

Abstract
The ether-à-go-go (EAG) family of voltage-gated K+ channels contains three subfamilies, EAG, ether-à-go-go related (ERG) and ether-à-go-go like (ELK). The human ether-à-go-go related gene (hERG) K+ channel has been of significant interest because loss of function in the hERG channel is associated with a markedly increased risk of cardiac arrhythmias. The hERG channel has unusual kinetics with slow activation and deactivation but very rapid and voltage-dependent inactivation. The outer pore region of the hERG K+ channel is predicted to be different from that of other members of the voltage-gated K+ channel family. HERG has a much longer linker between the fifth transmembrane domain (SS) and the pore helix (S5P linker) compared to other families of voltage-gated K+ channels (43 amino acids compared to 14–23 amino acids). Further, the S5P linker contains an amphipathic α-helix that in hERG channels probably interacts with the mouth of the pore to modulate inactivation. The human EAG and rat ELK2 channels (hEAG and rELK2) show reduced or no inactivation in comparison to hERG channels, yet both channels are predicted to contain a similarly long S5P linker to that of hERG. In this study, we have constructed a series of chimaeric channels consisting of the S1–S6 of hERG but with the S5P α-helical region of either hEAG or rELK2, and one consisting of the S1–S6 of rELK2 but with the S5P α-helical region of hERG to investigate the role of the S5P linker in inactivation. Our studies show that charged residues on the α-helix of the S5P linker contribute significantly to the differences in inactivation characteristics of the EAG family channels. Further, individually mutating each of the hydrophilic residues on the S5P α-helix of hERG to a charged residue had significant effects on the voltage dependence of inactivation and the two residues with the greatest affect when mutated to a lysine, N588 and Q592, both lie on the same face of the S5P α -helix. We suggest that inactivation of hERG involves the interaction of this face of the S5P α-helix with a charged residue on the remainder of the outer pore domain of the channel.
The human ether-à-go-go related gene (hERG) K+ channel is a member of the voltage-gated K+ channel family (Warmke & Ganetzky, 1994) and encodes the pore-forming α-subunit of the rapid delayed rectifier K+ channel (Sanguinetti et al. 1995). The hERG channel has unusual functional characteristics under physiological conditions. During the plateau phase of the cardiac action potential, hERG passes little outward current but during repolarization passes a significantly larger outward current (Spector et al. 1996; Zhou et al. 1998; Hancox et al. 1998; Lu et al. 2001). This apparent inward rectification of hERG current is due to slow activation and deactivation kinetics but very rapid and voltage-dependent inactivation kinetics (Sanguinetti et al. 1995; Schönherr & Heinemann, 1996; Smith et al. 1996; Spector et al. 1996; Zhou et al. 1998).
Inactivation of voltage-gated potassium channels can occur by many mechanisms (Yellen, 1998). Inactivation in hERG K+ channels most closely resembles C-type (‘collapse’ of the pore) inactivation in Shaker K+ channels (Smith et al. 1996). Both are slowed by an increase in the permeant ion (K+) concentration or by extracellular tetraethylammonium (TEA) (Hoshi et al. 1991; Yellen et al. 1994; Smith et al. 1996), and both are affected by mutations in the outer pore region (Schönherr & Heinemann, 1996; Smith et al. 1996; Ficker et al. 1998). However, in contrast to C-type inactivation in Shaker channels, inactivation in hERG channels is very rapid and voltage-dependent (Schönherr & Heinemann, 1996; Smith et al. 1996; Spector et al. 1996).
Voltage-gated potassium channels are composed of four subunits, each with six transmembrane domains (S1–S6). The S5 and S6 domains along with the intervening pore loop (P domain) from each of the four subunits form the ion conductance pathway (Yellen, 2002). In early studies of the molecular basis of inactivation in hERG K+ channels, two serine residues, Ser620 and Ser631, in the P domain were shown to be crucial for inactivation (Schönherr & Heinemann, 1996; Smith et al. 1996; Ficker et al. 1998; Herzberg et al. 1998). The outer pore region of the ether-á-go-go subfamily of voltage-gated K+ channels is unique among the voltage-gated ion channel family in that it has an extra domain (∼40 residues) located between the S5 domain and the P domain (i.e. the S5P linker; Warmke & Ganetzky, 1994). The structure of the S5P linker domain was recently determined using two-dimensional NMR spectroscopy and shown to contain an amphipathic α-helix (Torres et al. 2003). Furthermore, it was shown that this amphipathic α-helix was crucial for normal inactivation (Torres et al. 2003). Thus there appear to be at least two distinct regions of the protein (i.e. the P domain and the S5P linker) that contribute to inactivation in hERG channel.
The human ether-à-go-go (EAG) family of channels consists of three subtypes: EAG, ERG and the EAG-like or ELK channels. The human ELK2 and EAG channels (hELK2 and hEAG) show reduced or no C-type-like inactivation in comparison to hERG channels (Becchetti et al. 2002; Meyer & Heinemann, 1998; Camacho et al. 2000), yet both channels contain the long S5P helix that is important for fast inactivation of hERG. To investigate the role of the S5P linker in inactivation of EAG family channels we have constructed a series of chimaeric channels consisting of the S1–S6 of hERG but with the S5P region of either hEAG or rat ELK2 (rELK2), and a chimaera consisting of the S1–S6 of rELK2 but with the S5P region of hERG. These studies show that charged residues that lie adjacent to the hydrophobic face of the α-helix of the S5P linker are important determinants of the inactivation characteristics of the EAG channel family.
Methods
Molecular biology
HERG cDNA (a gift from Gail Robertson, University of Wisconsin) was subcloned into a pBluescript vector containing the 5′ untranslated region (UTR) and 3′ UTR of the Xenopus laevisβ-globin gene (a gift from Robert Vandenberg, University of Sydney). rELK2 cDNA in pcDNA3 was a gift from Birgit Engeland (Zentrum fur Molekulare Neurobiologie, Hamburg) and hEAG cDNA in pcDNA3 was a gift from Laurent Bernheim (University of Geneva). Mutagenesis was carried out on a BstEII/Bgl II fragment (1119–1937 base pairs) of hERG using the megaprimer PCR method. Briefly, an oligonucleotide primer with the introduced mutation was used as the 5′ primer and a primer covering the Bgl II restriction site in hERG was used as the 3′ primer in the first round PCR reaction. The product of this reaction (the megaprimer) was used as the 3′ primer and a primer covering the BstE II restriction site in hERG was used as the 5′ primer in the second-round PCR reaction. The product of the second PCR reaction was digested with BstE II and Bgl II and ligated into the hERG cDNA construct. Mutation constructs were confirmed by bi-directional sequencing. cRNA was synthesized, after linearizing the plasmid with BamHI, using the mMessage mMachine kit (Ambion) according to the manufacturer's protocols. From here on chimaeric channels are referred to by the channel which forms the main body of the chimaera, followed by the channel from which the S5P sequence was taken (e.g. hERG/hEAG S5P).
Oocyte preparation
Xenopus laevis oocytes were prepared as previously described (Clarke et al. 2000). Briefly, female Xenopus laevis frogs were anaesthetized in 0.17% w/v tricaine and segments of the ovarian lobes were removed through a small abdominal incision. The follicular layer was removed by digestion for 2–3 h with 2 mg ml−1 collagenase A (Boehringer Mannheim USA) in OR-2 buffer containing (mm): NaCl 82.5, KCl 2.0, MgCl2 1.0 and Hepes 5 (pH adjusted to 7.5 with NaOH), and then rinsed with ND96 containing (mm): KCl 2.0, NaCl 96.0, CaCl2 1.8, MgCl2 1.0 and Hepes 5.0 (pH adjusted to 7.5 with NaOH). Stage V and VI oocytes were isolated, stored in tissue culture dishes containing ND96, 2.5 mm pyruvic acid sodium salt and 0.5 mm theophylline supplemented with 10 μg ml−1 gentamicin, adjusted to pH 7.5 with NaOH and incubated at 18°C. All experiments were approved by the Animal Ethics Committee of the University of Sydney. The animals were humanely killed following the final collection of oocytes.
Electrophysiology
Xenopus oocytes were injected with 5–10 ng cRNA and incubated at 18°C for 24–72 h prior to electrophysiological recordings. All experiments were undertaken at room temperature (21–22°C). Two-electrode, voltage-clamp experiments were performed using a Geneclamp 500B amplifier (Axon Instruments). Glass microelectrodes had tip resistances of 0.3–1.0 MΩ when filled with 3 m KCl. Oocytes were perfused with ND96 solution (see above). In all protocols a 10-mV step from the holding potential of −90 to −100 mV was applied for 20 ms at the start of each sweep to enable off-line leak correction. We assumed that the leak was linear in the voltage range −140 to +40 mV. Data acquisition and analysis were performed using pCLAMP software (Version 9.0, Axon Instruments) and Excel software (Microsoft Corporation) as previously described (Subbiah et al. 2004). All data are shown as means ± s.e.m
Steady-state inactivation
The voltage-dependence of inactivation was assessed using a two-step voltage protocol. The cell was depolarized from a resting membrane potential of −90 to +40 mV for 400 ms, to fully activate and inactivate the channels, and then stepped to potentials in the range +40 to −140 mV for 2 s. For high-impact charge mutants that caused large positive shifts in the half-inactivation voltage (V0.5), cells were depolarized from −90 to +80 mV before stepping from +80 to −120 mV. A single-exponential function was fitted to the deactivation phase of the tail current recordings and extrapolated back to the start of the test pulse to obtain the peak current at each voltage (see Fig. 1). Conductance values were obtained by dividing the current values by the electrical driving force (i.e. test voltage minus reversal potential) and the conductance–voltage data were fitted with a Boltzmann function:
| (1) |
Figure 1. Steady-state inactivation of EAG family channels.

A, typical example of currents recorded from wt hERG (a), wt rELK2 (b) and wt hEAG (c) channels during a two-step voltage protocol (see top trace) to measure steady-state inactivation. Arrows indicate zero current. Single exponential functions were fitted to the deactivation phase of the current traces and extrapolated back to the start of the test pulse to estimate the peak current at each voltage for both wt hERG and rELK2 (see dashed lines and asterisks in a). These data were then used to calculate conductance–voltage curves. B, conductance–voltage curve for wt hERG (▪) and wt rELK2 (•) inactivation. Data points are mean ± s.e.m and the curve fitted to the data is a Boltzmann function (see eqn (1) in the Methods) with a V0.5 of inactivation of −73 mV and slope factor of −29 mV (n = 9) for wt hERG and a V0.5 of 32 mV and a slope factor of −9 mV (n = 5) for wt rELK2.
Where g/gmax is the relative conductance, Vt is the test potential and k is the slope factor.
Steady-state activation
The voltage-dependence of activation was measured using a standard isochronal tail current protocol (Sanguinetti et al. 1995). Cells at a holding potential of −90 mV were subject to 4-s depolarizing steps to voltages in the range 80 to +50 mV before stepping the voltage to −70 mV where tail currents were recorded. Tail current data were normalized to the maximum current value (Imax) and fitted with a Boltzmann function (see eqn (1) above).
Results
Inactivation of wild-type EAG family channels
Injection of wild-type (wt) hERG into Xenopus oocytes resulted in expressed channels leading to resting membrane potentials of −56.6 ± 1.4 mV (n = 9) compared to −22 ± 1.6 mV (n = 7) in non-injected oocytes. Figure 1Aa shows a representative family of current traces from an oocyte injected with wt hERG channels during a 400-ms depolarization to +40 mV from a holding potential of −90 mV and subsequent repolarization to voltages in the range +40 to −140 mV in 10-mV steps. There is relatively small current flow at +40 mV, as hERG channels open slowly and inactivate rapidly. During the subsequent repolarization steps there is rapid recovery from inactivation and hence an increase in current is seen, followed by a slower decay in the current as the channels deactivate.
Injection of wt rELK2 and wt hEAG into Xenopus oocytes resulted in expressed channels leading to resting membrane potentials of −51.8 ± 2.7 mV (n = 12) and −60.9 ± 4.2 mV (n = 9), respectively. Although the negative resting membrane potential of injected oocytes indicated good expression of wt rELK2, currents were very small (e.g. see Fig. 1Ab) with a maximal outward recorded current (at 10 mV) of 245.4 ± 62.4 nA (n = 6) compared to 2.2 ± 0.4 μA (n = 10) maximal outward current (at −40 mV) for wt hERG and 25.1 ± 8.0 μA at +40 mV (n = 6) for wt hEAG (Fig. 1Ac). It was therefore difficult to obtain data for wt rELK2 channels, particularly at positive holding potentials. Nevertheless, there is noticeably much less inactivation seen in wt rELK2 channel recordings (Fig. 1Ab). There was no inactivation visible in wt hEAG currents (Fig. 1Ac). These results are consistent with previous studies of rELK2 and hEAG channels (Engeland et al. 1998; Occhiodoro et al. 1998; Ganetzky et al. 1999; Bauer & Schwarz, 2001; Becchetti et al. 2002).
Steady-state inactivation curves were derived from the current recordings of wt hERG and wt rELK2 by first fitting single exponentials to the deactivation phase of the tail currents (see dashed lines in Fig. 1Aa) and extrapolating these fits back to the start of the test pulse to obtain the peak current at each voltage (see asterisks in Fig. 1Aa). The resulting current–voltage relationships can be converted to conductance–voltage relationships by dividing the current by the driving force (test voltage minus reversal potential). The curve fitted to the data in Fig. 1B is a Boltzmann function eqn (1)). Plotting the conductance of the channel against voltage clearly highlights the fact that maximal conductance of wt hERG is seen at very negative voltages while the channel is almost non-conducting at +40 mV (see Fig. 1B). Inactivation in wt rELK2 is greatly reduced compared to wt hERG with an apparent V0.5 of 31.8 ± 3.4 mV and a slope factor of −9.2 ± 1.7 mV (n = 5) compared to V0.5 of −73.0 ± 3.9 mV and a slope of −29.9 ± 1.3 mV (n = 9) for wt hERG. The mean (± s.e.m) data for the V0.5 and slope factors for steady-state inactivation (and activation, see below) for all channels investigated in this study are summarized in the Supplemental material.
Inactivation of hERG/hEAG and hERG/rELK2 chimaeras
We (Torres et al. 2003) and others (Liu et al. 2002; Jiang et al. 2005) have suggested a role for the long S5P extracellular linker, and in particular an α-helical region of 10 amino acids within the linker (see Fig. 2), in hERG inactivation. HERG, hEAG and rELK2 are all predicted to contain an α-helical region within their S5P linkers (see upper panel in Fig. 2), yet each of these EAG family members do not inactivate in the same manner. To study the exact role of the S5P linker region in hERG, hEAG and rELK2 channel inactivation, we created chimaeras consisting of the S1–S6 regions of wt hERG and the S5P linker region of either hEAG (hERG/hEAG S5P) or rELK2 (hERG/rELK2 S5P).
Figure 2. EAG family S5P chimaera characteristics.

Upper panel, alignment of the residues of the S5P linker of hERG, rELK2 and hEAG is shown at the top of the figure below a cartoon depicting wt hERG structure. The demonstrated α-helical region (Torres et al. 2003) is boxed. The charged residues of interest within the α-helical region of rELK2 are highlighted. Aa, steady-state inactivation of hERG/hEAG S5P chimaera. Typical example of currents recorded from hERG/hEAG S5P channels during a steady-state inactivation voltage protocol (see Fig. 1A) are shown. Arrow indicates zero current. Ab, the conductance–voltage plot of hERG/hEAG S5P (•). The curve fitted to the hERG/hEAG S5P data is a Boltzmann function with a V0.5 of inactivation of −57 mV and a slope factor of −31 mV (n = 5). The dotted line shows the Boltzmann fit to the wt hERG data from Fig. 1B. Ba, typical example of currents recorded from hERG/rELK2 S5P chimaera channels during a steady-state inactivation voltage protocol (see Fig. 1A). Arrow indicates zero current. Bb, the conductance–voltage plot of hERG/rELK2 S5P (•) compared with wt hERG (dotted line) and wt rELK2 (•). The curve fitted to the hERG/rELK2 S5P data is a Boltzmann function with a V0.5 of inactivation of 27 mV and a slope factor of −34 mV (n = 4). The data for wt hERG and wt rELK2 are reproduced from Fig. 1B.
hERG/hEAG S5P
Injection of hERG/hEAG S5P cRNA into Xenopus oocytes resulted in channel expression leading to resting membrane potentials of −59.0 ± 2.2 mV (n = 9). Steady-state inactivation protocols produced currents that resembled wt hERG (see Fig. 2Aa and compare to Fig. 1Aa), with reversal potentials of −93 ± 3.7 mV (n = 5), although the voltage dependence of inactivation was slightly shifted to the right (Fig. 2Ab). The currents rapidly inactivated at positive potentials with a V0.5 of −57.4 ± 4.1 mV and a slope of −30.8 ± 1.6 mV (n = 5) compared to V0.5 of −70.3 ± 6.1 mV and a slope of − 27.9 ± 0.8 mV (n = 6) for wt hERG (Fig. 2Ab).
These data suggest that changes in the residues of the S5P region of hEAG compared to those of hERG are not the reason for the lack of inactivation seen in wt hEAG channels, which is consistent with previous observations by Ficker et al. (1998) and Herzberg et al. (1998).
hERG/rELK2 S5P
Injection of hERG/rELK2 S5P RNA into Xenopus oocytes resulted in channel expression leading to resting membrane potentials of −48.8 ± 1 mV (n = 6). Steady-state inactivation protocols produced currents that looked essentially like wt rELK2 (see Fig. 2Ba and compare to Fig. 1Ab) with reversal potentials of −87 ± 2.5 mV (n = 5). The currents were larger than that observed for wt rELK2 (maximal current at −10 mV was 0.58 ± 0.04 μA, n = 5, compared to 0.25 ± 0.6 μA, for wt rELK2) although still not as large as seen with wt hERG (2.2 ± 0.4 μA). Inactivation in hERG/rELK2 S5P showed a V0.5 of 27.3 ± 0.3 mV and a slope of − 34.4 ± 1.2 mV (n = 4). These values are much more similar to that observed for wt rELK2 (see Fig. 1B) and clearly very different to that for wt hERG (see Fig. 1A and Supplemental material). These data suggest that residues within the S5P linker of rELK2 channels are importantly involved in its altered inactivation characteristics.
Rapid inactivation is ‘gained’ in the rELK2/hERG S5P chimaera
To investigate whether the inactivation characteristics of hERG could be transferred to rELK2, we created a chimaera consisting of the S1–S6 of rELK2 with the S5P linker of hERG. Injection of the rELK2/hERG S5P chimaera into Xenopus oocytes resulted in channel expression leading to resting membrane potentials of −76.7 ± 4.0 mV (n = 5). Currents were small (maximal current, 48 ± 2 nA at −50 mV n = 5) and showed retained K+ selectivity with reversal potentials of −99 ± 3.5 mV. Steady-state inactivation protocols produced currents that more closely resembled wt hERG than wt rELK2 (compare Fig. 3A with Fig. 1Aa and Ab). The currents rapidly inactivated but with two apparent components to inactivation. This can be seen from the ‘m’ shaped current–voltage curve in Fig. 3B, with the first component spanning −140 to ∼0 mV, and accounting for over 90%, and the second component from ∼0 to +40 mV (Fig. 3B). The first component closely resembled wt hERG (Fig. 3C) with a V0.5 of −80.0 ± 4.2 mV and a slope factor of −22.6 ± 2.3 mV (n = 5). While the second component, resembled wt rELK2 with an apparent V0.5 of 28.5 ± 2.2 mV and a slope factor of −8.7 ± 4.1 mV (n = 5, Fig. 3D).
Figure 3. Steady-state inactivation of rELK2/hERG S5P.
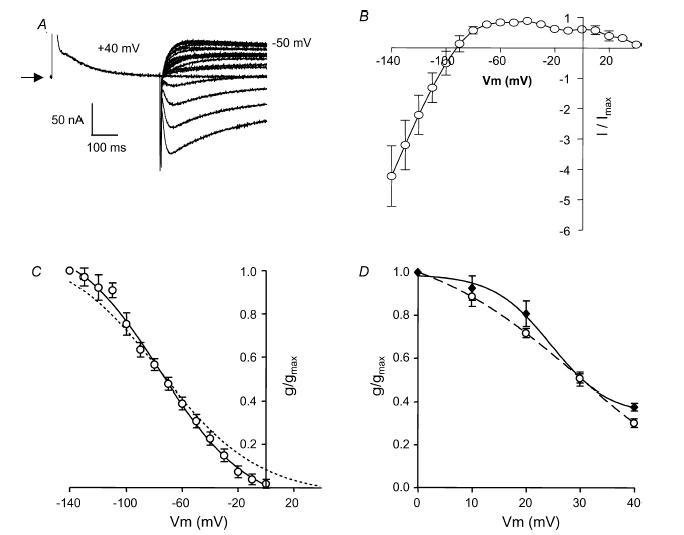
A, typical example of currents recorded from rELK2/hERG S5P channels during a steady-state inactivation voltage protocol (see Fig. 1A). Arrow indicates zero current. B, steady-state inactivation curve for rELK2/hERG S5P indicated the presence of more than one component to inactivation (n = 5); that is the curve appears to be ‘m’-shaped rather than ‘n’-shaped as is the case for wt hERG (see Fig. 6 below). The first component spans the region −140 to ∼0 mV and the second component from 0 to +40 mV C, the conductance–voltage plot of the ‘first component’ of rELK2/hERG S5P inactivation (○) compared to that of wt hERG (dotted line, for data from Fig. 1B). The curve fitted to the data is a Boltzmann function with a V0.5 of inactivation of −80 mV and a slope factor of −23 mV (n = 5). D, the conductance–voltage plot of the ‘second component’ of rELK2/hERG S5P inactivation (open circles) compared to that of wt rELK2 (♦, reproduced from Fig. 1b). The curve fitted to the rELK2/hERG S5P data is a Boltzmann function with a V0.5 of inactivation of 29 mV and slope factor of −9 mV (n = 5).
The results from this rELK2/hERG S5P chimaera suggest that there is more than one component to C-type-like inactivation in EAG family K+ channels, with one that involves the S5P linker and is voltage sensitive in the negative membrane potential range and another as yet unidentified component that occurs at much more positive potentials.
The structure of the amphipathic helix in the S5P linker is predicted to be the same in the hERG/rELK2 S5P chimaera as in wt hERG (JPred prediction: http://www.compbio.dundee.ac.uk, data not shown) and the hydrophobic residues in this segment are highly conserved (see upper panel in Fig. 2). There are, however, significant variations in the hydrophilic residues. N588 in hERG is replaced with a glutamate in rELK2 and D591 and Q592 are replaced with arginines (see Fig. 2). To test whether these differences could account for the different inactivation characteristics conferred by the S5P linker of hERG and rELK2, we created a hERG N588E point mutant and a hERG D591R/Q592R double mutant and studied their inactivation characteristics.
Charged residues on the S5P linker affect inactivation
Expression of hERG N588E produced rapidly inactivating currents (Fig. 4Aa). Steady-state current–voltage plots showed that the mutation shifted the voltage dependence of inactivation to the left compared to wt hERG, with an apparent V0.5 of −109 ± 5.2 mV and slope factor of −17.3 ± 1.5 mV (n = 5; Fig. 4Ab), while K+ selectivity was retained (reversal potential, −98 ± 1.8 mV, n = 5).
Figure 4. Steady-state inactivation of hERG N588E and hERG D591R/Q592R.
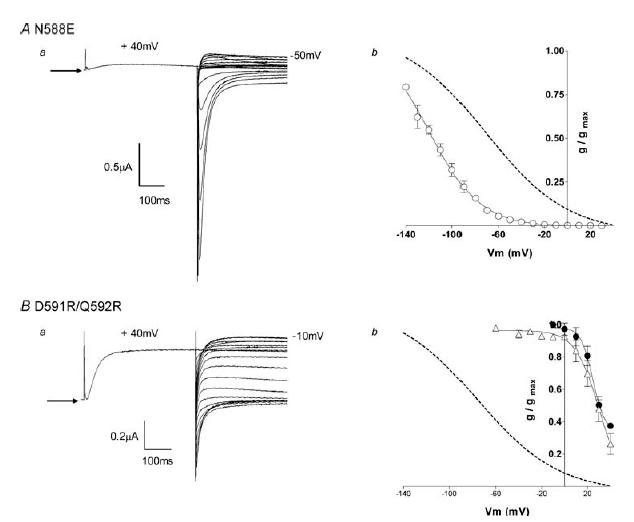
Aa, typical example of currents recorded from hERG N588E channels during a steady-state inactivation voltage protocol (see Fig. 1A). Arrow indicates zero current. Ab, the conductance–voltage plot of hERG N588E (○) compared with wt hERG (dotted line). The curve fitted to the hERG N588E data is a Boltzmann function with a V0.5 of inactivation of −109 mV and slope factor of −17 mV (n = 5). Data for wt hERG (dotted line) are reproduced from Fig. 1B. Ba, typical example of currents recorded from hERG D591R/Q592R channels during a steady-state inactivation voltage protocol (see Fig. 1A). Arrow indicates zero current. Bb, the conductance–voltage plot of D591R/Q592R (▵) compared with wt hERG (dotted line) and wt rELK2 (•). The curve fitted to the hERG D591R/Q592R data is a Boltzmann function with a V0.5 of inactivation of 29 mV and a slope factor of −10 mV (n = 5). The data for wt hERG and wt rELK2 are reproduced from Fig. 1B.
The hERG D591R/Q592R double mutant, when expressed in Xenopus oocytes, displayed current characteristics similar to both wt rELK2 and hERG/rELK2 S5P (compare Fig. 4Ba with Fig. 1Ab and Fig. 2Ba). Inactivation was shifted to the right in comparison with wt hERG and resembled wt rELK with an apparent V0.5 of inactivation of +28.7 ± 3.3 mV and slope factor of −10.2 ± 1.4 mV (n = 5, Fig. 4Bb). Reversal potentials indicated retained K+ selectivity (−89 ± 4.4 mV, n = 5).
The origin of the voltage sensitivity of inactivation in hERG channels is a hotly debated topic. Until recently it was thought that the voltage sensitivity of inactivation was not linked to the voltage sensitivity of activation. For example, the mutation S631A causes a marked shift in the voltage sensitivity of inactivation but no change in the voltage sensitivity of activation (Zou et al. 1998). Similarly, changes in [Ca2+]o (Johnson et al. 1999) and temperature (Vandenberg et al. 2006) have very distinct effects on activation and inactivation. However, based on a combination of gating-current measurements and computer modelling, Piper et al. (2003) have suggested that the voltage sensitivity of inactivation is linked to the voltage sensitivity of activation. Given the marked shifts in the voltage sensitivity of inactivation in the S5P chimaera channels, we investigated the effects these chimaeras have on steady-state activation. Figure 5 summarizes mean data for the voltage dependence of activation for wt hERG, hERG/hEAG S5P, hERG/rELK2 S5P, and N588E and D591R/Q592R hERG channels. The values for the V0.5 of activation were not statistically different for any of the chimaera channels compared to wt hERG.
Figure 5. Steady-state activation for hERG/hEAG/rELK2 chimaeras.
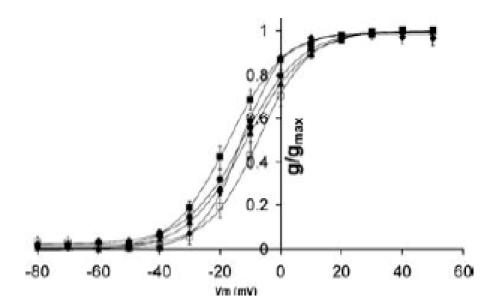
Conductance–voltage curve for wt hERG (▪), hERG/hEAG S5P chimera (▴), hERG/rELK2 S5P chimera (♦), hERG N588E (•) and hERG D591R/Q592R (□) activation. Data points are mean ± s.e.m and the curves fitted to the data are Boltzmann functions. None of the chimaeras caused a significant shift in steady-state activation compared to wt hERG. Full data for V0.5 and slope of the Boltzmann fits are listed in the Supplemental material.
Charge scanning mutagenesis of the hERG S5P α-helix
The data from the chimaera channels presented above strongly suggest that altering charge on the α-helix segment of the S5P linker in hERG affects the voltage dependence of inactivation. However, given the small currents associated with some of the mutant hERG channels, especially the hERG/rELK2 S5P chimaera, it is difficult to make quantitative predictions based on these data. To investigate the influence of charge on the hERG S5P α-helix in more detail we mutated each of the hydrophilic residues in the α-helix to either a negatively charged amino acid (aspartate) or a positively charged amino acid (lysine). Aspartate and lysine were chosen as they are the least bulky of the negatively and positively charged amino acids. Figure 6 illustrates typical examples of currents recorded from each of the hERG S5P α-helix lysine mutants, H587K, N588K, D591K and Q592K. In the case of N588K and Q592K, the voltage dependence of inactivation was shifted to the right by over +100 mV, necessitating a change in the voltage protocol to measure inactivation (see Methods and legend to Fig. 6). The effect of each of the mutations (both lysine and aspartate) on steady-state inactivation are summarized in Fig. 6Ba and Bb. The values for V0.5 and the slope factors for steady-state inactivation are shown in the Supplemental material. Mutation of N588 had the most marked effects with a shift of −34 mV in the V0.5 of inactivation for the aspartate mutation and of +140 mV for the lysine mutation. Mutation of D591 showed an intermediate phenotype; no effect for the neutral glutamine mutation but a shift of +84 mV for the lysine mutation. Mutation of Q592 to lysine resulted in a shift of +135 mV, which is similar to that shown above for the hERG/rELK2 chimaera (compare Figs 2B and 6B), whereas mutation to aspartate resulted in no significant change in the V0.5 of inactivation. Mutation of H587 to either lysine or aspartate caused small but significant negative shifts in the V0.5 of steady-state inactivation. It is worth noting that mutation of N588 to aspartate or glutamate had very similar effects on steady-state inactivation. Similarly, mutation of Q592 to lysine had a very similar effect to the Q592R/D591R mutation on steady-state inactivation. Thus it would appear that in these instances it is charge, rather than the side chain of the mutant that is important.
Figure 6. Steady-state inactivation for charge scan mutants of hERG S5P α-helix.
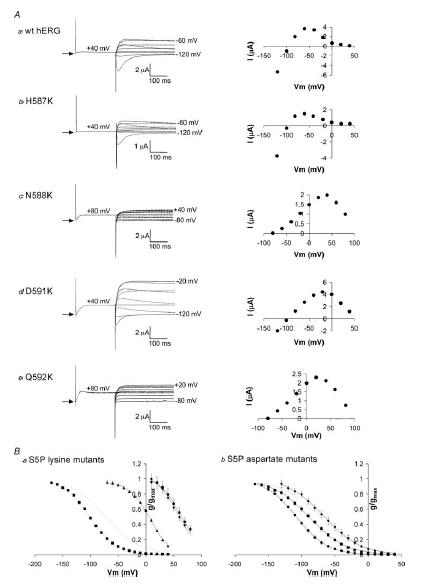
A, typical examples of currents recorded from wt hERG, H587K, N588K, D591K and Q592K channels during a steady-state inactivation voltage protocol (see Fig. 1A). Arrow indicates zero current. Each representative trace is accompanied by a graph illustrating the current–voltage relationship for that mutant. B, conductance–voltage curves for charge scan lysine mutants (a) and aspartate mutants (b). Data points are mean ± s.e.m for H587 (▪), N588 (•), D591 (▴) and Q592 (♦) mutants. The dotted line represents wt hERG steady-state inactivation. Curves fitted to the data are Boltzmann functions. Full data for V0.5 and slope of the Boltzmann fits are listed in the Supplemental material.
In contrast to the very marked effect some of the S5P α-helix charge scan mutants had on the voltage dependence of inactivation, these mutants had only very small effects on the voltage dependence of steady-state activation (see Fig. 7). With the exception of D591Q channel, which had a modest positive shift in the reversal potential, the S5P α-helix charge scan mutants did not show significant shifts in reversal potential (see Supplemental material).
Figure 7. Steady-state activation for charge scan mutants of hERG S5P α-helix.
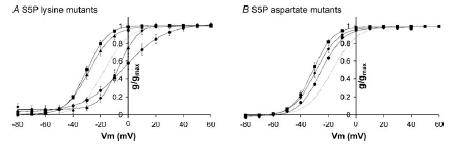
Conductance–voltage curves for charge scan lysine mutants (A) and aspartate mutants (B). Data points are mean ± s.e.m for H587 (▪), N588 (•), D591 (▴) and Q592 (♦) mutants. The dotted line represents wt hERG steady-state activation. Curves fitted to the data are Boltzmann functions. Mutation of S5P hydrophilic residues to lysine or aspartate caused only small shifts in V0.5 of steady-state activation. The largest shifts were observed with the N588K and H587D mutants which shifted steady-state activation +15 and −15 mV, respectively, compared to wt hERG. Full data for V0.5 and slope of the Boltzmann fits are listed in the Supplemental material.
Overall, the effect of the charge scan mutants indicate that N588 and Q592 were high-impact residues, D591 was an intermediate-impact residue and H587 a low-impact residue. Figure 8 illustrates a molecular model of the hERG S5P a-helix, using the NMR structure co-ordinates (PDB: accession number 1UJL, Torres et al. 2003) where the hydrophilic residues are colour-coded according to the impact of the lysine mutant on the V0.5 of steady-state inactivation (red corresponds to a V0.5 of +70 mV, and yellow to a V0.5 of −100 mV). Mutation of any of the hydrophobic residues (shown in white in Fig. 8) results in non-functional channels or severe disruption of inactivation (Liu et al. 2002). The two orthogonal views of the S5P α-helix structure show that the high-impact residues clearly line up on one face of the helix adjacent to the hydrophobic face and opposite to the low-impact face of the helix. D591 (intermediate impact) lies between the high- and low-impact surfaces.
Figure 8. Distribution of the impact of charge mutants on the three-dimensional structure of hERG S5P α-helix.
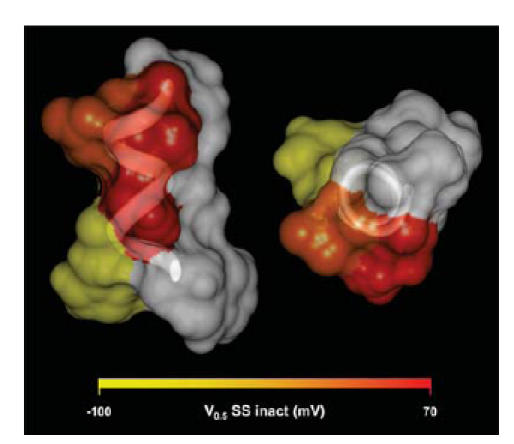
Orthogonal views of a space filling model of the hERG S5P α-helix, based on NMR structure of S5P linker (PDB: accession number 1JUL), with side chains colour-coded according to the effect lysine mutants have on the V0.5 of steady-state inactivation. Hydrophobic residues are shown in white.
Discussion
Inactivation of EAG channel family members is affected by the S5P linker
The human EAG family of channels consists of EAG, ERG and the EAG-like or ELK channels. The hELK2 and hEAG show reduced or no C-type-like inactivation in comparison to hERG channels (Meyer & Heinemann, 1998; Camacho et al. 2000; Becchetti et al. 2002), yet both channels contain the long S5P linker that is important for fast inactivation of hERG (Liu et al. 2002; Torres et al. 2003).
Previous cysteine scanning mutagenesis studies of the S5P α-helix of hERG have shown that this region is crucial for inactivation (Liu et al. 2002). The importance of the hydrophobic residues in this region was also suggested by the fact that they are almost completely conserved among all family members (see Fig. 2 and Torres et al. 2003). In this study, replacement of the S5P α-helix of hERG channels with the homologous region from hEAG led to a chimaera with inactivation characteristics very similar to those of wt hERG. This suggests that the lack of inactivation in hEAG channels is not due to residues within the S5P α-helix, as also suggested by Ficker et al. (1998) and Herzberg et al. (1998). Rather, the differences in inactivation between hERG and hEAG appear to be due to differences in pore-helix residues, namely: Ser620 which is a threonine in hEAG and Ser631 which is an alanine in hEAG (Schönherr & Heinemann, 1996; Herzberg et al. 1998; Ficker et al. 1998, 2001).
In contrast to the results with the hERG/hEAG chimaera, the replacement of hERG S5P α-helix with the homologous region of the rELK2 channel led to a chimaera with inactivation characteristics more closely resembling wt rELK2 channels than wt hERG. Furthermore, replacement of the rELK2 S5P α-helix with that of hERG led to a chimaera with inactivation characteristics more closely resembling wt hERG. The residues in rELK2 that correspond to Ser620 and Ser631 of hERG are both serines. This suggests that in the presence of these serines the hERG S5P α-helix can have significant effects on inactivation. However, in hEAG channels, the absence of those serines has a dominant effect over the S5P α-helix.
Voltage-dependence of inactivation of EAG family channels is modulated by charged residues on S5P α-helix
Most of the residue changes that occur within the α-helical region when replacing hERG S5P with rELK2 S5P involve changes in charge. These are asparagine (N) 588 to a glutamate (E) and the presence of two positively charged arginine residues (R) rather than an aspartate (D) and a glutamine (Q) at positions 591 and 592, respectively. Mutation of hERG D591 and Q592 to arginines (the homologous residues in wt rELK2) alone led to functional channels that closely resembled wt rELK2 channels. Mutation of N588 to a negatively charged glutamate (the homologous residue in rELK2 channels) gave rapidly inactivating currents, with the voltage dependence of inactivation shifted to more negative potentials. These changes for hERG N588E are in the opposite direction to those observed when the entire α-helical region of hERG was replaced with that of rELK2, suggesting that the presence of positive charges in the C-terminal half of the S5P α-helix has the dominant influence on inactivation.
To investigate in more detail the possible role of charge on the hERG S5P α-helix on the inactivation characteristics of the channel, we systematically mutated each charged hydrophilic residue in the α-helix to either a positive charge (lysine) or a negative charge (aspartate). These results indicate that altering the charge at residue N588 or Q592 had a major impact on the voltage dependence of inactivation, altering charge on D591 had an intermediate impact but altering charge on H587 had little impact (see Fig. 6). These data are entirely consistent with the hERG/hEAG and hERG/rELK2 chimaera data. In the hERG/rELK2 chimaeras, residues were changed to arginine and glutamate but in the charge scan mutagenesis residues were changed to lysine or aspartate. The similarity of the results therefore suggests that it is the charge rather than the particular side chain that is important.
It is significant that the two residues in the hERG S5P α-helix that, when mutated, had the highest impact on inactivation are located on the same face of the three-dimensional structure of the α-helix (Fig. 8). Conversely, H587, the residue where mutations had the least impact on inactivation, points in the opposite direction. Furthermore, these two faces are separated by the hydrophobic face of the hERG S5P α-helix. Based on this distribution of residues we propose that inactivation of hERG channels involves the interaction of the S5P α-helix with the remainder of the pore domain of the channel, with the surface formed by N588, Q592 and the adjacent hydrophobic residues forming the interaction surface on the S5P α-helix (see Fig. 8).
There are three additional points of interest from the charge scanning mutagenesis data. Firstly, with the exception of the D591Q channel, which had a modest positive shift in the reversal potential, the S5P α-helix charge scan mutants did not show significant shifts in reversal potential (see Supplemental material). This is in contrast to the majority of the S5P α-helix cysteine that had marked changes in reversal potential (Liu et al. 2002). It has subsequently been shown that the majority of these S5P α-helix cysteine mutants form intersubunit disulphide bonds (Jiang et al. 2005) and thus have significantly disrupted outer pore structure. In the charge scan mutants, the maintenance of selectivity for K+ over Na+ suggests that none of these mutants have caused a major perturbation to the outer pore structure of the channel.
Secondly, in our hands we found that mutation of H587 to lysine had only a small impact on inactivation, whereas a previous study reported that this mutation resulted in abolition of inactivation (Dun et al. 1999). We have since tested the H587K clone used by Dun et al. (1999) and found that it contains a second mutation G604D, which, based on a previous analysis by this group of the entire S5P linker region, is a high-impact position (Liu et al. 2002) and so could account for the phenotype in their clone. However, it is important to point out that whether H587K has a small negative shift in the V0.5 of steady-state inactivation (our data) or disrupts inactivation (Dun et al. 1999) does not impact on any of the previous conclusions with regard to the role of H587 in HERG function (i.e. the S5P linker is important for inactivation; Dun et al. 1999; Liu et al. 2002) and H587 is not involved in H+ sensing (Jiang et al. 1999). The nature of the phenotype for H587K does, however, have important consequences for our study. The distribution of the high- and low-impact positions on the three-dimensional structure of the S5P α-helix is clearly much more consistent with H587K having little effect on steady-state inactivation (see Fig. 8).
Thirdly, charge mutations of N588 have been reported in patients with long QT syndrome (N588D, Splawski et al. 1998) and short QT syndrome (N588K, Brugada et al. 2004). There are no previously reported data for the cellular phenotype of N588D. Our data are consistent with a loss of function; that is, the negative shift in V0.5 of inactivation would mean that the channels would be almost completely inactivated at the normal resting membrane potential. The most significant effect we observed for the N588K mutation, a large positive shift in V0.5 of inactivation, is consistent with previous reports (Cordeiro et al. 2005; McPate et al. 2005) and again consistent with the clinical phenotype, in this case short QT syndrome.
Summary
The data presented here, clearly confirm an important role for the S5P α-helix in inactivation of hERG K+ channels. Clearly, this cannot be the whole story as Ser631 (Schönherr & Heinemann, 1996; Smith et al. 1996) and Ser620 (Ficker et al. 1998; Herzberg et al. 1998) are also very important for inactivation. There is no doubt that the mechanisms underlying the rapid inactivation in hERG channels are complex and therefore still remain elusive. Although the structure of the α-helix of the S5P linker is known (Torres et al. 2003), its orientation and possible interaction with other regions of the protein remain unknown but must be elucidated before the role that the unique S5P linker plays in inactivation of hERG channels can be fully understood. Our data, however, provide some very important clues in this regard. The alignment of N588 and Q592, the two high-impact residues, on one side of the S5P α-helix strongly suggests that this face interacts with the remainder of the channel. Furthermore, we suggest that there is likely to be a charged residue close to this site of interaction. There are then two possibilities; this charge is either positive or negative. If it is a positive charge near the site of interaction between N588/Q592 and the remainder of the channel then the addition of positive charge to N588/Q592 is destabilizing an interaction that takes place in the inactive state. Conversely, if it is a negative charge near the site of interaction between N588/Q592 and the remainder of the channel then the addition of a positive charge to N588/Q592 is stabilizing an interaction in the non-inactivated state.
There are nine charged (five positive and four negative) residues present in the remainder of the outer pore region of hERG: E575, D580, R582, K595, K608, D609, K610, E637 and K638. Which of these charges are important for inactivation remains to be determined. However, mutations of R582, D609, E637 and K638 have all been identified as causes of congenital long QT syndrome (Jongbloed et al. 1999; Splawski et al. 2000; Hayashi et al. 2002) and therefore may lie adjacent to the point of interaction between the S5P α-helix and the remainder of the hERG channel.
Acknowledgments
We are grateful to Jane Bursill, Sue Ku and Ken Wyse for expert technical assistance and to Peter Riek for assitance with molecular graphics. J.I.V. is an National Health and Medical Research Council (NHMRC) Career Development Fellow. C.E.C. is an Australian Research Council (ARC) Postdoctoral Fellow. This work was supported by a project grant from the NHMRC (no. 209547) and a University of New South Wales Faculty of Medicine Research Grant to C.E.C.
Supplementary material
The online version of this paper can be accessed at:
DOI: 10.1113/jphysiol.2006.108332
http://jp.physoc.org/cgi/content/full/jphysiol.2006.108332/DC1 and contains supplemental material consisting of a table that summarises V0.5 and K values for Bottzmann fits to activation data for all Chimaeras and mutants analysed in this study.
This material can also be found as part of the full-text HTML version available from http://www.blackwell-synergy.com
References
- Bauer CK, Schwarz JR. Physiology of EAG K+ channels. J Membr Biol. 2001;182:1–15. doi: 10.1007/s00232-001-0031-3. [DOI] [PubMed] [Google Scholar]
- Becchetti A, De Fusco M, Crociani O, Cherubini A, Restano-Cassulini R, Lecchi M, Masi A, Arcangeli A, Casari G, Wanke E. The functional properties of the human ether-a-go-go-like (HELK2) K+ channel. Eur J Neurosci. 2002;16:415–428. doi: 10.1046/j.1460-9568.2002.02079.x. [DOI] [PubMed] [Google Scholar]
- Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, et al. Sudden death associated with short-QT syndrome linked to mutations in hERG. Circ. 2004;109:30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- Camacho J, Sanchez A, Stuhmer W, Pardo LA. Cytoskeletal interactions determine the electrophysiological properties of human EAG potassium channels. Pflugers Arch. 2000;441:167–174. doi: 10.1007/s004240000420. [DOI] [PubMed] [Google Scholar]
- Clarke CE, Benham CD, Bridges A, George AR, Meadows HJ. Mutation of histidine 286 of the human P2X4 purinoceptor removes extracellular pH sensitivity. J Physiol. 2000;523:697–703. doi: 10.1111/j.1469-7793.2000.00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro JM, Brugada R, Wu YS, Hong K, Dumaine R. Modulation of I(Kr) inactivation by mutation N588K in KCNH2: a link to arrhythmogenesis in short QT syndrome. Cardiovasc Res. 2005;67:498–509. doi: 10.1016/j.cardiores.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Dun W, Jiang M, Tseng GN. Allosteric effects of mutations in the extracellular S5P loop on the gating and ion permeation properties of the hERG potassium channel. Pflugers Arch. 1999;439:141–149. doi: 10.1007/s004249900101. [DOI] [PubMed] [Google Scholar]
- Engeland B, Neu A, Ludwig J, Roeper J, Pongs O. Cloning and functional expression of rat ether-à-go-go-like K+ channel genes. J Physiol. 1998;513:647–654. doi: 10.1111/j.1469-7793.1998.647ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker E, Jarolimek W, Brown AM. Molecular determinants of inactivation and dofetilide block in ether a-go-go (EAG) channels and EAG-related K+ channels. Mol Pharmacol. 2001;60:1343–1348. doi: 10.1124/mol.60.6.1343. [DOI] [PubMed] [Google Scholar]
- Ficker E, Jarolimek W, Kiehn J, Baumann A, Brown AM. Molecular determinants of dofetilide block of hERG K+ channels. Circ Res. 1998;82:386–395. doi: 10.1161/01.res.82.3.386. [DOI] [PubMed] [Google Scholar]
- Ganetzky B, Robertson GA, Wilson GF, Trudeau MC, Titus SA. The eag family of K+ channels in Drosophila and mammals. Ann N Y Acad Sci. 1999;868:356–369. doi: 10.1111/j.1749-6632.1999.tb11297.x. [DOI] [PubMed] [Google Scholar]
- Hancox JC, Levi AJ, Witchel HJ. Time course and voltage-dependence of expressed hERG current compared with native ‘rapid’ delayed rectifier K current during the cardiac ventricular action potential. Pflugers Arch. 1998;436:843–853. doi: 10.1007/s004240050713. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Shimizu M, Ino H, Yamaguchi M, Mabuchi H, Hoshi N, Higashida H. Characterization of a novel missense mutation E637K in the pore-S6 loop of HERG in a patient with long QT syndrome. Cardiovasc Res. 2002;54:67–76. doi: 10.1016/s0008-6363(02)00240-7. [DOI] [PubMed] [Google Scholar]
- Herzberg IM, Trudeau MC, Robertson GA. Transfer of rapid inactivation and sensitivity to the class III antiarrhythmic drug E-4031 from hERG to M-eag channels. J Physiol. 1998;511:3–14. doi: 10.1111/j.1469-7793.1998.003bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation on Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Jiang M, Dun W, Tseng GN. Mechanism for the effects of extracellular acidification on HERG-channel function. Am J Physiol. 1999;277:H1283–H1292. doi: 10.1152/ajpheart.1999.277.4.H1283. [DOI] [PubMed] [Google Scholar]
- Jiang M, Zhang M, Maslennikov IV, Liu J, Wu DM, Korolkova YV, Arseniev AS, Grishin EV, Tseng GN. Dynamic conformational changes of extracellular S5P linkers in the hERG channel. J Physiol. 2005;569:75–89. doi: 10.1113/jphysiol.2005.093682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JP, Jr, Mullins FM, Bennett PB. Human ether a-go-go-related gene K+ channel gating probed with extracellular Ca2+: evidence for two distinct voltage sensors. J Gen Physiol. 1999;113:565–580. doi: 10.1085/jgp.113.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongbloed AJ, Wilde AA, Geelen JL, Doevendans P, Schaap van Langen I, van Tintelen JP, Cobben JM, Beaufoft-krol, Geraedts JP, Smeets HJ. Novel KCNQ1 and HE nuissence mutations in Dutch long-RT families. Hum Mutat. 1999;13:301–310. doi: 10.1002/(SICI)1098-1004(1999)13:4<301::AID-HUMU7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang M, Jiang M, Tseng GN. Structural and functional role of the extracellular S5P linker in the hERG potassium channel. J Gen Physiol. 2002;120:723–737. doi: 10.1085/jgp.20028687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Mahaut-Smith MP, Varghese A, Huang CL, Kemp PR, Vandenberg JI. Effects of premature stimulation on hERG K+ channels. J Physiol. 2001;537:843–851. doi: 10.1111/j.1469-7793.2001.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPate MJ, Duncan RS, Milnes JT, Witchel HJ, Hancox JC. The N588K-hERG K+ channel mutation in the ‘short QT syndrome’: mechanism of gain-in-function determined at 37 degrees C. Biochem Biophys Res Commun. 2005;334:441–449. doi: 10.1016/j.bbrc.2005.06.112. [DOI] [PubMed] [Google Scholar]
- Meyer R, Heinemann SH. Characterization of an eag-like potassium channel in human neuroblastoma cells. J Physiol. 1998;508:49–56. doi: 10.1111/j.1469-7793.1998.049br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Occhiodoro T, Bernheim L, Liu JH, Bijlenga P, Sinnreich M, Bader CR, Fischer-Lougheed J. Cloning of a human ether-a-go-go potassium channel expressed in myoblasts at the onset of fusion. FEBS Lett. 1998;434:177–182. doi: 10.1016/s0014-5793(98)00973-9. [DOI] [PubMed] [Google Scholar]
- Piper DR, Varghese A, Sanguinetti MC, Tristani-Firouzi M. Gating currents associated with intramembrane charge displacement in hERG potassium channels. Proc Natl Acad Sci U S A. 2003;100:10534–10539. doi: 10.1073/pnas.1832721100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: hERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Schönherr R, Heinemann SH. Molecular determinants for activation and inactivation of hERG, a human inward rectifier potassium channel. J Physiol. 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the hERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. J Gen Physiol. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in long-QT syndrome genes. Kvlqt1, HERG, Scn5a, Kcne1, KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Vincent GM, Lehmann MH, Keating MT. Genomic structure of three long QT syndrome genes. Kvlqt1, HERG, KCNE1. Genomics. 1998;51:86–97. doi: 10.1006/geno.1998.5361. [DOI] [PubMed] [Google Scholar]
- Subbiah RN, Clarke CE, Smith DJ, Zhao J, Campbell TJ, Vandenberg JI. Molecular basis of slow activation of the human ether-a-go-go related gene (hERG) potassium channel: critical role of lysine 525. J Physiol. 2004;558:417–431. doi: 10.1113/jphysiol.2004.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres A, Bansal PS, Sunde M, Clarke CE, Bursill JA, Smith DJ, et al. Structure of the hERG K+ channel S5P extracellular linker: role of an amphipathic α-helix in c-type inactivation. J Biol Chem. 2003;278:42136–42148. doi: 10.1074/jbc.M212824200. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Varghese A, Lu Y, Bursill JA, Mahaut-Smith MP, Huang CL. Temperature dependence of human Ether-a-go-go Related Gene (hERG) K+ currents. Am J Physiol Cell Physiol. 2006 doi: 10.1152/ajpcell.00596.2005. in press. [DOI] [PubMed] [Google Scholar]
- Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci U S A. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. The moving parts of voltage-gated ion channels. Q Rev Biophys. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- Yellen G. The voltage-gated potassium channels and their relatives. Nature. 2002;419:35–42. doi: 10.1038/nature00978. [DOI] [PubMed] [Google Scholar]
- Yellen G, Sodickson D, Chen T-Y, Jurman ME. An engineered cysteine in the external mouth of a K+ channel allows inactivation to be modulated by metal binding. Biophys J. 1994;66:1068–1075. doi: 10.1016/S0006-3495(94)80888-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT. Properties of hERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J. 1998;74:230–241. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou A, Xu QP, Sanguinetti MC. A mutation in the pore region of hERG K+ channels expressed in Xenopus oocytes reduces rectification by shifting the voltage dependence of inactivation. J Physiol. 1998;509:129–137. doi: 10.1111/j.1469-7793.1998.129bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The online version of this paper can be accessed at:
DOI: 10.1113/jphysiol.2006.108332
http://jp.physoc.org/cgi/content/full/jphysiol.2006.108332/DC1 and contains supplemental material consisting of a table that summarises V0.5 and K values for Bottzmann fits to activation data for all Chimaeras and mutants analysed in this study.
This material can also be found as part of the full-text HTML version available from http://www.blackwell-synergy.com