound to glutathione-agarose, washed, incubated with 2 μg of maltose binding protein (MBP)-fused Vav1-SH2 proteins, rotated at 4°C for 20 min, and then washed four times with lysis buffer.
Preparation and Phosphorylation of Recombinant HS1
His-Tagged HS1 was expressed in E. coli. After IPTG induction, bacteria were lysed with phosphate buffer (20 mM NaPO4, 1% NP40, 500 mM NaCl, 20 mM Imidazole [pH 7.2]) with EDTA-free protease inhibitors (Roche). Lysates were clarified by centrifugation and loaded onto a HiTrap column (Amersham), precharged with 100 mM NiSO4 on an AKTA FPLC. The column was washed with phosphate buffer and eluted with a gradient to 500 mM Imidazole. The appropriate fraction was polished over a HiTrap Q HP column and eluted with a gradient to 1 M KCl. 20 μg of recombinant HS1 was incubated with or without 0.5 μg recombinant Syk (Upstate) in 50 mM Tris (pH 7.5), 100 μM NaVO4, 0.1% β-mercaptoethanol, 5 mM MgCl2,100 μM ATP for 10 min at 30°C.
Mass Spectrometry
Recombinant HS1 or HS1 immunoprecipitated from Jurkat cells was resolved by SDS-PAGE, stained with Coomassie R-250 (Pierce), excised, and washed extensively with 50:50 (acetonitrile/25 mM ammonium bicarbonate [pH 8.0]). Rehydrated gel pieces were infused with trypsin (Promega; 12.5 ng/μl) or Glu-C (Roche; 20 ng/μl) at pH 8.0 and digested at 37°C for 18–24 hr. Reactions were stopped by addition of 5% TFA. Nonpeptide impurities were removed with C18 or SCX ziptips (Millipore), and analysis was carried out in 0.1% phosphoric acid. MALDI-TOF spectra were acquired on a Ciphergen SELDI mass spectrometer in the linear mode. vMALDI Ion-Trap spectra were acquired on a Finnegan LTQ mass spectrometer. After the MS scan, a data-dependent ‘‘Top 9’’ experiment was acquired such that 1 full MS spectrum was first acquired followed by 9 MS2 spectra. For database searching, we used Bioworks 3.2 employing the SEQUEST algorithm, against the SWISSPROT database. To confirm that phosphopeptides were present, a neutral loss-dependent MS3 experiment was performed, and peaks that showed loss of 98 Daltons were subjected to further MS/MS.
Single-Cell Calcium Analysis and Conjugate Assay
Jurkat and mouse T cells were loaded with the cell-permeant Ca2+ indicator Fura-2 AM (3.0 μM, Molecular Probes), and Ca2+ mobilization was measured as previously described (Nolz et al., 2006). Conjugate assays were performed as described previously (Gomez et al., 2005).
Immunofluorescence Microscopy
Immunofluorescence of fixed conjugates was performed as described (Cannon et al., 2001; Gomez et al., 2005). For analysis of actin responses in primary mouse T cells, P815 cells were incubated with anti-CD3 and/or anti-CD28 for 30 min and allowed to interact with freshly isolated LN T cells for 30 min at 37°C while settling onto poly-L-lysine coverslips. Cells were imaged on a Zeiss LSM-510 confocal or a Zeiss Axiovert 200M microscope. Quantitation of protein polarization was performed by randomly selecting conjugates containing a T cell (GFP+ or mouse) contacting CMAC-labeled APC and scoring for the presence of a distinct band of the protein of interest at the cell-cell contact site. At least 50 conjugates were scored in each of three experiments. Data represent average ± SD.
Live Cell Imaging
Time-lapse images of actin dynamics were collected on an inverted Nikon Eclipse TE300 with a 60x Plan Apo long-working-distance objective, with an UltraVIEW LCI spinning-disk confocal imaging system (Perkin-Elmer). Delta-T cover dishes (Bioptechs) were treated with 1 mg/ml PLL for 1 hr at RT and rinsed with water. Prior to use, purified OKT3 (10 μg/ml in PBS) was bound overnight at 4°C. Dishes were rinsed in PBS, covered with 500 μl of serum-free-RPMI containing 25 mM HEPES, and kept at 37°C during imaging with a Bioptechs TC-3 stage and objective heater. Spreading was initiated by adding 15–20 μl of cell suspension (2 × 106 cells/ml in serum-free-RPMI). Stacks of 8–10 images spaced at 0.3 μm, starting from the plane of the coverglass, were collected, with a new stack initiated every 10 s. Data were analyzed with Slidebook v4.0.33. Projections were created from the 4D data sets, masks created for each cell at each time point, and the areas were determined in pixels. For radial variance calculations, 64 radii were struck from the centroid of each mask, and the standard deviation of the radii was calculated for each cell at each time point. Video sequences from several cells were aligned based on initial contact with the coverslip, and the average of the cell area and of the radial variance were calculated for the population of cells at each time point.
Acknowledgments
This work was supported by the Mayo Foundation, a Cancer Research Institute Investigator award, and NIH grant R01-AI065474 to D.D.B., by NIH grant F31-AI068624 to T.S.G., by a Leukemia and Lymphoma Scholar Award and NIH grant HD37091 to D.J.R., and by R01-AI44835 and a grant with the Pennsylvania Department of Health to J.K.B. The Pennsylvania Department of Health disclaims responsibility for any analyses, interpretations, or conclusions. We thank Shuixing Li and Renell Morgan for technical assistance, Morris Birnbaum and Edward Williamson for access to and assistance with spinning disk confocal microscopy, Mark Roden (3I) for help with morphometry, and Lynn Spruce and Mike Rosenblatt (CHOP Protein Core) for help with Mass Spectrometry. The authors declare that they have no competing financial interests.
References
- Badour K, Zhang J, Siminovitch KA. Involvement of the Wiskott-Aldrich syndrome protein and other actin regulatory adaptors in T cell activation. Semin Immunol. 2004;16:395–407. doi: 10.1016/j.smim.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Billadeau DD, Mackie SM, Schoon RA, Leibson PJ. The Rho family guanine nucleotide exchange factor Vav-2 regulates the development of cell-mediated cytotoxicity. J Exp Med. 2000a;192:381–392. doi: 10.1084/jem.192.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billadeau DD, Mackie SM, Schoon RA, Leibson PJ. Specific subdomains of Vav differentially affect T cell and NK cell activation. J Immunol. 2000b;164:3971–3981. doi: 10.4049/jimmunol.164.8.3971. [DOI] [PubMed] [Google Scholar]
- Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- Brunati AM, Donella-Deana A, James P, Quadroni M, Contri A, Marin O, Pinna LA. Molecular features underlying the sequential phosphorylation of HS1 protein and its association with c-Fgr protein-tyrosine kinase. J Biol Chem. 1999;274:7557–7564. doi: 10.1074/jbc.274.11.7557. [DOI] [PubMed] [Google Scholar]
- Bryce NS, Clark ES, Leysath JL, Currie JD, Webb DJ, Weaver AM. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr Biol. 2005;15:1276–1285. doi: 10.1016/j.cub.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Bubeck Wardenburg J, Pappu R, Bu JY, Mayer B, Chernoff J, Straus D, Chan AC. Regulation of PAK activation and the T cell cytoskeleton by the linker protein SLP-76. Immunity. 1998;9:607–616. doi: 10.1016/s1074-7613(00)80658-5. [DOI] [PubMed] [Google Scholar]
- Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE. Dynamic actin polymerization drives T cell receptor-induced spreading: a role for the signal transduction adaptor LAT. Immunity. 2001;14:315–329. doi: 10.1016/s1074-7613(01)00112-1. [DOI] [PubMed] [Google Scholar]
- Cannon JL, Burkhardt JK. Differential roles for Wiskott-Aldrich syndrome protein in immune synapse formation and IL-2 production. J Immunol. 2004;173:1658–1662. doi: 10.4049/jimmunol.173.3.1658. [DOI] [PubMed] [Google Scholar]
- Cannon JL, Labno CM, Bosco G, Seth A, McGavin MH, Siminovitch KA, Rosen MK, Burkhardt JK. Wasp recruitment to the T cell:APC contact site occurs independently of Cdc42 activation. Immunity. 2001;15:249–259. doi: 10.1016/s1074-7613(01)00178-9. [DOI] [PubMed] [Google Scholar]
- Cao Y, Janssen EM, Duncan AW, Altman A, Billadeau DD, Abraham RT. Pleiotropic defects in TCR signaling in a Vav-1-null Jurkat T-cell line. EMBO J. 2002;21:4809–4819. doi: 10.1093/emboj/cdf499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello PS, Walters AE, Mee PJ, Turner M, Reynolds LF, Prisco A, Sarner N, Zamoyska R, Tybulewicz VL. The Rho-family GTP exchange factor Vav is a critical transducer of T cell receptor signals to the calcium, ERK, and NF-kappaB pathways. Proc Natl Acad Sci USA. 1999;96:3035–3040. doi: 10.1073/pnas.96.6.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly RJ. Cortactin signalling and dynamic actin networks. Biochem J. 2004;382:13–25. doi: 10.1042/BJ20040737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- Dombroski D, Houghtling RA, Labno CM, Precht P, Takesono A, Caplen NJ, Billadeau DD, Wange RL, Burkhardt JK, Schwartzberg PL. Kinase-independent functions for Itk in TCR-induced regulation of Vav and the actin cytoskeleton. J Immunol. 2005;174:1385–1392. doi: 10.4049/jimmunol.174.3.1385. [DOI] [PubMed] [Google Scholar]
- Fuller CL, Braciale VL, Samelson LE. All roads lead to actin: the intimate relationship between TCR signaling and the cytoskeleton. Immunol Rev. 2003;191:220–236. doi: 10.1034/j.1600-065x.2003.00004.x. [DOI] [PubMed] [Google Scholar]
- Gomez TS, Hamann MJ, McCarney S, Savoy DN, Lubking CM, Heldebrant MP, Labno CM, McKean DJ, McNiven MA, Burkhardt JK, Billadeau DD. Dynamin 2 regulates T cell activation by controlling actin polymerization at the immunological synapse. Nat Immunol. 2005;6:261–270. doi: 10.1038/ni1168. [DOI] [PubMed] [Google Scholar]
- Hao JJ, Carey GB, Zhan X. Syk-mediated tyrosine phosphorylation is required for the association of hematopoietic lineage cell-specific protein 1 with lipid rafts and B cell antigen receptor signalosome complex. J Biol Chem. 2004;279:33413–33420. doi: 10.1074/jbc.M313564200. [DOI] [PubMed] [Google Scholar]
- Hao JJ, Zhu J, Zhou K, Smith N, Zhan X. The coiled-coil domain is required for HS1 to bind to F-actin and activate Arp2/3 complex. J Biol Chem. 2005;280:37988–37994. doi: 10.1074/jbc.M504552200. [DOI] [PubMed] [Google Scholar]
- Huang C, Ni Y, Wang T, Gao Y, Haudenschild CC, Zhan X. Down-regulation of the filamentous actin cross-linking activity of cortactin by Src-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:13911–13915. doi: 10.1074/jbc.272.21.13911. [DOI] [PubMed] [Google Scholar]
- Hutchcroft JE, Slavik JM, Lin H, Watanabe T, Bierer BE. Uncoupling activation-dependent HS1 phosphorylation from nuclear factor of activated T cells transcriptional activation in Jurkat T cells: differential signaling through CD3 and the costimulatory receptors CD2 and CD28. J Immunol. 1998;161:4506–4512. [PubMed] [Google Scholar]
- Irvin BJ, Williams BL, Nilson AE, Maynor HO, Abraham RT. Pleiotropic contributions of phospholipase C-gamma1 (PLC-gamma1) to T-cell antigen receptor-mediated signaling: reconstitution studies of a PLC-gamma1-deficient Jurkat T-cell line. Mol Cell Biol. 2000;20:9149–9161. doi: 10.1128/mcb.20.24.9149-9161.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnitz L, Sutor SL, Torigoe T, Reed JC, Bell MP, McKean DJ, Leibson PJ, Abraham RT. Effects of p56lck deficiency on the growth and cytolytic effector function of an interleukin-2-dependent cytotoxic T-cell line. Mol Cell Biol. 1992;12:4521–4530. doi: 10.1128/mcb.12.10.4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempiak SJ, Yamaguchi H, Sarmiento C, Sidani M, Ghosh M, Eddy RJ, Desmarais V, Way M, Condeelis J, Segall JE. A neural Wiskott-Aldrich Syndrome protein-mediated pathway for localized activation of actin polymerization that is regulated by cortactin. J Biol Chem. 2005;280:5836–5842. doi: 10.1074/jbc.M410713200. [DOI] [PubMed] [Google Scholar]
- Kinley AW, Weed SA, Weaver AM, Karginov AV, Bissonette E, Cooper JA, Parsons JT. Cortactin interacts with WIP in regulating Arp2/3 activation and membrane protrusion. Curr Biol. 2003;13:384–393. doi: 10.1016/s0960-9822(03)00107-6. [DOI] [PubMed] [Google Scholar]
- Kupfer A, Kupfer H. Imaging immune cell interactions and functions: SMACs and the immunological synapse. Semin Immunol. 2003;15:295–300. doi: 10.1016/j.smim.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Labno CM, Lewis CM, You D, Leung DW, Takesono A, Kamberos N, Seth A, Finkelstein LD, Rosen MK, Schwartzberg PL, Burkhardt JK. Itk functions to control actin polymerization at the immune synapse through localized activation of Cdc42 and WASP. Curr Biol. 2003;13:1619–1624. doi: 10.1016/j.cub.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Quiles N, Ho HY, Kirschner MW, Ramesh N, Geha RS. Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP. Mol Cell Biol. 2004;24:5269–5280. doi: 10.1128/MCB.24.12.5269-5280.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKean DJ, Huntoon CJ, Bell MP, Tai X, Sharrow S, Hedin KE, Conley A, Singer A. Maturation versus death of developing double-positive thymocytes reflects competing effects on Bcl-2 expression and can be regulated by the intensity of CD28 costimulation. J Immunol. 2001;166:3468–3475. doi: 10.4049/jimmunol.166.5.3468. [DOI] [PubMed] [Google Scholar]
- Morgan MM, Labno CM, Van Seventer GA, Denny MF, Straus DB, Burkhardt JK. Superantigen-induced T cell:B cell conjugation is mediated by LFA-1 and requires signaling through Lck, but not ZAP-70. J Immunol. 2001;167:5708–5718. doi: 10.4049/jimmunol.167.10.5708. [DOI] [PubMed] [Google Scholar]
- Nolz JC, Gomez TS, Zhu P, Li S, Medeiros RB, Shimizu Y, Burkhardt JK, Freedman BD, Billadeau DD. The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr Biol. 2006;16:24–34. doi: 10.1016/j.cub.2005.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzene M, Brunati AM, Marin O, Donella-Deana A, Pinna LA. SH2 domains mediate the sequential phosphorylation of HS1 protein by p72syk and Src-related protein tyrosine kinases. Biochemistry. 1996;35:5327–5332. doi: 10.1021/bi9528614. [DOI] [PubMed] [Google Scholar]
- Salomon AR, Ficarro SB, Brill LM, Brinker A, Phung QT, Ericson C, Sauer K, Brock A, Horn DM, Schultz PG, Peters EC. Profiling of tyrosine phosphorylation pathways in human cells using mass spectrometry. Proc Natl Acad Sci USA. 2003;100:443–448. doi: 10.1073/pnas.2436191100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto Y, Sato M, Furuta M, Hashimoto Y. Distinct binding patterns of HS1 to the Src SH2 and SH3 domains reflect possible mechanisms of recruitment and activation of downstream molecules. Int Immunol. 1996;8:1699–1705. doi: 10.1093/intimm/8.11.1699. [DOI] [PubMed] [Google Scholar]
- Taniuchi I, Kitamura D, Maekawa Y, Fukuda T, Kishi H, Watanabe T. Antigen-receptor induced clonal expansion and deletion of lymphocytes are impaired in mice lacking HS1 protein, a substrate of the antigen-receptor-coupled tyrosine kinases. EMBO J. 1995;14:3664–3678. doi: 10.1002/j.1460-2075.1995.tb00036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting AT, Karnitz LM, Schoon RA, Abraham RT, Leibson PJ. Fc gamma receptor activation induces the tyrosine phosphorylation of both phospholipase C (PLC)-gamma 1 and PLC-gamma 2 in natural killer cells. J Exp Med. 1992;176:1751–1755. doi: 10.1084/jem.176.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushin SA, Pennington KN, Carmona EM, Asin S, Savoy DN, Billadeau DD, Paya CV. Protein kinase Calpha (PKCalpha) acts upstream of PKCtheta to activate IkappaB kinase and NF-kappaB in T lymphocytes. Mol Cell Biol. 2003;23:7068–7081. doi: 10.1128/MCB.23.19.7068-7081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner M, Billadeau DD. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol. 2002;2:476–486. doi: 10.1038/nri840. [DOI] [PubMed] [Google Scholar]
- Uruno T, Liu J, Li Y, Smith N, Zhan X. Sequential interaction of actin-related proteins 2 and 3 (Arp2/3) complex with neural Wiscott-Aldrich syndrome protein (N-WASP) and cortactin during branched actin filament network formation. J Biol Chem. 2003a;278:26086–26093. doi: 10.1074/jbc.M301997200. [DOI] [PubMed] [Google Scholar]
- Uruno T, Zhang P, Liu J, Hao JJ, Zhan X. Haema-topoietic lineage cell-specific protein 1 (HS1) promotes actin-related protein (Arp) 2/3 complex-mediated actin polymerization. Biochem J. 2003b;371:485–493. doi: 10.1042/BJ20021791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT, Cooper JA. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol. 2001;11:370–374. doi: 10.1016/s0960-9822(01)00098-7. [DOI] [PubMed] [Google Scholar]
- Weaver AM, Heuser JE, Karginov AV, Lee WL, Parsons JT, Cooper JA. Interaction of cortactin and N-WASp with Arp2/3 complex. Curr Biol. 2002;12:1270–1278. doi: 10.1016/s0960-9822(02)01035-7. [DOI] [PubMed] [Google Scholar]
- Williams BL, Schreiber KL, Zhang W, Wange RL, Samelson LE, Leibson PJ, Abraham RT. Genetic evidence for differential coupling of Syk family kinases to the T-cell receptor: reconstitution studies in a ZAP-70-deficient Jurkat T-cell line. Mol Cell Biol. 1998;18:1388–1399. doi: 10.1128/mcb.18.3.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanashi Y, Okada M, Semba T, Yamori T, Umemori H, Tsunasawa S, Toyoshima K, Kitamura D, Watanabe T, Yamamoto T. Identification of HS1 protein as a major substrate of protein-tyrosine kinase(s) upon B-cell antigen receptor-mediated signaling. Proc Natl Acad Sci USA. 1993;90:3631–3635. doi: 10.1073/pnas.90.8.3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakaria S, Gomez TS, Savoy DN, McAdam S, Turner M, Abraham RT, Billadeau DD. Differential regulation of TCR-mediated gene transcription by Vav family members. J Exp Med. 2004;199:429–434. doi: 10.1084/jem.20031228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng R, Cannon JL, Abraham RT, Way M, Billadeau DD, Bubeck-Wardenberg J, Burkhardt JK. SLP-76 coordinates Nck-dependent Wiskott-Aldrich syndrome protein recruitment with Vav-1/Cdc42-dependent Wiskott-Aldrich syndrome protein activation at the T cell-APC contact site. J Immunol. 2003;171:1360–1368. doi: 10.4049/jimmunol.171.3.1360. [DOI] [PubMed] [Google Scholar]