

Summary
Background
The engagement of the T cell receptor results in actin cytoskeletal reorganization at the immune synapse (IS) and the triggering of biochemical signaling cascades leading to gene regulation and, ultimately, cellular activation. Recent studies have identified the WAVE family of proteins as critical mediators of Rac1-induced actin reorganization in other cell types. However, whether these proteins participate in actin reorganization at the IS or signaling pathways in T cells has not been investigated.
Results
By using a combination of biochemical, genetic, and cell biology approaches, we provide evidence that WAVE2 is recruited to the IS, is biochemically modified, and is required for actin reorganization and β-integrin-mediated adhesion after TCR crosslinking. Moreover, we show that WAVE2 regulates calcium entry at a point distal to PLCγ1 activation and IP3-mediated store release.
Conclusions
These data reveal a role for WAVE2 in regulating multiple pathways leading to T cell activation. In particular, this work shows that WAVE2 is a key component of the actin regulatory machinery in T cells and that it also participates in linking intracellular calcium store depletion to calcium release-activated calcium (CRAC) channel activation.
Introduction
The engagement of multisubunit immune recognition receptors on lymphocytes results in actin cytoskeletal reorganization and the triggering of biochemical signaling cascades, leading to gene regulation and, ultimately, cellular activation. Reorganization of the actin cytoskeleton is required for T cell activation and plays an important role in immunological synapse (IS) formation and ‘‘signalosome’’ assembly at the cell-cell contact site. In fact, treatment of T cells with the actin-destabilizing agent cytochalasin D inhibits T cell receptor (TCR)-mediated IL-2 gene transcription, demonstrating that reorganization of the actin cytoskeleton is a requisite event controlling T cell activation [1].
Rac1 and Cdc42, two Rho family GTPases, have been implicated in the regulation of multiple cellular events, including actin reorganization, after receptor activation in several cell types [2]. Indeed, dominant-negative versions of both Cdc42 and Rac1 have been shown to inhibit IL-2 gene transcription and actin cytoskeletal changes following TCR engagement [3]. However, our understanding of the relevant effector molecules downstream of Rac1 and Cdc42 is quite incomplete. Many studies have focused on the Cdc42 effector WASp (Wis-kott-Aldrich Syndrome protein), due to its association with human disease. Indeed, the activation of multiple hematopoietic cell types in response to receptor ligation is attenuated or abrogated in the absence of WASp[4]. However, recent data demonstrate that T cells from mice lacking WASp are still capable of forming conjugates and polymerizing actin at the IS under certain experimental conditions [5, 6]. Thus, other molecules must also be able to regulate actin polymerization during T cell activation. One likely candidate is the Rac1 effector molecule WAVE2.
All three WAVE isoforms (WAVE1–WAVE3) function to regulate de novo actin polymerization downstream of Rac1 [7]. Whereas WAVE1 and WAVE3 are expressed primarily in neuronal cells, WAVE2 shows a more ubiquitous distribution with high levels in hematopoietic cells [8]. Structurally, WAVE proteins contain a WAVE homology domain (WHD) at their N terminus, followed by a basic region (BR), a proline-rich region (PRR), and a C-terminal verprolincofilin-acidic (VCA) region involved in binding actin and the Arp2/3 complex. WAVE2 exists in a multimolecular complex with several other proteins, including PIR121 or Sra-1, Nap1, Abi-1/2, and HSCP300 [9, 12]. GTP bound Rac1 interacts with PIR121/Sra-1 [12] and thereby recruits WAVE2 to areas of cellular activation, where WAVE2 then promotes actin polymerization. Interestingly, mice deficient for WAVE2 die during gestation and display defects in development and cell migration [13, 14]. Importantly, fibroblasts from these animals show defects not only in Rac1-mediated migration but also in both lamellipodia formation and dorsal ruffling induced by Rac1 [13].
The role of WAVE2 in T cell function has not been addressed. Using a combination of biochemical, cellular, and genetic approaches, we asked if the WAVE2 signaling complex is involved in regulating actin reorganization at the IS during T cell activation. Our findings show that WAVE2 is involved in regulating actin reorganization, integrin function, and CRAC-mediated Ca2+ entry, ultimately leading to optimal T cell activation.
Results
The WAVE2 Complex Localizes to the Immunological Synapse
Though several studies show that WASp contributes to the regulation of actin response in T cells and NK cells [15–17], recent studies show that T cells from WASp-deficient mice are capable of polymerizing actin at the T cell-APC contact site under certain experimental conditions [5, 6]. This prompted us to explore other proteins expressed in T cells that have actin nucleating activity, more specifically, the WAVE/Scar family member WAVE2.
WAVE2 is expressed in multiple cell types, including peripheral blood lymphoctes [8], and is the major WAVE isoform expressed by T cells (Figure S1). In addition, WAVE2 exists in a complex with the proteins PIR121, Nap1, and Abi-1/2 in other cell types [9–12]. In order to initially determine whether WAVE2 forms a similar complex in hematopoietic cells, WAVE2 was immunoprecipitated from Jurkat and primary CD4+ T cells. We found that WAVE2 is associated with PIR121 and Abi-1/2 in both cell types (Figures 1A and 1B). Using both RT-PCR and immunoblotting, we could not detect expression of Nap1 in Jurkat T cells or other lymphocyte populations (T.S.G., J.C.N., and D.D.B., unpublished data). However, HEM-1, a hematopoietically expressed Nap1 homolog, was found to associate with the WAVE2 complex in both cell types (Figures 1A and 1B). Moreover, we found that the WAVE2 complex remained stably associated throughout the timecourse of TCR stimulation (Figure 1A). In addition, phosphorylation of WAVE2 could be detected by SDS-PAGE in response to ligation of the TCR (see the Supplemental Results section and Figure S2).
Figure 1.
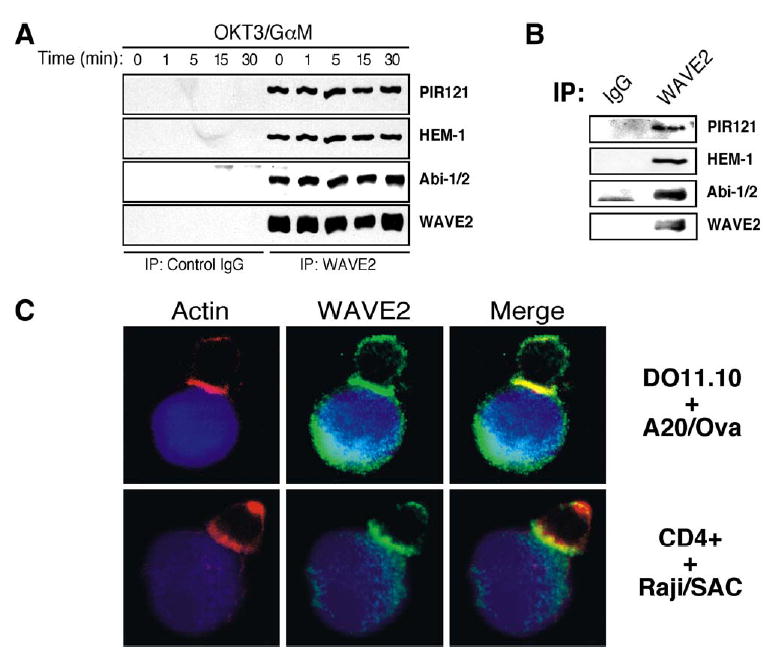
The WAVE2 Complex Localizes to the IS
(A) Jurkat T cells were stimulated for indicated time points (above lanes). After stimulation, cells were lysed, clarified, and subsequently immunoprecipitated with control IgG antibody or antibody against WAVE2. IPs were separated by SDS-PAGE, transferred, and subsequently blotted for PIR121, HEM-1, Abi-1/2, and WAVE2.
(B) Primary CD4+ T cells were isolated, lysed, immunoprecipitated, and subsequently blotted as in (A).
(C) Ovalbumin peptide-pulsed A20 B cells (blue) were incubated with secondary DO11.10 transgenic T cells (top). Conjugated cells were bound to poly-L-lysine coated coverslips, fixed, and stained with antibody against WAVE2 (green) and rhodamine-phalloidin (red). Also shown are primary CD4+ T cells conjugated to superantigen cocktail (SAC) pulsed Raji B cells (bottom).
To investigate the role of WAVE2 in T cell activation, we next asked if WAVE2 localizes to the T cell-APC interface, where actin polymerization takes place. Indeed, F-actin and WAVE2 are recruited to the contact site formed between DO11.10 T cells and ovalbumin peptide-pulsed A20 B cells (Figure 1C). Similar results were observed with primary human CD4+ T cells interacting with Raji B cells pulsed with a superantigen cocktail and Jurkat T cells interacting with SEE-pulsed NALM6 B cells (Figure 1C). Recruitment of other WAVE2 complex family members (Abi-2 and HEM-1) to the IS was also observed in human CD4+ T cells conjugated to antigen-pulsed Raji B cells (Figure S3). Recruitment of WAVE2 complex members and actin reorganization was not observed in T cell-B cell conjugates formed in the absence of ovalbumin peptide or superantigen (data not shown). Taken together, these data demonstrate that a WAVE2 complex exists in T cells and that WAVE2 colocalizes with sites of F-actin polymerization in response to TCR stimulation.
WAVE2 Protein Levels Are Reduced by the Elimination of Other WAVE2 Complex Members
Figures 1A and 1B clearly demonstrate that an intact WAVE2 complex is present in T lymphocytes. However, to further determine whether all of the WAVE2 complex members are found associated with WAVE2 or, alternatively, if there are free pools of WAVE2 complex proteins, we utilized serial immunoprecipitation. As shown in Figure 2A, three subsequent IPs for WAVE2 were performed to deplete all of the WAVE2 from 0.5 mg of T cell lysate. When the WAVE2-depleted lysates were subjected to a PIR121/HEM-1 IP, no residual PIR121 or HEM-1 protein was detectable (Figure 2A, lane 5). However, HEM-1 and PIR121 could clearly be detected by immunoprecipitation prior to WAVE2 depletion (Figure 2A, lane 6). These data demonstrate that the majority of PIR121 and HEM-1 protein in T lymphocytes is bound to WAVE2.
Figure 2.
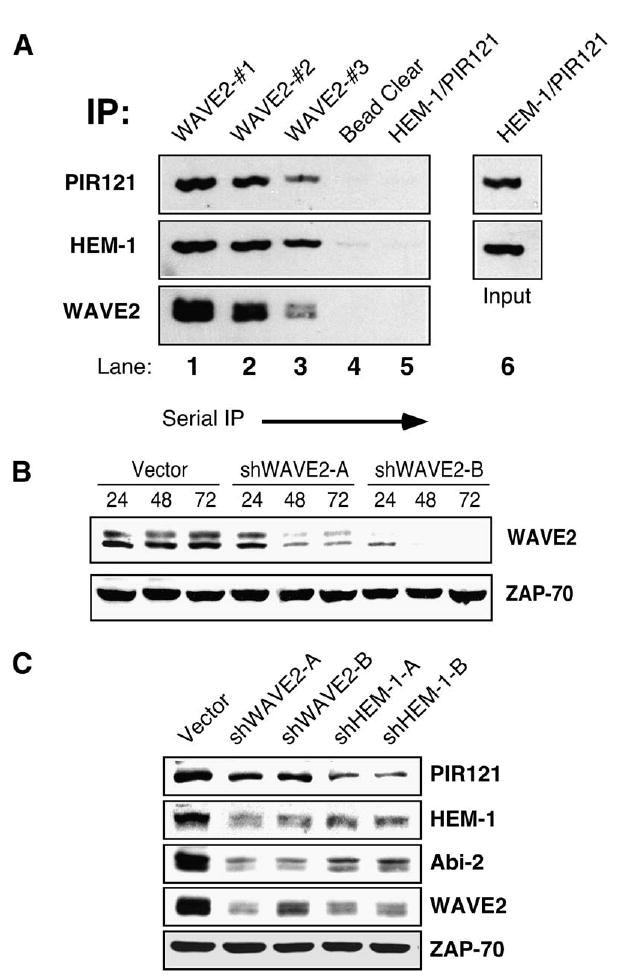
Depletion of WAVE2 Leads to Subsequent Degradation of Other Members of the Complex
(A) Three subsequent IPs for WAVE2 were performed on 0.5 mg of Jurkat T cell lysate (WAVE2#1-3, lanes 1–3). After preclearing the lysate of any residual WAVE2 antibody (bead clear, lane 4), an IP for HEM-1 and PIR121 was performed (PIR121/HEM-1, lane 5). HEM-1 and PIR121 was also directly IP’ed from 0.5 mg of Jurkat T cell lysate (PIR121/HEM-1, lane 6). Bound proteins from each IP were separated by SDS-PAGE, transferred, and subsequently blotted for WAVE2, HEM-1, and PIR121.
(B) Jurkat T cells were transfected with a control vector (vector) or one of two shWAVE2 suppression vectors (shWAVE2-A/-B). Cells were lysed at the 24, 48, and 72 hr time points. Equal amounts of protein were separated by SDS-PAGE, transferred, and blotted for WAVE2 and ZAP-70.
(C) Cells were transfected as in (B) with the addition of two suppression vectors against HEM-1 (shHEM-1-A/-B). After 72 hr, cells were harvested as in (B) and blotted for PIR121, HEM-1, Abi-2, WAVE2, and ZAP-70.
To further characterize the role of WAVE2 in T cell activation, we generated two shRNA targeting constructs against WAVE2. Both targeting constructs efficiently depleted WAVE2 from Jurkat T cells (Figure 2B). Quantitation of this suppression at 72 hr revealed that a transfection efficiency of >85% (as determined by flow cytometry for GFP expression) decreased WAVE2 levels by at least 80% for both shWAVE2-A and WAVE-B when compared to the vector control (J.C.N. and D.D.B., unpublished data). Experiments with either suppression vector consistently yielded similar results. For clarity, results are shown for shWAVE2-B only, except as otherwise indicated.
Previous studies have demonstrated that proteins of the WAVE2 complex are dependent upon each other for stability and that elimination of any of the complex members leads to subsequent degradation of other members of the complex [11, 12, 18–20]. Indeed, when WAVE2 levels were suppressed in Jurkat T cells, a decrease in the level of PIR121, HEM-1, and Abi-2 protein was also observed (Figure 2C). To further examine the stability of the WAVE2 complex in T cells, we generated suppression vectors to target the hematopoietic specific member of the complex, HEM-1. Similar to the effect seen with WAVE2 suppression, elimination of HEM-1 also led to subsequent degradation of other members of the WAVE2 complex (Figure 2C). Overall, these data support previous reports regarding architecture and stability of the WAVE2 complex.
Actin Polymerization and Conjugate Formation Are Decreased in the Absence of WAVE2
The ability of a T cell to recognize and conjugate with an APC is dependent on cytoskeletal dynamics and ‘‘inside-out’’ signaling leading to integrin-mediated adhesion. Activation of the β2 integrin LFA-1 leads to high avidity binding to ICAM proteins, which is crucial for stable T cell-APC conjugation [21]. To determine if there is a role for the WAVE2 complex in this process, we first asked if T cells lacking WAVE2 or HEM-1 are able to form conjugates with superantigen-pulsed B cells. Using a flow cytometric assay, we found that T cells deficient for either WAVE2 or HEM-1 show significant defects in conjugate formation relative to control-transfected cells (Figure 3A). Because Rho GTPase activation and cyto-skeletal remodeling are critical for regulating the multiple integrins expressed on T cells [21], we next determined if β1 integrin activation was also defective by examining the ability of WAVE2 suppressed cells to adhere to fibronectin in response to TCR stimulation. Consistent with the results shown above, WAVE2 and HEM-1 suppressed Jurkat T cells showed dramatic defects in TCR-dependent adhesion to fibronectin (Figure 3B and data not shown). Together, these experiments show that members of the WAVE2 complex are required for integrin-mediated adhesion in response to TCR stimulation.
Figure 3.
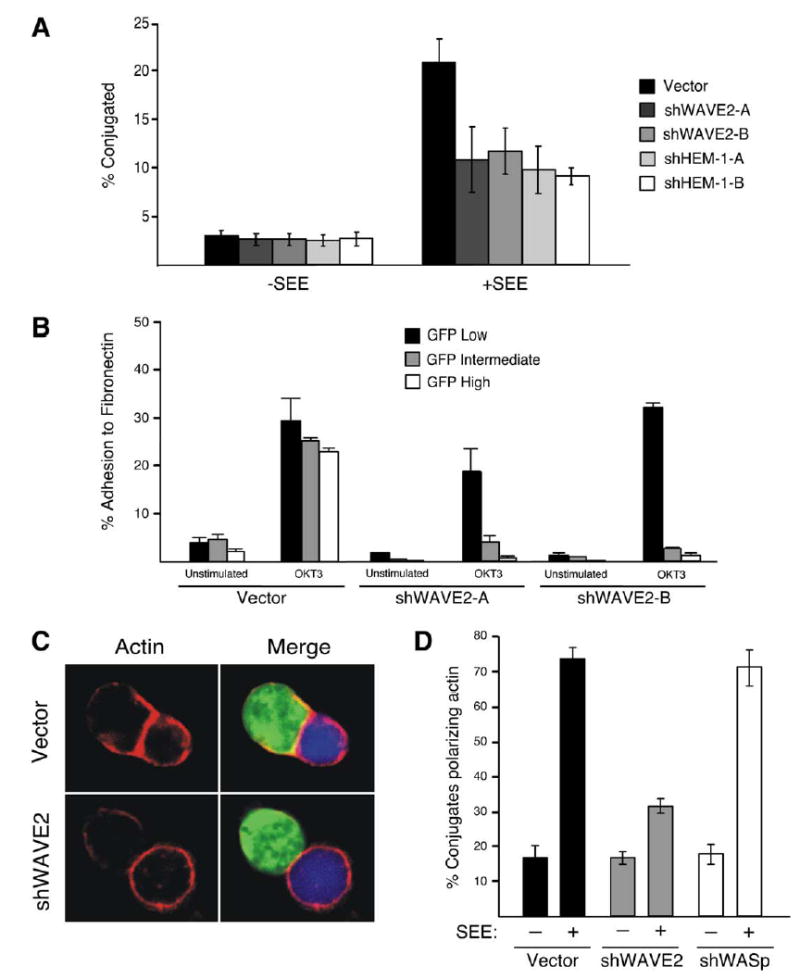
Suppression of WAVE2 Decreases Conjugate Formation and Actin Accumulation at the IS
(A) Cells were transfected with either vector control or suppression vectors targeting either WAVE2 or HEM-1. After 72 hr, transfected cells were incubated with NALM6 B cells stained red with ethidium bromide and pulsed with SEE (+SEE) or left unpulsed (−SEE). Using GFP expression as a marker for transfected cells, conjugate formation was determined using two-color flow cytometry. Shown is a representative example of three independent experiments.
(B) Jurkat T cells were transfected as in (A) and were analyzed for their ability to bind to fibronectin following no stimulation (unstimulated) or stimulation of the TCR (OKT3) for 10 min at 37ºC.After Following washing, adherent cells were collected and the adhesion of GFP-low, intermediate, and high cells in each sample was determined by flow cytometry.
(C) Cells were transfected as in (A) and after 72 hr were incubated with SEE-pulsed NALM6 B cells (blue). Conjugates were bound to poly-L-lysine coated coverslips, fixed, and stained with rhodamine-phalloidin (red) to visualize the accumulation of F-actin.
(D) Conjugates formed in (C) and Figure S4 were quantified on their ability to form F-actin at the IS. Shown is the average of three independent experiments.
Because some WAVE2-suppressed T cells did form conjugates, we next analyzed whether there was reduced actin polymerization at the IS in these cells. Using GFP expression to identify conjugates formed with T cells expressing the WAVE2 suppression vector, we analyzed the frequency of F-actin polymerization at the T-B interface. Indeed, many WAVE2-suppressed T cells contacting APCs showed little or no F-actin labeling at the IS (Figure 3C). Quantitation of this defect revealed that WAVE2 suppression decreased the frequency of conjugates exhibiting polarized F-actin by nearly 60%, whereas suppression of WASp had no affect (Figure 3D). Because this assay measures actin responses in only those cells that formed successful conjugates, it necessarily underestimates the overall actin defect. When considering both conjugate formation and the accumulation of F-actin at the IS, our results demonstrate that over 75% of WAVE2-suppressed cells show defects in actin-dependent responses leading to the formation of the IS. Some of the remaining responders are almost certainly cells in which WAVE2 suppression was inefficient. In addition, it is likely that other actin regulatory proteins contribute to the residual response. However, WASp is probably not a major contributor, because WASP-suppressed cells showed no significant defect in F-actin responses (Figure 3D and Figure S4) and because suppression of WAVE2 and WASp together showed no enhanced effect (data not shown). Taken together, these data show that WAVE2 is required for integrin-dependent adhesion and efficient actin polymerization at the IS.
WAVE2 Is Required for Lamellipodia Formation in T Cells Responding to TCR Stimulation
Spreading of T cells on anti-TCR coated coverslips requires the formation of stable actin structures and the generation of lamellipodia [22]. Because WAVE proteins have been implicated in regulating the formation of la-mellipodia downstream of Rac1 in other cell types, we analyzed the effect of WAVE2 suppression on the ability of T cells to spread onto anti-CD3 (OKT3)-coated cover-slips. As expected, control transfected Jurkat T cells expressing GFP-actin spead onto anti-CD3-coated coverslips in a highly ordered fashion, forming a round lamellipodial interface containing radially-arrayed F-actin-rich structures. Retrograde motion of GFP-actin was visible within the lamellar region (Figure 4A and Movie 1). In addition, DIC images showed membrane ruffling at the cell periphery (Movie 1). Maximal cell spreading was achieved by 2–3 min and cells began to retract by 7.5–10 min. In stark contrast to the behavior of control-transfected cells, cells expressing cherry-shWAVE2-B failed to spread on anti-CD3 coated coverslips. In fact, 25 min after contact with the coverslip, the WAVE2-suppressed cells still resembled control cells at the 20 s time point (Figures 4A and 4B). WAVE2-suppressed cells frequently sent out individual GFP-actin rich ruffles but never formed well-organized lamellipodia. The ruffles that were generated did not persist and were rapidly retracted (Figure 4B and Movie 2). In addition, these structures did not remain in contact with the coverslip. These data show that WAVE2 is required for the generation of stable lamellipodia in response to anti-TCR ligation.
Figure 4.
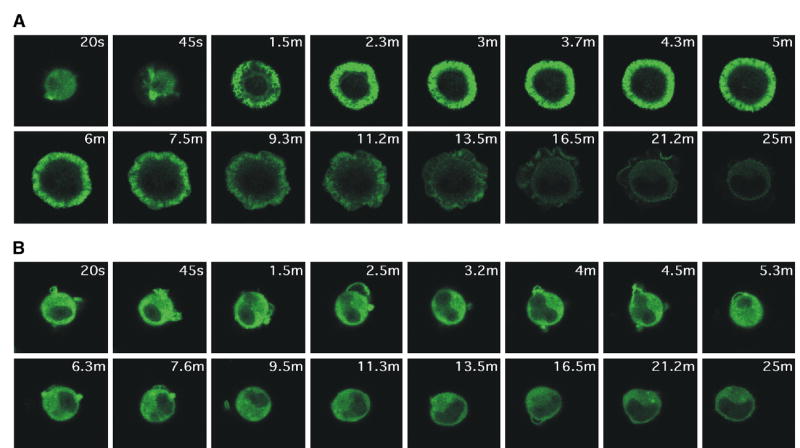
WAVE2-Suppressed T Cells Fail to Spread Properly in Response to TCR Ligation
Jurkat T cells stably expressing GFP-actin were transfected with (A) control shRNA-cherry vector or (B) cherry-shWAVE2-B. After 48 hr, the cells were placed onto OKT3 mAb-coated coverslips and activation events were recorded by taking images every 5 s, as explained in the Experimental Procedures. Shown are representative time-lapsed images occurring after initial contact with the OKT3-coated coverslips. More than 100 cherry-expressing cells were analyzed for each transfection. Images were taken at the same magnification. Movies are available online with the Supplemental Data.
WAVE2 Is Required for TCR-Stimulated IL-2p Activity
Because appropriate actin reorganization is required for T cell activation, we next explored whether WAVE2 depletion leads to defects in TCR-stimulated signaling events. Indeed, suppression of WAVE2 using either shWAVE2 construct resulted in a decrease in IL-2 promoter activity after anti-CD3 or anti-CD3/CD28 cross-linking (Figure 5A, and data not shown). This defect in IL-2p activity was not a result of a decrease in total or cell-surface TCR levels (J.C.N. and D.D.B., unpublished data) or decreased activation of proximal tyrosine kinases, because TCR-induced ZAP-70 phosphorylation was unaffected (Figure S5A). In addition, analysis of LAT immunoprecipitates from anti-TCR stimulated control or WAVE2-suppressed cells did not reveal any gross defects in the formation of the LAT signalosome (i.e., recruitment and phosphorylation of SLP76, Vav1, and PLCγ1) (Figure S5B). These data suggest that WAVE2 functions to regulate T cell activation independently of early signaling events.
Figure 5.
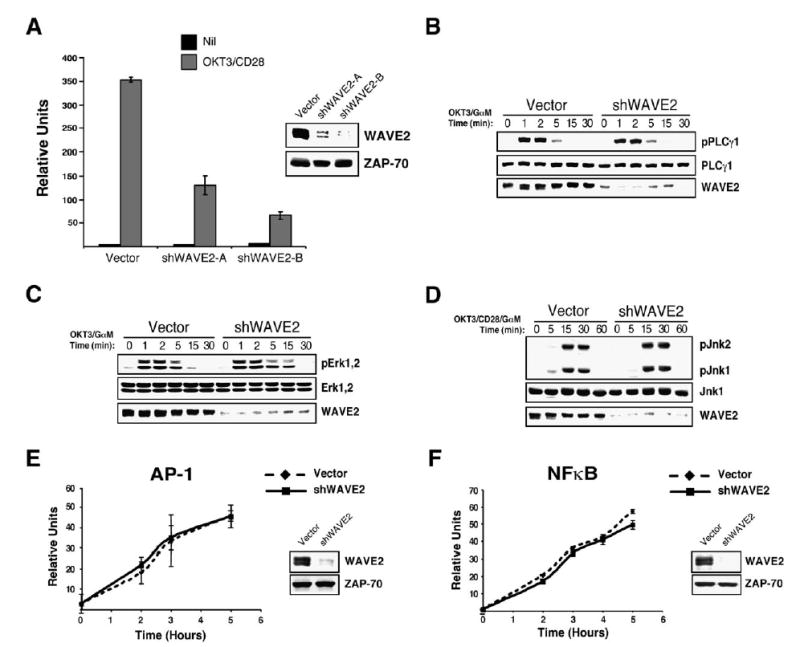
WAVE2 Regulates T Cell Activation Independently of PLCγ1, Erk, and Jnk Activation
(A) Jurkat T cells were transfected with IL-2p luciferase reporter along with either a control vector or one of two shWAVE2 suppression vectors (shWAVE2-A/-B). After 72 hr, cells were left unstimulated (Nil) or stimulated with OKT3 and anti-CD28 for 5 hr. Luciferase activity was measured and normalized to TK-Renilla readings.
(B) Jurkat T cells were transfected with vector control or shWAVE2 suppression vector. After 72 hr, cells were stimulated with OKT3 and goat anti-mouse antibody for the indicated time points. Whole-cell extracts were prepared and proteins were separated by SDS-PAGE, transferred, and analyzed for phosphorylation of PLCγ1 (Tyr 783) by immunoblot.
(C and D) Same as in (B) but blotted for phosphorylation of Erk1 and Erk2 and Jnk1 and Jnk2.
(E and F) Cells were transfected as in (A), but with a reporter construct for (E) AP-1 or (F) NFκB. Cells were stimulated for 2, 3, 4, or 5 hr, and luciferase activity was measured and normalized to TK-Renilla readings. Data shown are representative examples of three independent experiments for each.
WAVE2 Regulates Ca2+ Downstream of TCR Ligation Independently of PLCγ1
Because early activation events were apparently intact in WAVE2-suppressed cells, we next analyzed downstream signaling pathways that could regulate IL-2p activity. Interestingly, WAVE2 suppression did not affect TCR-induced phosphorylation of PLCγ1 on tyrosine 783 (Figure 5B). Consistent with the uncompromised activation of PLCγ1, WAVE2 suppressed cells were unaffected in their ability to activate both Erk and Jnk (Figures 5C and 5D), whereas T cells lacking PLCγ1 demonstrate defects in the activation of both pathways (Figures S6A and S6B). Consistent with our observation that Erk, Jnk, and PKC-stimulated pathways are intact in WAVE2-suppressed cells, these cells efficiently activated both isolated AP-1 and NFκB reporter constructs in response to TCR and CD28 ligation (Figures 5E and 5F). These data, along with the finding that functional PLCγ1 is required to activate AP-1-mediated gene transcription (Figure S6C), further support the notion that PLCγ1 activation is not compromised in WAVE2-suppressed T cells.
Because IL-2 transcription is both DAG and Ca2+ dependent, we next examined the effect of WAVE2 suppression on downstream steps involved in Ca2+ regulation. Surprisingly, bulk cell measurement of intracellular calcium using the flow cytometer indicated that WAVE2-suppressed cells have significantly diminished TCR-induced calcium signals (Figure 6A). Two distinct sources of Ca2+ feed into the cytoplasm after TCR-in-duced production of IP3. Initially, IP3 triggers Ca2+ release from intracellular stores, believed to be within the endoplasmic reticulum (ER). The resulting decrease in intraluminal ER Ca2+ concentration then triggers activation of calcium entry channels in the plasma membrane termed CRAC channels. Single cell calcium measurements were performed to pinpoint the location of the WAVE2 effect. First, lymphocytes were stimulated in Ca2+-free bath solution to evaluate intracellular release and then perfused with Ca2+-containing bath solution to examine the activation state of CRAC channels. Consistent with our data suggesting that PLCγ1 activation is not affected by WAVE2 suppression, TCR-induced (IP3- mediated) ER store release was indistinguishable between control and WAVE2-suppressed Jurkat T cells (Figure 6B, 0 mM Ca2+). However, the secondary elevation due to extracellular Ca2+ entry in WAVE2 suppressed T cells was significantly diminished (Figure 6B, 2 mM Ca2+). This diminished Ca2+ entry could result from alterations in the Ca2+ content or homeostasis of stores, defective coupling between intracellular stores and CRAC channels, or decreased Ca2+ permeation of CRAC channels. We examined this first mechanism by treating cells with anti-CD3 in Ca2+-free bath solution to physiologically induce Ca2+ release, and then thapsigargin, an inhibitor of the SERCA pump responsible for Ca2+ uptake into stores, to deplete any remaining Ca2+ in the ER. We found no significant thapsigargin-releasable calcium in control or WAVE2-suppressed cells after CD3 stimulation, suggesting that WAVE2 does not modulate the Ca2+ content of stores or its IP3-mediated release. Notably, thapsigargin also did not rescue Jurkat cells from WAVE2 suppression of CD3-induced Ca2+ entry (Figure 6C), suggesting that WAVE2 regulates CRAC channel activation subsequent to Ca2+ store depletion. In fact, when store depletion was pharmacologically induced (i.e., with thapsigargin alone), an identical defect in Ca2+ entry was seen in WAVE2-suppressed Jurkat cells after Ca2+ release from stores as with anti-CD3 (Figure 6D). These data indicate that WAVE2 regulates a step in TCR-stimulated calcium entry at a point distal to the activation of PLCγ1 and IP3-stimulated Ca2+ release from stores.
Figure 6.
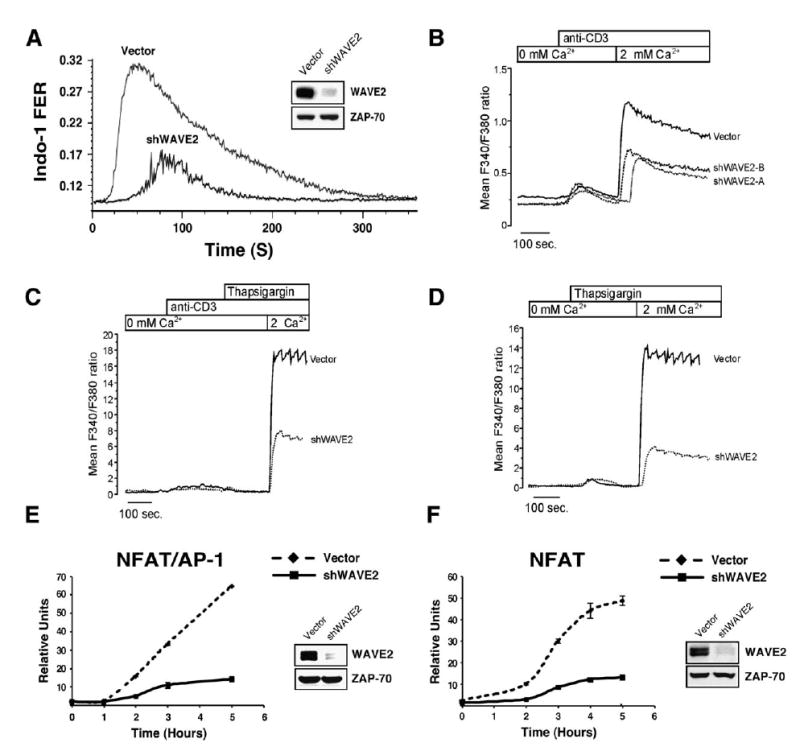
WAVE2 Regulates CRAC Channel Activation Leading to NFAT-Mediated Gene Transcription
(A) Jurkat T cells were transfected with vector control or shWAVE2 suppression vector. After 72 hr, calcium mobilization in response to OKT3/ goat anti-mouse was measured using Indo-1 staining and flow cytometry.
(B) Jurkat T cells were transfected as in (A). After 72 hr, calcium measurements were performed using single cell fluorescence ratio (Fura-2 imaging) of GFP positive control and WAVE2 suppressed Jurkat cells. In calcium-free bath solution, increases in Fura-2 ratio reflect Ca2+ release from intracellular stores. Extracellular Ca2+ entry via activated CRAC channels was subsequently assessed by reintroduction of extracellular calcium (2 mM Ca2+). Shown are representative examples of three independent experiments for each condition and each trace represents the average response of at least 100 cells in the recording chamber.
(C and D) Cells were prepared and analyzed as in (B) and stimulated as indicated above the graphs.
(E and F) Jurkat T cells were transfected with vector control or shWAVE2 suppression vector and either a (E) NFAT/AP-1 or (F) NFAT luciferase reporter. 72 hr post transfection, cells were stimulated with OKT3 and anti-CD28 for 2, 3, 4, and 5 hr. Luciferase activity was measured at each time point and normalized to TK-Renilla readings. Shown are representative examples of three independent experiments for each.
WAVE2 Regulates NFAT-Mediated Gene Transcription
In lymphocytes, NFAT activation requires sustained elevations in cytoplasmic calcium associated with extracellular calcium entry [23]. During T cell activation, a rise in intracellular calcium leads to activation of the phosphatase calcineurin, which mediates NFAT dephosphorylation and nuclear translocation, leading to gene transcription [24]. To verify the hypothesis that WAVE2 plays a role in Ca2+-regulated NFAT activation, we next conducted a luciferase reporter assay using the NFAT/AP-1 binding site of the IL-2p. In agreement with the defect in Ca2+ signaling, WAVE2-suppressed cells demonstrated a marked decrease in the activation of NFAT/AP-1 (Figure 6E). To further demonstrate that NFAT activation is decreased in the absence of WAVE2, we tested the activation of an NFAT-only binding site from the IFN-γ promoter. As expected, Jurkat T cells lacking WAVE2 failed to activate the NFAT-only reporter construct compared to control transfected cells (Figure 6F). These results indicate that WAVE2 is an important component of the T cell signaling pathway controlling calcium mobilization, leading to NFAT-mediated gene transcription.
Discussion
The actin cytoskeleton participates in organizing and maintaining signaling complexes and pathways emanating from cell surface receptors, including multisubunit immune recognition receptors on T and B lymphocytes. However, it remains unclear exactly how individual actin regulatory proteins contribute to this process and whether they participate in T cell activation apart from their prescribed roles as actin regulators. Herein, we describe a specific role for WAVE2, a protein closely related to WASp, in T cell activation. We demonstrate that WAVE2 plays an indispensable role in TCR-mediated actin cytoskeletal dynamics and integrin activation leading to proper conjugate formation. In addition, we define an unexpected role for WAVE2 in facilitating TCR-stimulated CRAC channel activation. Taken together, our data identify WAVE2 as a critical component for T cell activation.
The data presented here demonstrate that WAVE2 is an essential regulator of the actin cytoskeleton in T lymphocytes. In fact, although it is agreed that WASp plays a critical role in T cell activation [4], our data suggest that actin responses may be more dependent on the function of WAVE2 than WASp. Studies in WASp-deficient mice have yielded mixed results about the role of WASp in actin remodeling at the IS [5, 6, 16, 25, 26], and we show here that Jurkat T cells depleted of WASp with shRNA exhibit grossly normal actin repsonses. In contrast, parallel studies in WAVE2-suppressed T cells show defects in conjugate formation, TCR-activated binding to fibronectin, and actin polymerization in response to TCR ligation. WAVE proteins were initially identified as downstream effectors of Rac1 that localize to newly established lamellipodia and play a critical role in the formation of these structures [27, 28]. Similarly, we find that WAVE2 not only localizes to the leading edges of spreading T cells in response to TCR ligation (J.C.N. and D.D.B., unpublished data) but also is required for cell spreading, a phenomenon requiring F-actin polymerization.
Our studies also show that WAVE2 and HEM-1 are required for activation-dependent adhesion of T cells via both β1 and β2 integrins. The actin cytoskeleton is thought to impact integrin function by driving membrane remodeling and acting as a platform for integrins through interactions with actin-associated molecules, such as α-actinin, talin, vinculin, and filamin [29]. Indeed, T cells treated with the actin destabilizing agent cytochalasin D show an inability to form conjugates [30]. In addition, T cells lacking the Rho Family guanine nucleotide exchange factor (GEF) Vav1 fail to form stable, high-avidity conjugates and do not adhere efficiently to β1 integrin ligands such as fibronectin [6]. Vav1 is activated in response to TCR stimulation and can activate both Rac1 and Cdc42. Therefore, it is possible that activation of Rac1 by Vav1 is needed to direct WAVE2-induced lamellipodia, leading to integrinmediated conjugate formation in T cells. In fact, it was recently demonstrated that PIR121 is involved in T cell binding to fibronectin [31]. We note however, that not all hematopoietic actin regulatory molecules are required for conjugate formation. For example, Jurkat T cells lacking Dynamin 2 or the cortactin homolog HS1 show no defect in their ability to form stable T cell-APC conjugates, but these conjugates fail to exhibit the typical robust accumulation of F-actin at the IS [32] (T.S.Gomez et al., submitted). Similarly, under certain conditions, murine T cells lacking WASp aggregate integrins effectively and form stable conjugates with peptide specific APCs [6]. Thus, WAVE2 likely functions to regulate integrin-mediated adhesion via its effects on actin polymerization in a manner that is specific and distinct from other actin regulatory proteins. Further studies will be required to determine the mechanism by which WAVE2 regulates integrinmediated adhesion in T cells and to understand why some actin regulatory molecules affect integrin function, whereas others apparently do not.
WAVE proteins exist in a tight complex containing Nap1/HEM-1, Abi-1/2, Sra-1/PIR121, and HSPC300. The integrity of this complex is dependent upon all members because elimination of any single subunit leads to the subsequent degradation of WAVE protein [18–20, 33–35]. Interestingly, Nap1 appears to be absent from the WAVE2 complex in T lymphocytes. However, HEM-1 is a hematopoietic specific homolog of Nap1 [36], and little is known about its functional significance in T lymphocytes.
Although early studies suggested that Rac1 binding to Sra-1/PIR121 disrupted the complex leading to an active WAVE/HSPC300 subunit [9], our data demonstrate that the WAVE2 complex remains intact for at least 30 min after TCR stimulation. This finding is in agreement with recent evidence concluding that an intact WAVE complex is fully functional [11, 12] and that each of these proteins participates in the regulation of WAVE actin nucleating activity. However, the exact regulatory function that each of these proteins performs in T cells remains to be completely elucidated.
In addition to Rac-mediated activation, many modes of regulation have been suggested for the WAVE complex. Another possible mode of regulation involves phosphorylation. Phosphorylation of WAVE1 by Erk has been reported to occur downstream of growth factor receptors [37]. More recently, it was found that WAVE2’s actin polymerizing activity could be enhanced by c-Abl-mediated tyrosine phosphorylation, an event apparently dependent on Abi-1-mediated Abl recruitment to WAVE2 [38]. In spite of this, we have been unable to detect tyrosine phosphorylation of WAVE2 in response to TCR stimulation (J.C.N. and D.D.B., unpublished data). However, our data indicate that TCR-ligation or PMA stimulation leads to serine/threonine phosphorylation of WAVE2. Therefore, further investigation will be required to fully elucidate the functional significance of WAVE2 phosphorylation, and the regulatory mechanisms that control this process. In the long run, it seems likely that fine control of WAVE2 activation in T lymphocytes will prove to involve the convergence of several regulatory mechanisms.
The WAVE2 complex is also required for the activation of gene transcription in T cells. In particular, we find that WAVE2 is essential for calcium signaling and NFAT activation but dispensible for other pathways leading to activation of the AP-1 and NFκB transcription factors. In T cells lacking WAVE2, activation of PLCγ1 appears to be unaffected based on both phosphorylation at tyrosine 783 and full activation of the Erk and Jnk MAPK cascades. This phenotype is distinct from that of other actin regulatory proteins. For example, we recently demonstrated that the large GTPase Dynamin 2 coordinates actin reorganization at the IS. We found that T cells lacking Dynamin 2 fail to properly activate PLCγ1 and exhibit defects in calcium flux and signaling through Erk and Jnk [32]. Loss of a third actin regulatory protein, HS1, leads to normal PLCγ1 phosphorylation and Jnk activation but defects in IL-2p activation (T.S.Gomez et al., submitted). These results show that individual actin regulatory molecules play specific, nonredundant roles in TCR signaling pathways, leading to T cell activation.
One important phenotype of WAVE2-depleted cells is inhibition of CRAC channel activation in the face of normal Ca2+ release from stores. Although signaling events that link engagement of the TCR to calcium release from the ER stores is well established, how intracellular store depletion activates CRAC channels remains unresolved. This reflects the fact that the molecular identity of CRAC channels is not known. Significant evidence supports each of two different mechanisms of CRAC channel activation by store depletion [39, 40]. The first mechanism postulates that a soluble mediator, which is generated by intracellular Ca2+ store depletion, activates CRAC channels. The second mechanism involves the physical linkage of stores to CRAC channels in the plasma membrane. The most compelling data for this ‘‘conformation-coupling’’ mechanism comes from three recent studies, which suggest that STIM1 (stromal interaction molecule 1) is the long sought calcium sensor in T cells [41–43]. STIM1 suppression did not diminish TCR/IP3- mediated Ca2+ release from stores but abrogated CRAC channel activation [42]. This phenotype is similar to what we observe in WAVE2-suppressed Jurkat T cells, but how the WAVE2 complex regulates Ca2+ entry in T cells is unknown. Nonetheless, our results are consistent with the conformation-coupling model. Defining the mechanism by which WAVE2 regulates the coupling between calcium stores and CRAC channel activation is a focus of ongoing efforts.
In summary, this study demonstrates a requirement for WAVE2 in the regulation of T cell activation. We show that the WAVE2 complex not only controls actin reorganization and integrin-dependent adhesion but also is involved in signaling events coupling TCR-stimulated calcium store release to CRAC channel activation. Further analysis of this important protein and its interacting partners is likely to shed new light on the poorly understood events that coordinate cytoskeletal control, Ca2+ signaling, and activation of gene transcription during T cell activation.
Supplementary Material
Acknowledgments
This work was supported by the Mayo Foundation, a Cancer Research Institute Investigator award to D.D.B., and by National Institutes of Health grants R01-AI44835 to J.K.B., R01-AI31126 and R01-AI38474 to Y.S., R01-AI39678 and R01-AI060921 to B.D.F., and R01-AI065474 to D.D.B.
Footnotes
Experimental Procedures
See the Supplemental Data available with this article online.
References
- 1.Holsinger LJ, Graef IA, Swat W, Chi T, Bautista DM, Davidson L, Lewis RS, Alt FW, Crabtree GR. Defects in actin-cap formation in Vav-deficient mice implicate an actin requirement for lymphocyte signal transduction. Curr Biol. 1998;8:563–572. doi: 10.1016/s0960-9822(98)70225-8. [DOI] [PubMed] [Google Scholar]
- 2.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 3.Cantrell DA. GTPases and T cell activation. Immunol Rev. 2003;192:122–130. doi: 10.1034/j.1600-065x.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- 4.Thrasher AJ. WASp in immune-system organization and function. Nat Rev Immunol. 2002;2:635–646. doi: 10.1038/nri884. [DOI] [PubMed] [Google Scholar]
- 5.Cannon JL, Burkhardt JK. Differential roles for Wiskott-Aldrich syndrome protein in immune synapse formation and IL-2 production. J Immunol. 2004;173:1658–1662. doi: 10.4049/jimmunol.173.3.1658. [DOI] [PubMed] [Google Scholar]
- 6.Krawczyk C, Oliveira-dos-Santos A, Sasaki T, Griffiths E, Ohashi PS, Snapper S, Alt F, Penninger JM. Vav1 controls integrin clustering and MHC/peptide-specific cell adhesion to antigen-presenting cells. Immunity. 2002;16:331–343. doi: 10.1016/s1074-7613(02)00291-1. [DOI] [PubMed] [Google Scholar]
- 7.Takenawa T, Miki H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J Cell Sci. 2001;114:1801–1809. doi: 10.1242/jcs.114.10.1801. [DOI] [PubMed] [Google Scholar]
- 8.Suetsugu S, Miki H, Takenawa T. Identification of two human WAVE/SCAR homologues as general actin regulatory molecules which associate with the Arp2/3 complex. Biochem Biophys Res Commun. 1999;260:296–302. doi: 10.1006/bbrc.1999.0894. [DOI] [PubMed] [Google Scholar]
- 9.Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002;418:790–793. doi: 10.1038/nature00859. [DOI] [PubMed] [Google Scholar]
- 10.Gautreau A, Ho HY, Li J, Steen H, Gygi SP, Kirschner MW. Purification and architecture of the ubiquitous Wave complex. Proc Natl Acad Sci USA. 2004;101:4379–4383. doi: 10.1073/pnas.0400628101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Innocenti M, Zucconi A, Disanza A, Frittoli E, Areces LB, Steffen A, Stradal TE, Di Fiore PP, Carlier MF, Scita G. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004;6:319–327. doi: 10.1038/ncb1105. [DOI] [PubMed] [Google Scholar]
- 12.Steffen A, Rottner K, Ehinger J, Innocenti M, Scita G, Weh-land J, Stradal TE. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. EMBO J. 2004;23:749–759. doi: 10.1038/sj.emboj.7600084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan C, Martinez-Quiles N, Eden S, Shibata T, Takeshima F, Shinkura R, Fujiwara Y, Bronson R, Snapper SB, Kirschner MW, et al. WAVE2 deficiency reveals distinct roles in embryogenesis and Racmediated actin-based motility. EMBO J. 2003;22:3602–3612. doi: 10.1093/emboj/cdg350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamazaki D, Suetsugu S, Miki H, Kataoka Y, Nishikawa S, Fujiwara T, Yoshida N, Takenawa T. WAVE2 is required for directed cell migration and cardiovascular development. Nature. 2003;424:452–456. doi: 10.1038/nature01770. [DOI] [PubMed] [Google Scholar]
- 15.Badour K, Zhang J, Shi F, McGavin MK, Rampersad V, Hardy LA, Field D, Siminovitch KA. The Wiskott-Aldrich syndrome protein acts downstream of CD2 and the CD2AP and PSTPIP1 adaptors to promote formation of the immunological synapse. Immunity. 2003;18:141–154. doi: 10.1016/s1074-7613(02)00516-2. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Shi F, Badour K, Deng Y, McGavin MK, Siminovitch KA. WASp verprolin homology, cofilin homology, and acidic region domain-mediated actin polymerization is required for T cell development. Proc Natl Acad Sci USA. 2002;99 :2240–2245. doi: 10.1073/pnas.042686099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orange JS, Ramesh N, Remold-O’Donnell E, Sasahara Y, Koopman L, Byrne M, Bonilla FA, Rosen FS, Geha RS, Strominger JL. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc Natl Acad Sci USA. 2002;99:11351–11356. doi: 10.1073/pnas.162376099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogers SL, Wiedemann U, Stuurman N, Vale RD. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J Cell Biol. 2003;162:1079–1088. doi: 10.1083/jcb.200303023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunda P, Craig G, Dominguez V, Baum B. Abi, Sra1, and Kette control the stability and localization of SCAR/ WAVE to regulate the formation of actinbased protrusions. Curr Biol. 2003;13:1867–1875. doi: 10.1016/j.cub.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Grove M, Demyanenko G, Echarri A, Zipfel PA, Quiroz ME, Rodriguiz RM, Playford M, Martensen SA, Robinson MR, Wetsel WC, et al. ABI2-deficient mice exhibit defective cell migration, aberrant dendritic spine morphogenesis, and deficits in learning and memory. Mol Cell Biol. 2004;24:10905–10922. doi: 10.1128/MCB.24.24.10905-10922.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Kooyk Y, Figdor CG. Avidity regulation of integrins: the driving force in leukocyte adhesion. Curr Opin Cell Biol. 2000;12:542–547. doi: 10.1016/s0955-0674(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 22.Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE. Dynamic actin polymerization drives T cell receptor-induced spreading: a role for the signal transduction adaptor LAT. Immunity. 2001;14:315–329. doi: 10.1016/s1074-7613(01)00112-1. [DOI] [PubMed] [Google Scholar]
- 23.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 24.Feske S, Okamura H, Hogan PG, Rao A. Ca2+/ calcineurin signalling in cells of the immune system. Biochem Biophys Res Commun. 2003;311:1117–1132. doi: 10.1016/j.bbrc.2003.09.174. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Shehabeldin A, da Cruz LA, Butler J, Somani AK, McGavin M, Kozieradzki I, dos Santos AO, Nagy A, Grinstein S, et al. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein-deficient lymphocytes. J Exp Med. 1999;190:1329–1342. doi: 10.1084/jem.190.9.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cannon JL, Burkhardt JK. The regulation of actin remodeling during T-cell-APC conjugate formation. Immunol Rev. 2002;186:90–99. doi: 10.1034/j.1600-065x.2002.18609.x. [DOI] [PubMed] [Google Scholar]
- 27.Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998;17:6932–6941. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hahne P, Sechi A, Benesch S, Small JV. Scar/ WAVE is localised at the tips of protruding lamellipodia in living cells. FEBS Lett. 2001;492:215–220. doi: 10.1016/s0014-5793(01)02239-6. [DOI] [PubMed] [Google Scholar]
- 29.Sampath R, Gallagher PJ, Pavalko FM. Cytoskeletal interactions with the leukocyte integrin beta2 cytoplasmic tail. Activation-dependent regulation of associations with talin and alpha-actinin. J Biol Chem. 1998;273:33588–33594. doi: 10.1074/jbc.273.50.33588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivas FV, O’Keefe JP, Alegre ML, Gajewski TF. Actin cytoskeleton regulates calcium dynamics and NFAT nuclear duration. Mol Cell Biol. 2004;24:1628–1639. doi: 10.1128/MCB.24.4.1628-1639.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayne M, Moffatt T, Kong H, McLaren PJ, Fowke KR, Becker KG, Namaka M, Schenck A, Bardoni B, Bernstein CN, et al. CYFIP2 is highly abundant in CD4+ cells from multiple sclerosis patients and is involved in T cell adhesion. Eur J Immunol. 2004;34:1217–1227. doi: 10.1002/eji.200324726. [DOI] [PubMed] [Google Scholar]
- 32.Gomez TS, Hamann MJ, McCarney S, Savoy DN, Lubking CM, Heldebrant MP, Labno CM, McKean DJ, McNiven MA, Burkhardt JK, et al. Dynamin 2 regulates T cell activation by controlling actin polymerization at the immunological synapse. Nat Immunol. 2005;6:261–270. doi: 10.1038/ni1168. [DOI] [PubMed] [Google Scholar]
- 33.Blagg SL, Stewart M, Sambles C, Insall RH. PIR121 regulates pseudopod dynamics and SCAR activity in Dictyostelium. Curr Biol. 2003;13:1480–1487. doi: 10.1016/s0960-9822(03)00580-3. [DOI] [PubMed] [Google Scholar]
- 34.Bogdan S, Grewe O, Strunk M, Mertens A, Klambt C. Sra-1 interacts with Kette and Wasp and is required for neuronal and bristle development in Drosophila. Development. 2004;131:3981–3989. doi: 10.1242/dev.01274. [DOI] [PubMed] [Google Scholar]
- 35.Echarri A, Lai MJ, Robinson MR, Pendergast AM. Abl interactor 1 (Abi-1) wave-binding and SNARE domains regulate its nucleocytoplasmic shuttling, lamellipodium localization, and wave-1 levels. Mol Cell Biol. 2004;24:4979–4993. doi: 10.1128/MCB.24.11.4979-4993.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hromas R, Collins S, Raskind W, Deaven L, Kaushansky K. Hem1, a potential membrane protein, with expression restricted to blood cells. Biochim Biophys Acta. 1991;1090:241–244. doi: 10.1016/0167-4781(91)90109-y. [DOI] [PubMed] [Google Scholar]
- 37.Miki H, Fukuda M, Nishida E, Takenawa T. Phosphorylation of WAVE downstream of mitogen-activated protein kinase signaling. J Biol Chem. 1999;274:27605–27609. doi: 10.1074/jbc.274.39.27605. [DOI] [PubMed] [Google Scholar]
- 38.Leng Y, Zhang J, Badour K, Arpaia E, Freeman S, Cheung P, Siu M, Siminovitch K. Abelson-interactor-1 promotes WAVE2 membrane translocation and Abelson-mediated tyrosine phosphorylation required for WAVE2 activation. Proc Natl Acad Sci USA. 2005;102:1098–1103. doi: 10.1073/pnas.0409120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prakriya M, Lewis RS. CRAC channels: activation, permeation, and the search for a molecular identity. Cell Calcium. 2003;33 :311–321. doi: 10.1016/s0143-4160(03)00045-9. [DOI] [PubMed] [Google Scholar]
- 40.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 41.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.