

Abstract
The objective of this study was to investigate the involvement of tyrosine phosphorylation in the regulation of the cardiac slowly activating delayed-rectifier K+ current (IKs) that is important for action potential repolarization. Constitutive IKs recorded from guinea-pig ventricular myocytes was suppressed by broad-spectrum tyrosine kinase (TK) inhibitors tyrphostin A23 (IC50, 4.1 ± 0.6 μm), tyrphostin A25 (IC50, 12.1 ± 2.1 μm) and genistein (IC50, 64 ± 4 μm), but was relatively insensitive to the inactive analogues tyrphostin A1, tyrphostin A63, daidzein and genistin. IKs was unaffected by AG1478 (10 μm), an inhibitor of epidermal growth factor receptor TK, and was strongly suppressed by the Src TK inhibitor PP2 (10 μm) but not by the inactive analogue PP3 (10 μm). The results of experiments with forskolin, H89 and bisindolylmaleimide I indicate that the suppression of IKs by TK inhibitors was not mediated via inhibition of (IKs-stimulatory) protein kinases A and C. To evaluate whether the suppression was related to lowered tyrosine phosphorylation, myocytes were pretreated with TK inhibitors and then exposed to the phosphotyrosyl phosphatase inhibitor orthovanadate (1 mm). Orthovanadate almost completely reversed the suppression of IKs induced by broad-spectrum TK inhibitors at concentrations around their IC50 values. We conclude that basal IKs is strongly dependent on tyrosine phosphorylation of Ks channel (or channel-regulatory) protein.
The slowly activating delayed-rectifier K+ current (IKs) plays an important role in the repolarization of the cardiac action potential. Consequently, changes in IKs density and gating have far-reaching effects on cardiac function (Sanguinetti & Jurkiewicz, 1990; Lei et al. 2000; Terrenoire et al. 2005). The Ks channels that carry IKs are formed by the co-assembly of α-subunit KCNQ1 and β-subunit KCNE1, and mutations in these proteins slow repolarization and are implicated in severe cardiac disorders, including long QT syndrome (Wang et al. 1996; Sanguinetti, 2000).
It is important to understand the intracellular factors that modulate IKs in cardiac cells. Among these are three serine/threonine kinases: protein kinase A (PKA), protein kinase C (PKC) and protein kinase G (PKG). Studies on guinea-pig ventricular myocytes have established that an acute stimulation of PKA activity, whether via activation of the β-adrenergic pathway or more directly, results in stimulation of IKs (Walsh & Kass, 1988, 1991; Harvey & Hume, 1989; Freeman et al. 1992). Similarly, both indirect and direct stimulation of PKC lead to increases in the amplitude of the current (Tohse et al. 1987, 1992; Habuchi et al. 1992; Asai et al. 1996). Conversely, stimulation of PKG via muscarinic receptors has little effect on basal IKs, but antagonizes PKA-mediated stimulation of the current (Harvey & Hume, 1989; Yazawa & Kameyama, 1990).
It is now generally recognized that in addition to regulation by serine/threonine kinases, ion channels may be acutely regulated by tyrosine kinase (TK) (Davis et al. 2001), an enzyme with activity that is affected by a wide array of signals, including angiotensin II, insulin, growth factors, mechanical perturbation and anisosmotic stress (Sadoshima et al. 1996; Pawson & Scott, 1997; Browe & Baumgarten, 2004; Cohen, 2005; Gavi et al. 2006). Prominent among the channels regulated by TK are a diverse group of voltage-gated K+ channels (Holmes et al. 1996; Davis et al. 2001; Cayabyab & Schlichter, 2002; Gamper et al. 2003). Whether cardiac Ks channels belong within that group is an open question. Zhou et al. (1997) found that the TK inhibitor genistein decreased the amplitude of IKs in canine ventricular myocytes, and suggested that TK may be a positive regulator of cardiac IKs. However, this view was discounted in subsequent studies on guinea-pig ventricular myocytes because (i) broad-spectrum TK inhibitors lavendustin A and tyrphostin 51 had little effect on IKs (Washizuka et al. 1998), and (ii) equimolar concentrations of genistein and the analogue daidzein had roughly equivalent inhibitory effects on the current (Matsubayashi et al. 1999).
We have investigated the role of TK in the regulation of IKs in guinea-pig ventricular myocytes by performing three groups of experiments. Firstly, we compared the effects of four TK inhibitors (tyrphostin A23, tyrphostin A25, genistein and PP2) with those of five inactive analogues (tyrphostins A1 and A63, daidzein, genistin and PP3) (Akiyama et al. 1987; Gazit et al. 1989; Hanke et al. 1996; Bain et al. 2003). Secondly, we evaluated whether the actions of TK inhibitors on IKs were likely to be mediated by serine/threonine kinases and Gs protein. Thirdly, we determined whether the actions of TK inhibitors on IKs were antagonized by orthovanadate, a phosphotyrosyl phosphatase (PTP) inhibitor (Swarup et al. 1982).
Methods
Preparation of myocytes
Adult guinea-pigs (250–300 g) were killed by cervical dislocation in accordance with Canadian and Dalhousie University regulations on animal experimentation. Hearts were quickly removed, mounted on a Langendorff column, and perfused through the coronary artery for 10–15 min. The Ca2+-free perfusate (37°C) contained (mm): NaCl 125, KCl 5, MgCl2 1.2, taurine 20, glucose 20 and N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (Hepes) 5 (pH 7.4), as well as 0.08–0.12 mg ml−1 collagenase (Yakult Pharmaceutical Co., Tokyo, Japan). On completion of collagenase digestion, the heart tissue was minced, and myocytes were dispersed in a high-K+, nutrient-supplemented storage solution (22°C) that contained (mm): KCl 30, KOH 80, KH2PO4 30, MgSO4 3, glutamic acid 50, taurine 20, glucose 20, ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) 0.5 and Hepes 10 (pH adjusted to 7.4 with KOH).
Electrophysiology
A few drops of myocyte suspension were placed in a 0.3-ml chamber mounted on the stage of an inverted phase-contrast microscope, and the chamber was perfused with bathing solution (see below) at a flow rate of 2 ml min−1. Rod-shaped quiescent myocytes were voltage clamped using the standard ruptured-patch method. Recording pipettes were fabricated from thick-walled borosilicate glass capillaries (H15/10/137, Jencons Scientific Ltd, Leighton Buzzard, UK), and had resistances of 2–3 MΩ when filled with pipette solution. Pipette offsets were nulled prior to patch formation, and liquid junction potentials (approximately −10 mV) were offset during data analysis. Series resistance ranged between 3 and 5 MΩ, and was compensated by 60–80%. Membrane currents were recorded with an EPC-9 amplifier (HEKA Electronics, Mahone Bay, Nova Scotia, Canada), filtered at 3 kHz, and digitized with Pulse software (HEKA Electronics) at a sampling rate of 12 kHz. Data files were converted from Pulse to Axon (Axon Instruments, Union City, CA, USA) format, and analysed with Clampfit electrophysiology software (Axon Instruments). All experiments were conducted at 36°C.
Bathing and pipette solutions
The standard K+-free, Ca2+-free bathing solution contained (mm): NaCl 140, MgCl2 1, glucose 10 and Hepes 5 (pH 7.4), as well as 0.2 mm Cd2+ and 3 μm glibenclamide (Sigma-Aldrich, Oakville, Ontario, Canada). The standard pipette-filling solution contained (mm): KCl 40, potassium aspartate 106, MgCl2 4.8, K2ATP 4, MgATP 1, EGTA 5 and Hepes 5 (pH adjusted to 7.2 with KOH).
Chemicals and drugs
All chemicals used in making solutions were purchased from Sigma-Aldrich and were of the highest purity grade available. N-[2-((p-romocinnamyl)amino)ethyl]-5-isoquinolinesulphonamide (H89), AG1478, bisindolylmaleimide I, forskolin, daidzein, genistein, genistin, PD98059, PP2, PP3 and tyrphostins A1, A23, A25 and A63 were purchased from Calbiochem (La Jolla, CA, USA), prepared as stock solutions in dimethyl sulphoxide (DMSO) (Sigma-Aldrich), stored in the dark at −20°C and added to experimental solutions as required. The highest eventual concentration of DMSO in the bathing solutions was 0.1%, a concentration that has little effect on IKs in guinea-pig ventricular myocytes (Ogura et al. 1995). Daidzein, genistein and genistin (Calbiochem) were prepared as stock solutions as above, except when the desired final concentration was ≥ 100 μm; in which case, both the control and test bathing solutions were slightly alkaline (pH ∼7.6) and contained 0.2% DMSO to minimize the precipitation of drug in the test bathing solutions. Guanosine 5′-O-(2-thiodiphosphate) trilithium (GDPβS) was dissolved in pipette solution with reduced potassium aspartate concentration (95 mm). Sodium orthovanadate (Fisher Scientific, Nepeon, Ontario, Canada) was freshly prepared in water before each experiment, and added to the superfusate and then the pH was adjusted to 7.4 with HCl. E4031 (Eisai, Tokyo, Japan) and thiopentone sodium (Abbott Laboratories, Saint Laurent, Quebec, Canada) were dissolved in the bathing solution.
Statistics
Experimental data are expressed as means ±s.e.m.; n represents the number of experiments. Statistical comparisons were made using Student's paired or unpaired t test. Differences were considered significant when P < 0.05.
Results
Myocytes were bathed in K+-free solution that contained 0.2 mm Cd2+ to suppress inward cation current, and depolarized from −30 to +50 mV for 500 ms at 0.05–0.1 Hz. Under these experimental conditions, the time-dependent outward current that developed during depolarization and the outward tail current that deactivated during repolarization were almost exclusively composed of IKs. As indicated by the representative data in the time plot of Fig. 1A, the amplitude of the tail current was unaffected by acute application of the rapidly activating K+ channel blocker E4031 (5 μm). However, it was decreased in a concentration-dependent manner by thiopentone, a compound that has been reported to abolish IKs in guinea-pig ventricular myocytes at 100–300 μm (Heath &, Terrar, 1996, 2000). In the present study, 20 μm thiopentone reduced the amplitude of the tail current by 51 ± 4% (n = 6), and 100 μm reduced it by 90 ± 3% (n = 4). As a result, it was reasonable to quantify the effects of pharmacological interventions on IKs by expressing tail current amplitude during quasi-steady-state drug action as a percentage of control (predrug) amplitude.
Figure 1. Effects of E4031 and thiopentone onIKs in a representative myocyte.
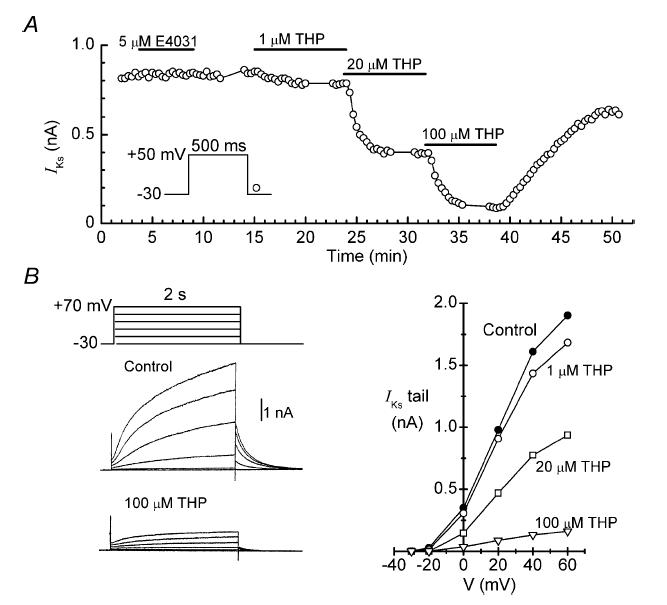
A, time course of changes in the amplitude of the IKs tail during cumulative applications of thiopentone (THP). Time on this and similar plots is referenced to patch-breakthrough time. B, left: two of the sets of current records obtained on the sequences of 2-s depolarizations applied at the times indicated by the data breaks in A. Right: tail current–voltage relationships obtained in this experiment.
In most of the experiments conducted for this work, regular pulsing from −30 to +50 mV was interrupted at appropriate times for determination of isochronic (2 s) current–voltage (I–V) relationships. As indicated by the current records and I–V relationships obtained during the example thiopentone experiment (Fig. 1B), the amplitudes of time-dependent current on depolarization, and associated tail current on repolarization, were small below 0 mV, increased sharply between 0 and +40 mV, and saturated at more positive potentials. Boltzmann function fits to full-scale tail I–V relationships (−30 to +90 mV) from seven control myocytes had a half-maximal voltage of 21.4 ± 2.1 mV, and a slope of 14.2 ± 1.3 mV. These values are in good agreement with those determined in earlier studies on IKs activation in guinea-pig ventricular myocytes (Heath & Terrar, 1996; Matsubayashi et al. 1999).
Effects of TK inhibitors and inactive analogues on IKs
Broad-spectrum tyrphostin inhibitors and inactive analogues
Figure 2A shows the effects of 20 μm tyrphostin A25 on the amplitude of the IKs tail in a representative myocyte. The broad-spectrum TK inhibitor (Gazit et al. 1989; Davis et al. 2001) decreased the amplitude of the current by ∼70%, and this effect was almost fully reversed by a 10-min washout period. The families of current traces obtained while recording the I–V relationship in this experiment indicate that the inhibitory action of the drug was exerted at voltages up to +90 mV, and that decreases in the amplitude of time-dependent currents on depolarization were matched by decreases in the amplitude of tail currents on repolarization (Fig. 2B, left). Measurements of tail amplitudes and time courses of IKs before and ∼8 min after application of tyrphostin A25 (n = 8) indicate that the inhibitor had little effect on either the voltage dependence of IKs activation (e.g. Fig. 2B, right) or the kinetics of IKs activation and deactivation (data not shown).
Figure 2. Inhibition of IKs by tyrphostin A25.
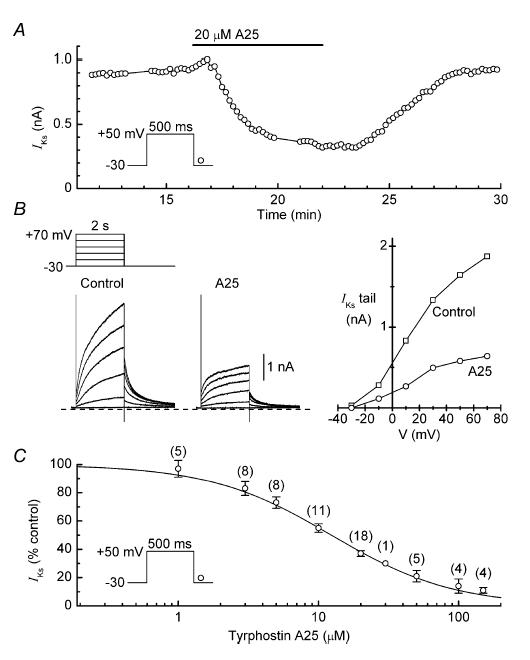
A, reversible inhibitory effect of 20 μm tyrphostin A25 (A25) on the amplitude of the IKs tail in a representative myocyte. B, records and I–V relationships obtained at the times indicated by the data breaks in A. C, dependence of IKs inhibition on the concentration of tyrphostin A25. Myocytes were treated with single concentrations of the inhibitor for ∼8 min, and the amplitude of the tail current at that time was expressed as a percentage of the control (predrug) amplitude. The Hill equation fitting the data has an IC50 of 12.3 ± 2.1 μm and a coefficient of 1.0 ± 0.2. Number of myocytes is shown in parentheses.
Myocytes were treated with single concentrations of tyrphostin A25 (1–150 μm) to determine the dependence of IKs inhibition on the concentration of the drug. The lowest concentration tested had little effect on the amplitude of the current, whereas the highest almost completely abolished the current. The full set of data is well described by the Hill equation with an IC50 of 12.3 ± 2.1 μm and a coefficient of 1.0 ± 0.2 (Fig. 2C).
To obtain information on whether the inhibition of IKs by tyrphostin A25 was likely to be caused by an inhibition of TK, we investigated the effects of a related broad-spectrum TK inhibitor, tyrphostin A23, and two inactive compounds, tyrphostin A1 and A63 (Gazit et al. 1989). Tyrphostin A23 reversibly decreased the amplitude of IKs with an IC50 of 4.1 ± 0.6 μm and a coefficient of 0.9 ± 0.1 (Fig. 3A and B). In marked contrast, neither tyrphostin A1 (10–200 μm) nor A63 (50–100 μm) affected the amplitude of the current (Fig. 3A and B).
Figure 3. Effects of TK-inhibitor tyrphostin A23 and the inactive tyrphostins A1 and A63 on IKs.
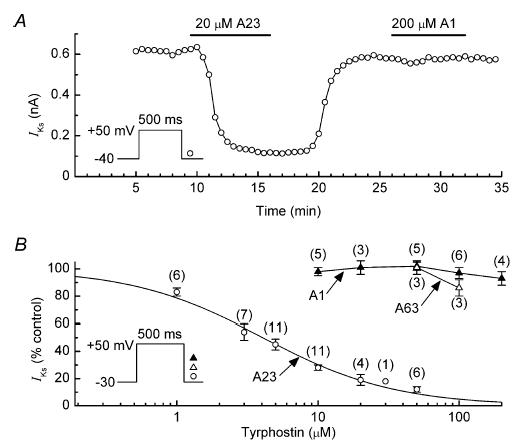
A, comparison of the effects of 20 μm tyrphostin A23 (A23) and 200 μm tyrphostin A1 (A1) on the amplitude of IKs in a representative myocyte. B, dependence of inhibition of IKs on the concentrations of tyrphostins A1, A23 and A63. Myocytes were treated with single concentrations of the drugs for ∼8 min. The Hill equation fitting the tyrphostin A23 data has an IC50 of 4.1 ± 0.6 μm, and a coefficient of 0.93 ± 0.1. Number of myocytes is shown in parentheses.
Genistein, genistin and daidzein
Genistein is a broad-spectrum TK inhibitor (Akiyama et al. 1987) that is structurally and mechanistically different from tyrphostins A23 and A25 (Davis et al. 2001). Genistein had a rapid inhibitory effect on IKs, and this action was fully reversed by washout of the drug (Fig. 4A and B). To determine the dependence of IKs inhibition on the concentration of genistein, myocytes were treated with single concentrations of the drug for ∼8 min. The data are well described by the Hill equation with an IC50 of 64 ± 4 μm and a coefficient of 0.8 ± 0.1 (Fig. 4C).
Figure 4. Effects of TK inhibitor genistein and the inactive analogues genistin and daidzein on IKs.
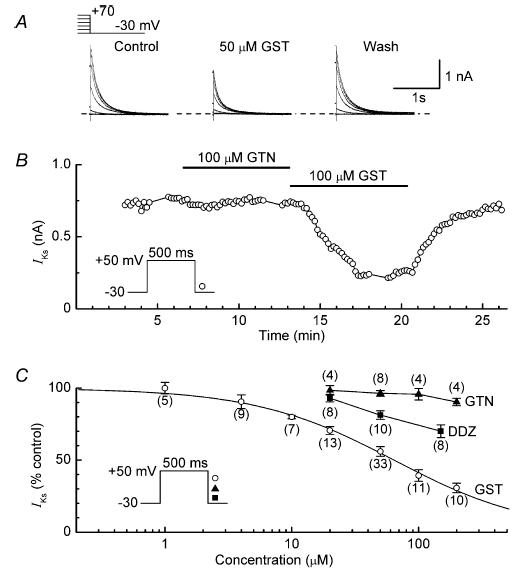
A, example records of IKs tails recorded before (control), 6 min after addition of 50 μm genistein (GST) and 8 min after washout of the drug (Wash). B, comparison of the effects of 100 μm genistin (GTN) and 100 μm genistein on the amplitude of IKs in a representative myocyte. C, dependence of inhibition of IKs on the concentrations of genistein, genistin and daidzein (DDZ). Myocytes were treated with single concentrations of the drugs for ∼8 min. The Hill equation fitting the genistein data has an IC50 of 64 ± 4 μm, and a coefficient of 0.8 ± 0.1. Number of myocytes is shown in parentheses.
For comparison with the results obtained with genistein, experiments were conducted with two inactive analogues, genistin and daidzein (Akiyama et al. 1987). Although genistin at concentrations up to 200 μm had little effect on IKs, daidzein inhibited the current in a concentration-dependent manner (Fig. 4B and C). However, at any given concentration, the degree of inhibition produced by daidzein was substantially smaller than that produced by genistein (Fig. 4C).
PP2, PP3 and tyrphostin AG1478
PP2, a potent inhibitor of Src TK (nanomolar IC50 value; Hanke et al. 1996), is a useful pharmacological tool for detection of Src TK involvement in ion-channel regulation (e.g. Du et al. 2004). In the present study, we compared the effects of PP2 with those of PP3, a structurally related molecule that has little effect on Src TK (Bain et al. 2003). PP2 (10 μm) decreased the amplitude of IKs by 69 ± 2% (n = 9), whereas PP3 (10 μm) only decreased it by an insignificant 2 ± 4% (n = 8; P < 0.001 versus PP2) (Fig. 5A and C). Similarly, AG1478 (10 μm), a selective inhibitor of epidermal growth factor receptor (EGFR) TK (IC50, ∼3nm) (Liu et al. 1999), had little effect on IKs (Fig. 5B and C).
Figure 5. Effects of Src TK inhibitor PP2, inactive analogue PP3 and EGFR TK inhibitor AG1478 on IKs.
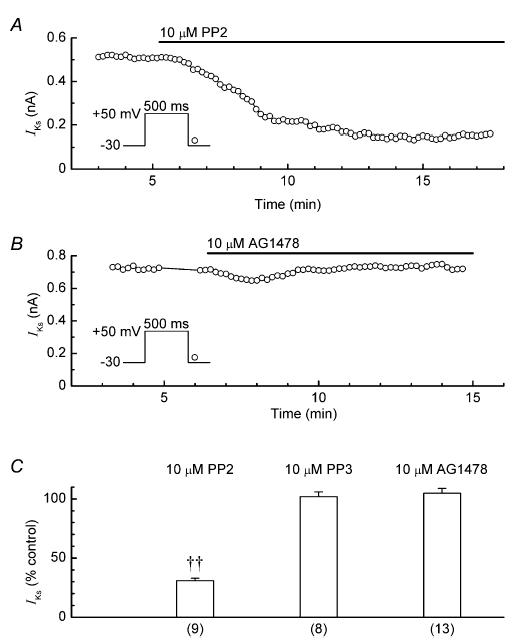
A, data obtained from a myocyte treated with 10 μm PP2. B, data obtained from a myocyte treated with 10 μm AG1478. C, summary of the results. Myocytes were treated for ∼10 min; ††P < 0.001. Number of myocytes is shown in parentheses.
Investigation of potential mediators of TK-inhibitor action on IKs
The results obtained with broad-spectrum TK inhibitors and their inactive analogues suggest that inhibition of IKs by TK inhibitors is related to a decrease of tyrosine phosphorylation. The pertinent tyrosine phosphorylation might be on Ks channel protein or, alternatively, on channel regulatory protein that directly or indirectly affects channel activity. In the latter case, the most likely end effectors are serine/threonine kinases and direct-acting G-proteins. The involvement of these potential mediators was investigated as described below.
Serine/threonine kinases
Two types of experiments were conducted to evaluate whether inhibition of IKs by a TK inhibitor was mediated via an inhibition of PKA. In the first, we investigated whether pretreatment of myocytes with 20 μm tyrphostin A25 suppressed the stimulatory effects of a relatively low (0.2 μm) concentration of forskolin; Fig. 6A and B shows that this was not the case. In the second set of experiments, we investigated whether inhibition of basal PKA activity caused an inhibition of IKs comparable to that caused by TK inhibitors. The PKA-inhibitor H89 (IC50, ∼50 nm) (Chijiwa et al. 1990) was applied either externally (10 μm in the bathing solution) or internally (100 μm in the pipette solution) for ∼12 min. As neither of these H89 treatments reduced the amplitude of IKs by more than 10% (Fig. 6C and D), it seems unlikely that the inhibitor action of TK on IKs is mediated via inhibition of PKA (or via stimulation of phosphatases that act on channel PKA sites). It is also unlikely that the inhibitor action of TK is mediated via inhibition of PKG and protein kinase B (PKB) because H89 inhibits these kinases with IC50 values of ∼0.5 μm and ∼2.6 μm, respectively (Chijiwa et al. 1990; Davies et al. 2000).
Figure 6. Effects of modulators of PKA, PKC and G-protein activity on IKs.
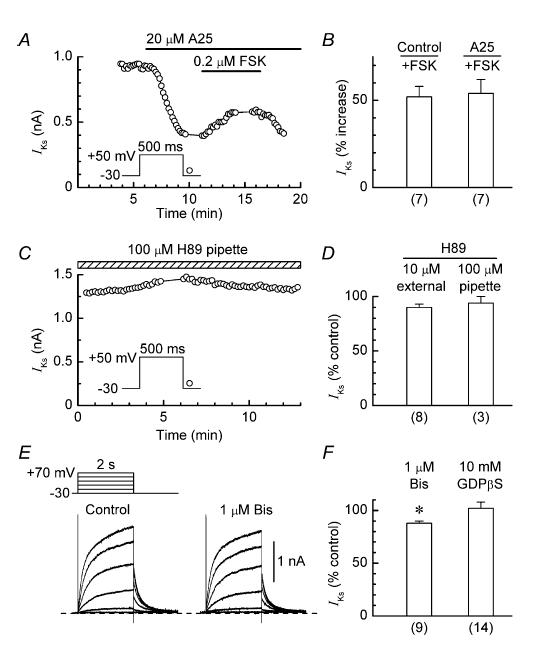
A, stimulation of IKs by 0.2 μm forskolin (FSK) in a myocyte pretreated with 20 μm tyrphostin A25 (A25). B, comparison of the stimulatory effects of 0.2 μm forskolin on IKs in control and tyrphostin-pretreated myocytes. C, lack of effect of H89 (100 μm) in the pipette solution on IKs in a representative myocyte. D, summary of the results obtained with myocytes that were treated with external H89 (10 μm) or internal H89 (100 μm) for ∼12 min. For evaluation of the effects of internal H89, the average amplitude of the IKs tail during the first minute after patch formation was taken as the control amplitude. E, records obtained from a myocyte before and 20 min after addition of 1 μm bisindolylmaleimide I (Bis). F, summary of the effects of treatments for 10–20 min with external 1 μm bisindolylmaleimide I and internal 10 mm GDPβS on the amplitude of the IKs tail; *P < 0.05. Number of myocytes is shown in parentheses.
The PKC inhibitor bisindolylmaleimide I (IC50, ∼70 nm) (Toullec et al. 1991) was used to evaluate possible mediation via inhibition of basal PKC activity. This compound has been used at external concentrations of 0.1–0.2 μm in earlier investigations on ion channel regulation by PKC in cardiac cells (Ward & Giles, 1997; Lei et al. 2000; Zhang et al. 2003). In the present study, treatment of myocytes with a concentration of 1 μM for up to 20 min only lowered the amplitude of IKs by a moderate 12 ± 2% (n = 9; P < 0.05; Fig. 6E and F). This finding suggests that the inhibitor action of TK on IKs does not involve either an inhibition of PKC or a stimulation of phosphatases that act on channel PKC sites.
Mitogen-activated protein kinases (MAPKs) have been shown to have positive regulatory actions on Ca2+ (Ma et al. 1996), Cl− (Crepel et al. 1998) and K+ channels (Schrader et al. 2005). Possible involvement of MAPKs in the inhibitory effects of TK inhibitors on IKs was investigated by treating myocytes with PD98059 and SB202190. PD98059 is an inhibitor of the upstream kinase MEK1 that is specific for extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) (Alessi et al. 1995), whereas SB202190 is a specific inhibitor of stress-activated protein kinase (SAPK) 2a (p38) (Davies et al. 2000). In the first series of experiments, myocytes were exposed to a relatively high (50 μm) concentration of PD98059 for 8–12 min. This exposure had little effect on the amplitude of IKs (reduction of 5 ± 4%, n = 5). In the second series, myocytes were pretreated with 50 μm PD98059, 20 μm SB202190 or vehicle for 2 h, and the amplitude of IKs was measured at 5 min after patch formation. The amplitudes in these myocytes were 0.48 ± 0.05 nA (n = 11), 0.49 ± 0.13 nA (n = 3) and 0.46 ± 0.04 nA (n = 18), respectively, suggesting lack of involvement of ERK1/2, p38 and corresponding phosphatases in the inhibition of IKs by TK inhibitors.
G-proteins
The possibility of G-protein involvement in TK inhibitor action arises because tyrosine phosphorylation of G-protein α-subunits can up-regulate subunit activity (Hausdorff et al. 1992; Poppleton et al. 1996; Umemori et al. 1997) which, in turn, could cause direct (membrane-delimited) up-regulation of Ks channels (Freeman et al. 1992). Thus, inhibition of basal TK activity could suppress basally active G-protein and cause down-regulation of the channels. To investigate this possibility, myocytes were dialysed with a pipette solution that contained a high (10 mm) concentration of inhibitory GDPβS (Gilman, 1987). In 14 myocytes dialysed with GDPβS solution, the amplitude of IKs measured at ∼15 min after patch formation was 102 ± 6% of that measured at ∼30 s after patch formation (Fig. 6F).
Effects of orthovanadate
The finding that inhibitors of TK inhibited IKs raised the possibility that the PTP inhibitor orthovanadate would have a stimulatory effect on the current. In fact, application of a 1 mM orthovanadate for 8–10 min only increased the amplitude of IKs by 6 ± 3% (n = 18; P < 0.05). However, the effect of orthovanadate was quite different when myocytes were pretreated with a broad-spectrum TK inhibitor. Figure 7A shows the results obtained in an experiment with 5 μm A25 and orthovanadate. The TK inhibitor decreased the amplitude of IKs to 70% of its control value, and the addition of orthovanadate increased it to 95% control. In nine experiments of this type, 5 μm A25 reduced the amplitude of IKs to 71 ± 3% control, and addition of orthovanadate increased it to 100 ± 3% (P < 0.001; Fig. 7B).
Figure 7. Effects of 1 mm orthovanadate on IKs in myocytes pretreated with TK inhibitors.
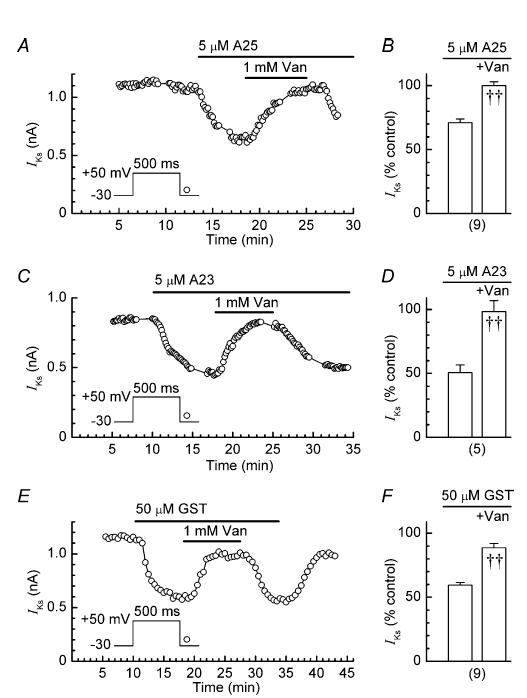
A, C and E, examples of reversible stimulation of IKs by orthovanadate (Van). B, D and F, Summaries of the effects of TK inhibitor pretreatment (left) and subsequent cotreatment with orthovanadate (right) on the amplitude (% control) of the IKs tail; ††P < 0.001 versus before application of orthovanadate. Number of myocytes is shown in parentheses. A23, tyrphostin A23; A25, tyrphostin A25; GST, genistein; Van, orthovanadate.
Similar trials were conducted with concentrations of A23 and genistein around their IC50 values. In five experiments with 5 μm A23, the inhibitor reduced the amplitude of IKs to 51 ± 7% of its control value, and addition of orthovanadate increased it to 98 ± 10% (P < 0.001; Fig. 7C and D). The corresponding inhibitor-induced and orthovanadte-induced values in nine experiments with 50 μm genistein were 59 ± 2% and 89 ± 3%, respectively (P < 0.001; Fig. 7E and F).
An interpretation of these results is that orthovanadate caused restoration of IKs by permitting a build-up of tyrosine phosphorylation mediated by ‘residual’ (non-inhibited) TK. To investigate whether stronger inhibition of TK might lower residual TK activity and thereby weaken restoration of IKs, myocytes were treated with relatively high concentrations of A25 or A23 and then cotreated with 1 mm orthovanadate. Figure 8A shows the results obtained in an experiment with 20 μm A25. The inhibitor rapidly reduced the amplitude of IKs to about 35% of its control value, and subsequent addition of orthovanadate increased it to about 90% of control. In eight myocytes, 20 μm A25 reduced the current to 38 ± 4% of its control amplitude, and addition of 1 mm orthovanadate increased it to 86 ± 7% (P < 0.001). However, the restoration of IKs was less impressive when myocytes were pretreated with 50–100 μm A25. In these myocytes, the orthovanadate-induced restoration of IKs was slower and did not exceed 70% of control amplitude (n = 3) (e.g. Fig. 8B). Similarly, the response of IKs to orthovanadate in three myocytes pretreated with a high concentration (50 μm) of A23 was muted in comparison to that in myocytes pretreated with 5 μm A23 (e.g. Fig. 8C).
Figure 8. Slowing of the stimulatory effect of 1 mm orthovanadate in myocytes pretreated with high concentrations of TK inhibitors.
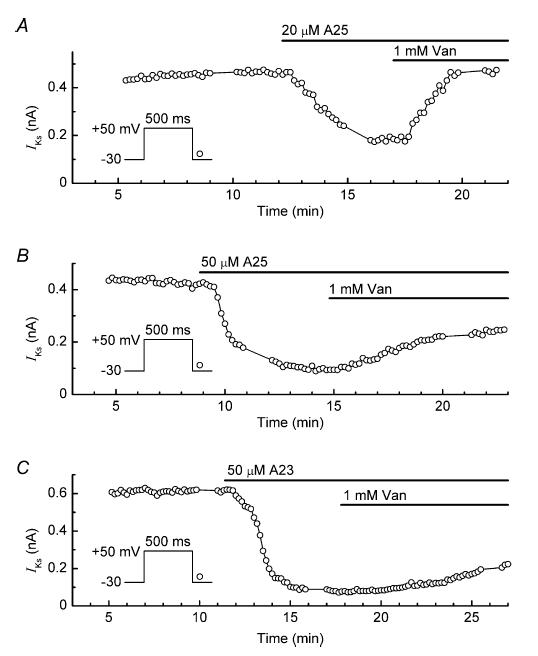
A–C, effects of orthovanadate (Van) on the amplitude of IKs in myocytes pretreated with 20 μm tyrphostin A25 (A25, A), 50 μm tyrphostin A25 (B) and 50 μm tyrphostin A23 (A23, C).
A potential problem with the use of orthovanadate is that in addition to inhibiting PTP, it might also inhibit serine/threonine phosphatases (Shenolikar & Nairn, 1991) and thereby cloud the interpretation of experimental results (Herzig & Neumann, 2000). To evaluate the severity of that potential problem in the present study, we measured (i) the degree of stimulation of IKs by 1 μm forskolin in control and orthovanadate-pretreated myocytes, and (ii) the half-time of decay of stimulation following removal of forskolin in these experiments. An increase in the degree of stimulation and a pronounced slowing of the decay in the orthovanadate-pretreated myocytes would point to an inhibition of serine/threonine phosphatase activity. Figure 9A shows the data obtained from a control myocyte that was treated with forskolin for ∼5 min. The stimulant increased the amplitude of IKs by 120%, and the half-time for decay of stimulation after re-admission of control solution was 165 s. Figure 9B shows the data obtained from a myocyte that was pretreated with orthovanadate for 5 min, and then cotreated with forskolin for ∼5 min. In this myocyte, forskolin increased the amplitude of IKs by 110%, and the stimulation decayed with a half-time of 180 s. Overall, seven control myocytes were treated with forskolin for 4.8 ± 0.2 min, and seven test myocytes were pretreated with orthovanadate for 5 ± 0.3 min and then cotreated with forskolin for 4.8 ± 0.1 min. The orthovanadate pretreatment did not increase the degree of stimulation of IKs by forskolin (control, 111 ± 12%; forskolin, 114 ± 17%), and did not prolong the decay of stimulation (half-time: control, 161 ± 12 s; forskolin, 162 ± 11 s; Fig. 9C and D).
Figure 9. Lack of effect of 1 mm orthovanadate on either the degree of stimulation of IKs by 1 μm forskolin, or the half-time of decay of stimulation following the removal of forskolin.
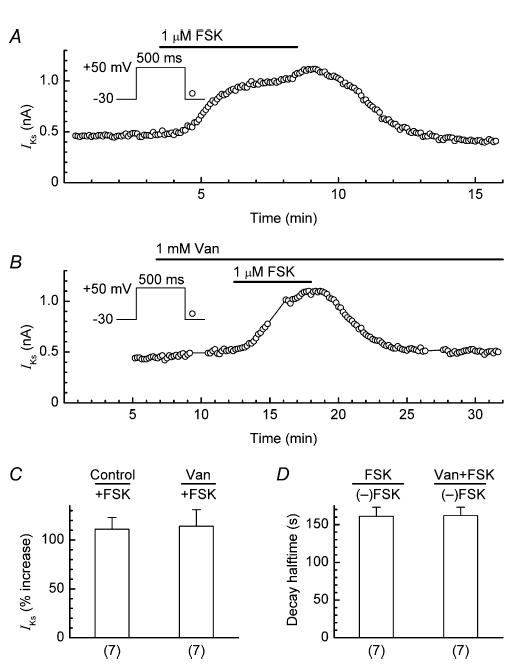
A, example of stimulation and decay of IKs in a control myocyte treated with forskolin (FSK). B, example of stimulation and decay of IKs in a myocyte pretreated with orthovanadate (Van) and subsequently cotreated with forskolin. C, comparison of the degree of stimulation of IKs induced by forskolin in control and orthovanadate-pretreated myocytes. D, comparison of the half-times of decay of forskolin-induced stimulation upon withdrawal of forskolin ((–)FSK) in control and orthovanadate-pretreated myocytes. Number of myocytes is shown in parentheses.
Discussion
We have investigated the involvement of tyrosine phosphorylation in the regulation of Ks channels in guinea-pig ventricular myocytes by measuring the responses of IKs to application of TK inhibitors, inactive analogues and the PTP inhibitor orthovanadate. Four of the five TK inhibitors examined had strong inhibitory effects on IKs, whereas four of the five analogues examined had little or no effect on the current. These results, and the finding that the TK inhibitor action was reversed by orthovanadate, suggest that tyrosine phosphorylation is an important factor in the regulation of cardiac Ks channels. In the discussion that follows, we compare our observations on TK inhibitors and analogues with those reported in earlier studies on cardiac IKs, evaluate the effects of orthovanadate, and consider mechanisms that can account for TK inhibitor and PTP inhibitor actions on IKs.
Comparison with earlier findings on TK inhibitors and inactive analogues
Genistein and daidzein have been employed as pharmacological tools in three earlier studies concerned with the regulation of cardiac IKs by TK. Zhou et al. (1997) observed that 50 μm genistein decreased the amplitude of IKs in canine ventricular myocytes by 46%, whereas 50 μm daidzein decreased it by 33%. They concluded that genistein inhibits the current in both a TK-dependent and TK-independent manner. By contrast, Washizuka et al. (1998) and Matsubayashi et al. (1999) concluded that IKs in guinea-pig ventricular myocytes was not regulated by TK because both genistein and daidzein inhibited the current.
In the present study, we compared the inhibitory effects of genistein with those of analogues daidzein and genistin. Genistein inhibited IKs with an IC50 of 64 ± 4 μm, daidzein was far less effective and genistin (≤ 200 μm) was completely inactive (Fig. 4C). Thus, these results suggest that inhibition of IKs by genistein is predominantly related to inhibition of TK, and that inhibition by daidzein may be due to weak inhibition of TK, and/or to an action that is independent of TK (and possibly shared, to some degree, by genistein). Direct channel block is a strong possibility, whereas inhibition of PKA is an unlikely one in view of the insensitivity of IKs to the PKA inhibitor H89.
Washizuka et al. (1998) investigated the effects of genistein and two other broad-spectrum TK inhibitors, lavendustin A and tyrphostin 51, on IKs in guinea-pig ventricular myocytes. They found that 50 μm genistein inhibited IKs by 48%, but that neither 10 μm lavendustin A nor 100 μm tyrphostin 51 affected the current. In the present study, we elected to investigate the effects of two other broad-spectrum TK inhibitors, tyrphostin A23 and A25. Although these compounds have been utilized in earlier studies on TK regulation of ion channels (e.g. Wijetunge et al. 1992; Wu & Cohen, 1997; Ogura et al. 1999; Du et al. 2004), they have not previously been utilized to evaluate involvement of TK in the acute regulation of cardiac Ks channels. We found that tyrphostins A23 and A25 inhibited IKs with IC50 values of 4.1 ± 0.6 and 12.3 ± 2.1 μm, respectively, whereas inactive tyrphostins A1 and A63 had little effect on the current. These results complement those obtained with genistein and its analogues, and appear to rule out direct channel block as the mechanism responsible for the inhibition of IKs by broad-spectrum TK inhibitors. Rather, they point to inhibition of constitutively active TK as the likely mechanism (see also below).
To hone in on the type of TK that may be involved in the regulation of Ks channels, we measured the responses of IKs to the EGFR TK inhibitor AG1478, the Src TK inhibitor PP2, and the inactive analogue PP3 (all in the 10 μm range). IKs was unaffected by AG1478, suggesting that Ks channels are little influenced by suppression of basal EGFR TK activity. On the other hand, the current was strongly inhibited by PP2, and this inhibition was not replicated by its analogue PP3. These observations suggest that in common with a wide variety of other ion channels (e.g. neuronal Kv1.3 (Holmes et al. 1996), smooth muscle L-type Ca2+ (Hu et al. 1998), cardiac volume-sensitive Cl− (Ren & Baumgarten, 2005), and cardiac Na+ (Ahern et al. 2005) channels), cardiac Ks channels are regulated by Src TK. Identification of the type of Src TK involved in the regulation, and investigation of the modulation of its activity by signalling molecules such as COOH terminal Src kinase (Csk), PTPs, receptor TKs, focal adhesion kinase, and integrins (see Cohen, 2005), are challenging future tasks.
Effects of orthovanadate on IKs
Orthovanadate is a PTP inhibitor that has frequently been used to help evaluate whether a particular electrophysiological action of a TK inhibitor is likely to be due to an inhibition of tyrosine phosphorylation (e.g. Ogura et al. 1999; Gamper et al. 2003; Ren & Baumgarten, 2005). In the present study, we found that orthovanadate had a strong stimulatory effect on IKs in myocytes that were pretreated with TK inhibitors. Depending on the type and concentration of the TK inhibitor, the stimulation elicited by orthovanadate was as pronounced as the stimulation elicited by 1 μm forskolin in control myocytes (Figs 7–9). These findings raise the question of whether the stimulation by orthovanadate was due to an enhancement of PKA-mediated phosphorylation of Ks channel protein. In theory, such an enhancement could be due to stimulation of PKA activity or to inhibition of serine/threonine phosphatase activity. As the data in Fig. 9 appear to rule out the latter possibility, the question narrows to whether orthovanadate stimulates PKA activity. The main argument against this possibility is that orthovanadate only augmented the amplitude of basal IKs by 6 ± 3%, a very modest increase by comparison with the 52 ± 6% increase induced by a low concentration (0.2 μm) of forskolin. It could be argued that the weak PKA-stimulatory action of orthovanadate was potentiated in TK inhibitor-pretreated myocytes due to a (masked) concomitant PKA-stimulatory action of TK inhibitor (such as an inhibition of phosphodiesterase (Nichols & Morimoto, 2000)). However, we found no evidence of a masked facilitatory effect of TK inhibitor on PKA activity; that is, the response of IKs to a low concentration of forskolin was the same in TK inhibitor-pretreated myocytes as in control myocytes (Fig. 6A and B).
Possible mechanisms underlying TK and PTP inhibitor actions on IKs
The results obtained with TK inhibitors, inactive analogues and orthovanadate suggest that a decrease in basal tyrosine phosphorylation causes a decrease in basal IKs. The pertinent tyrosine phosphorylation could be on protein that directly or indirectly affects Ks channel activity, or on the Ks channel protein itself. With regard to the first possibility, we investigated whether inhibition of IKs by TK inhibitors was likely to be mediated via inhibition of serine/threonine kinases that have known or potential regulatory actions on Ks channels. However, of the four kinase inhibitors tested (PKA/PKG/PKB inhibitor H89, PKC inhibitor bisindolylmaleimide I, MEK1 inhibitor PD98059 and p38 inhibitor SB202190), not one was nearly as effective as the TK inhibitors in suppressing basal IKs. This suggests that TK inhibitor action on IKs was not mediated by inhibition of any of the six aforementioned serine/threonine kinases (or by stimulation of the corresponding phosphatases). While mediation of TK inhibitor action via other potential channel-regulatory mechanisms has not been ruled out, the most straightforward explanation of the present results is that inhibition of basal IKs by TK inhibition was due to a lowering of basal tyrosine phosphorylation on Ks channel protein. Application of Net Phos software (Blom et al. 1999) indicates that there are at least six high-score tyrosine phosphorylation sites on KCNQ1, and mutations of four of these (Y111A (Jespersen et al. 2004), Y184S (Jongbloed et al. 1999; Tranebjaerg et al. 1999), Y281C (Tranebjaerg et al. 1999; Bianchi et al. 2000) and Y315S (Jongbloed et al. 1999; Chouabe et al. 2000)) have been associated with impaired channel function and long QT syndrome. Site-directed mutagenesis and electrophysiological characterization of mutants is required to establish the relative importance of phosphorylation of these and other tyrosine sites in the regulation of cardiac Ks channels.
Acknowledgments
The authors thank Gina Dickie for excellent technical assistance. Funding for the study was provided by the Heart and Stroke Foundation of New Brunswick and the Canadian Institutes of Health Research (CIHR).
References
- Ahern CA, Zhang JF, Wookalis MJ, Horn R. Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circ Res. 2005;96:991–998. doi: 10.1161/01.RES.0000166324.00524.dd. [DOI] [PubMed] [Google Scholar]
- Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem. 1987;262:5592–5595. [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Asai T, Shuba LM, Pelzer DJ, McDonald TF. PKC-independent inhibition of cardiac L-type Ca2+ channel current by phorbol esters. Am J Physiol. 1996;270:H620–H627. doi: 10.1152/ajpheart.1996.270.2.H620. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi L, Priori SG, Napolitano C, Surewicz KA, Dennis AT, Memmi M, Schwartz PJ, Brown AM. Mechanisms of IKs suppression in LQT1 mutants. Am J Physiol Heart Circ Physiol. 2000;279:H3003–H3011. doi: 10.1152/ajpheart.2000.279.6.H3003. [DOI] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Browe DM, Baumgarten CM. Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl− current elicited by beta1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol. 2004;124:273–287. doi: 10.1085/jgp.200409040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayabyab FS, Schlichter LC. Regulation of an ERG K+ current by Src tyrosine kinase. J Biol Chem. 2002;277:13673–13681. doi: 10.1074/jbc.M108211200. [DOI] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino) ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- Chouabe C, Neyroud N, Richard P, Denjoy I, Hainque B, Romey G, Drici MD, Guicheney P, Barhanin J. Novel mutations in KvLQT1 that affect IKs activation through interactions with Isk. Cardiovasc Res. 2000;45:971–980. doi: 10.1016/s0008-6363(99)00411-3. [DOI] [PubMed] [Google Scholar]
- Cohen DM. SRC family kinases in cell volume regulation. Am J Physiol Cell Physiol. 2005;288:C483–C493. doi: 10.1152/ajpcell.00452.2004. [DOI] [PubMed] [Google Scholar]
- Crepel V, Panenka W, Kelly ME, MacVicar BA. Mitogen-activated protein and tyrosine kinases in the activation of astrocyte volume-activated chloride current. J Neurosci. 1998;18:1196–1206. doi: 10.1523/JNEUROSCI.18-04-01196.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Gui P, Hill MA, Wilson E. Regulation of ion channels by protein tyrosine phosphorylation. Am J Physiol Heart Circ Physiol. 2001;281:H1835–H1862. doi: 10.1152/ajpheart.2001.281.5.H1835. [DOI] [PubMed] [Google Scholar]
- Du XL, Gao Z, Lau CP, Chiu SW, Tse HF, Baumgarten CM, Li GR. Differential effects of tyrosine kinase inhibitors on volume-sensitive chloride current in human atrial myocytes: evidence for dual regulation by Src and EGFR kinases. J Gen Physiol. 2004;123:427–439. doi: 10.1085/jgp.200409013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman LC, Kwok WM, Kass RS. Phosphorylation-independent regulation of cardiac IK by guanine nucleotides and isoproterenol. Am J Physiol. 1992;262:H1298–H1302. doi: 10.1152/ajpheart.1992.262.4.H1298. [DOI] [PubMed] [Google Scholar]
- Gamper N, Stockand JD, Shapiro MS. Subunit-specific modulation of KCNQ potassium channels by Src tyrosine kinase. J Neurosci. 2003;23:84–95. doi: 10.1523/JNEUROSCI.23-01-00084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavi S, Shumay E, Wang HY, Malbon CC. G-protein-coupled receptors and tyrosine kinases: crossroads in cell signaling and regulation. Trends Endocrinol Metab. 2006;17:48–54. doi: 10.1016/j.tem.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Gazit A, Yaish P, Gilon C, Levitzki A. Tyrphostins I: synthesis and biological activity of protein tyrosine kinase inhibitors. J Med Chem. 1989;32:2344–2352. doi: 10.1021/jm00130a020. [DOI] [PubMed] [Google Scholar]
- Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Habuchi Y, Tanaka H, Furukawa T, Tsujimura Y, Takahashi H, Yoshimura M. Endothelin enhances delayed potassium current via phospholipase C in guinea pig ventricular myocytes. Am J Physiol. 1992;262:H345–H354. doi: 10.1152/ajpheart.1992.262.2.H345. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Harvey RD, Hume JR. Autonomic regulation of delayed rectifier K+ current in mammalian heart involves G proteins. Am J Physiol. 1989;257:H818–H823. doi: 10.1152/ajpheart.1989.257.3.H818. [DOI] [PubMed] [Google Scholar]
- Hausdorff WP, Pitcher JA, Luttrell DK, Linder ME, Kurose H, Parsons SJ, Caron MG, Lefkowitz RJ. Tyrosine phosphorylation of G protein alpha subunits by pp60c-src. Proc Natl Acad Sci U S A. 1992;89:5720–5724. doi: 10.1073/pnas.89.13.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath BM, Terrar DA. Separation of the components of the delayed rectifier potassium current using selective blockers of IKr and IKs in guinea-pig isolated ventricular myocytes. Exp Physiol. 1996;81:587–603. doi: 10.1113/expphysiol.1996.sp003961. [DOI] [PubMed] [Google Scholar]
- Heath BM, Terrar DA. Protein kinase C enhances the rapidly activating delayed rectifier potassium current, IKr, through a reduction in C-type inactivation in guinea-pig ventricular myocytes. J Physiol. 2000;522:391–402. doi: 10.1111/j.1469-7793.2000.t01-2-00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev. 2000;80:173–210. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- Holmes TC, Fadool DA, Levitan IB. Tyrosine phosphorylation of the Kv1.3 potassium channel. J Neurosci. 1996;16:1581–1590. doi: 10.1523/JNEUROSCI.16-05-01581.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XQ, Singh N, Mukhopadhyay D, Akbarali HI. Modulation of voltage-dependent Ca2+ channels in rabbit colonic smooth muscle cells by c-Src and focal adhesion kinase. J Biol Chem. 1998;273:5337–5342. doi: 10.1074/jbc.273.9.5337. [DOI] [PubMed] [Google Scholar]
- Jespersen T, Rasmussen HB, Grunnet M, Jensen HS, Angelo K, Dupuis DS, Vogel LK, Jorgensen NK, Klaerke DA, Olesen SP. Basolateral localisation of KCNQ1 potassium channels in MDCK cells: molecular identification of an N-terminal targeting motif. J Cell Sci. 2004;117:4517–4526. doi: 10.1242/jcs.01318. [DOI] [PubMed] [Google Scholar]
- Jongbloed RJ, Wilde AA, Geelen JL, Doevendans P, Schaap C, Van Langen I, van Tintelen JP, Cobben JM, Beaufort-Krol GC, Geraedts JP, Smeets HJ. Novel KCNQ1 and HERG missense mutations in Dutch long-QT families. Hum Mutat. 1999;13:301–310. doi: 10.1002/(SICI)1098-1004(1999)13:4<301::AID-HUMU7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Lei M, Brown HF, Terrar DA. Modulation of delayed rectifier potassium current, Ik, by isoprenaline in rabbit isolated pacemaker cells. Exp Physiol. 2000;85:27–35. [PubMed] [Google Scholar]
- Liu W, Akhand AA, Kato M, Yokoyama I, Miyata T, Kurokawa K, Uchida K, Nakashima I. 4-hydroxynonenal triggers an epidermal growth factor receptor-linked signal pathway for growth inhibition. J Cell Sci. 1999;112:2409–2417. doi: 10.1242/jcs.112.14.2409. [DOI] [PubMed] [Google Scholar]
- Ma H, Matsunaga H, Li B, Schieffer B, Marrero MB, Ling BN. Ca2+ channel activation by platelet-derived growth factor-induced tyrosine phosphorylation and Ras guanine triphosphate-binding proteins in rat glomerular mesangial cells. J Clin Invest. 1996;97:2332–2341. doi: 10.1172/JCI118676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubayashi T, Matsuura H, Ehara T. On the mechanism of the enhancement of delayed rectifier K+ current by extracellular ATP in guinea-pig ventricular myocytes. Pflugers Arch. 1999;437:635–642. doi: 10.1007/PL00008090. [DOI] [PubMed] [Google Scholar]
- Nichols MR, Morimoto BH. Differential inhibition of multiple cAMP phosphodiesterase isozymes by isoflavones and tyrphostins. Mol Pharmacol. 2000;57:738–745. doi: 10.1124/mol.57.4.738. [DOI] [PubMed] [Google Scholar]
- Ogura T, Shuba LM, McDonald TF. Action potentials, ionic currents and cell water in guinea pig ventricular preparations exposed to dimethyl sulfoxide. J Pharmacol Exp Ther. 1995;273:1273–1286. [PubMed] [Google Scholar]
- Ogura T, Shuba LM, McDonald TF. L-type Ca2+ current in guinea pig ventricular myocytes treated with modulators of tyrosine phosphorylation. Am J Physiol. 1999;276:H1724–H1733. doi: 10.1152/ajpheart.1999.276.5.H1724. [DOI] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Poppleton H, Sun H, Fulgham D, Bertics P, Patel TB. Activation of Gsd by the epidermal growth factor receptor involves phosphorylation. J Biol Chem. 1996;271:6947–6951. doi: 10.1074/jbc.271.12.6947. [DOI] [PubMed] [Google Scholar]
- Ren Z, Baumgarten CM. Antagonistic regulation of swelling-activated Cl− current in rabbit ventricle by Src and EGFR protein tyrosine kinases. Am J Physiol Heart Circ Physiol. 2005;288:H2628–H2636. doi: 10.1152/ajpheart.00992.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoshima J, Qiu Z, Morgan JP, Izumo S. Tyrosine kinase activation is an immediate and essential step in hypotonic cell swelling-induced ERK activation and c-fos gene expression in cardiac myocytes. EMBO J. 1996;15:5535–5546. [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC. Long QT syndrome: ionic basis and arrhythmia mechanism in long QT syndrome type 1. J Cardiovasc Electrophysiol. 2000;11:710–712. doi: 10.1111/j.1540-8167.2000.tb00035.x. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader LA, Birnbaum SG, Nadin BM, Ren Y, Bui D, Anderson AE, Sweatt JD. ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am J Physiol Cell Physiol. 2006;290:C852–861. doi: 10.1152/ajpcell.00358.2005. [DOI] [PubMed] [Google Scholar]
- Shenolikar S, Nairn AC. Protein phosphatases: recent progress. Adv Second Messenger Phosphoprotein Res. 1991;23:1–121. [PubMed] [Google Scholar]
- Swarup G, Cohen S, Garbers DL. Inhibition of membrane phosphotyrosyl-protein phosphatase activity by vanadate. Biochem Biophys Res Commun. 1982;107:1104–1109. doi: 10.1016/0006-291x(82)90635-0. [DOI] [PubMed] [Google Scholar]
- Terrenoire C, Clancy CE, Cormier JW, Sampson KJ, Kass RS. Autonomic control of cardiac action potentials: role of potassium channel kinetics in response to sympathetic stimulation. Circ Res. 2005;96:e25–34. doi: 10.1161/01.RES.0000160555.58046.9a. [DOI] [PubMed] [Google Scholar]
- Tohse N, Kameyama M, Irisawa H. Intracellular Ca2+ and protein kinase C modulate K+ current in guinea pig heart cells. Am J Physiol. 1987;253:H1321–H1324. doi: 10.1152/ajpheart.1987.253.5.H1321. [DOI] [PubMed] [Google Scholar]
- Tohse N, Nakaya H, Kanno M. Alpha 1-adrenoceptor stimulation enhances the delayed rectifier K+ current of guinea pig ventricular cells through the activation of protein kinase C. Circ Res. 1992;71:1441–1446. doi: 10.1161/01.res.71.6.1441. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Tranebjaerg L, Bathen J, Tyson J, Bitner-Glindzicz M. Jervell and Lange–Nielsen syndrome: a Norwegian perspective. Am J Med Genet. 1999;89:137–146. [PubMed] [Google Scholar]
- Umemori H, Inoue T, Kume S, Sekiyama N, Nagao M, Itoh H, Nakanishi S, Mikoshiba K, Yamamoto T. Activation of the G protein Gq/11 through tyrosine phosphorylation of the alpha subunit. Science. 1997;276:1878–1881. doi: 10.1126/science.276.5320.1878. [DOI] [PubMed] [Google Scholar]
- Walsh KB, Kass RS. Regulation of a heart potassium channel by protein kinase A and C. Science. 1988;242:67–69. doi: 10.1126/science.2845575. [DOI] [PubMed] [Google Scholar]
- Walsh KB, Kass RS. Distinct voltage-dependent regulation of a heart-delayed IK by protein kinases A and C. Am J Physiol. 1991;261:C1081–C1090. doi: 10.1152/ajpcell.1991.261.6.C1081. [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Ward CA, Giles WR. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J Physiol. 1997;500:631–642. doi: 10.1113/jphysiol.1997.sp022048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washizuka T, Horie M, Obayashi K, Sasayama S. Genistein inhibits slow component delayed-rectifier K currents via a tyrosine kinase-independent pathway. J Mol Cell Cardiol. 1998;30:2577–2590. doi: 10.1006/jmcc.1998.0815. [DOI] [PubMed] [Google Scholar]
- Wijetunge S, Aalkjaer C, Schachter M, Hughes AD. Tyrosine kinase inhibitors block calcium channel currents in vascular smooth muscle cells. Biochem Biophys Res Commun. 1992;189:1620–1623. doi: 10.1016/0006-291x(92)90262-j. [DOI] [PubMed] [Google Scholar]
- Wu JY, Cohen IS. Tyrosine kinase inhibition reduces If in rabbit sinoatrial node myocytes. Pflugers Arch. 1997;434:509–514. doi: 10.1007/s004240050430. [DOI] [PubMed] [Google Scholar]
- Yazawa K, Kameyama M. Mechanism of receptor-mediated modulation of the delayed outward potassium current in guinea-pig ventricular myocytes. J Physiol. 1990;421:135–150. doi: 10.1113/jphysiol.1990.sp017937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Wang J, Han H, Nattel S, Wang Z. Normal function of HERG K+ channels expressed in HEK293 cells requires basal protein kinase B activity. FEBS Lett. 2003;534:125–132. doi: 10.1016/s0014-5793(02)03804-8. [DOI] [PubMed] [Google Scholar]
- Zhou YY, Yao JA, Tseng GN. Role of tyrosine kinase activity in cardiac slow delayed rectifier channel modulation by cell swelling. Pflugers Arch. 1997;433:750–757. doi: 10.1007/s004240050341. [DOI] [PubMed] [Google Scholar]