

Abstract
T lymphocytes encounter hypoxia when they migrate to pathological sites such as tumours and wounds. The inability of T cells to provide an efficient defence at these sites can in part be explained by the hypoxic environment. Kv1.3 channels, important components of the T cell activation process are inhibited by hypoxia and their inhibition accounts for a hypoxia-induced decrease in T cell proliferation. Although Kv1.3 channels play a key role in T cell O2 sensing, the signalling mechanisms mediating their response to hypoxia are still not understood. In this study, we show that the src-protein tyrosine kinase p56Lck (Lck) is required for Kv1.3 channel response to hypoxia. Pre-exposure to the src inhibitor PP2 abolished the hypoxia-induced inhibition of Kv1.3 channels in primary human T lymphocytes. Moreover, Kv1.3 channel sensitivity to hypoxia was lost in Lck-deficient Jurkat T cells. Further studies with recombinant Kv1.3 channels showed that Kv1.3 channels lack intrinsic O2 sensitivity, but delivery of Lck into the cells and transfection of a constitutively active Lck (Y505FLck) restored their sensitivity to hypoxia. Although Lck is necessary for the Kv1.3 channel response to hypoxia, it does not directly inhibit Kv1.3 channels. Indeed, under normal oxygen tension, delivery of active Lck into L929 cells and overexpression of Y505FLck did not decrease recombinant Kv1.3 currents. On the contrary, activation of endogenous src kinases increased wild-type Kv1.3 currents in T lymphocytes. Our findings indicate that Lck is required for the acute response to hypoxia of human T lymphocytes as it is necessary to confer O2 sensitivity on Kv1.3 channels.
The microenvironment at pathological sites such as tumours and wounds is characterized by particular physico-chemical conditions (hypoxia, low pH and low glucose) capable of affecting the function of the different cell types involved in the progression of the disease. Immune cells are important determinants of disease progression and it has been proposed that the decreased immune surveillance observed in solid tumours could be explained in part by the effects of the tumour microenvironment on responding immune cells (Whiteside, 1998; Marincola et al. 2000). Numerous studies have indicated that hypoxia inhibits the function of T lymphocytes, thus decreasing their defence capabilities (Loeffler et al. 1990, 1991, 1992; Zuckerberg et al. 1994; Naldini & Carraro, 1999; Caldwell et al. 2001; Conforti et al. 2003). Still, the mechanisms by which hypoxia inhibits T cell function have not been fully elucidated. We have shown that Kv1.3 channels mediate the response to hypoxia of human T lymphocytes (Conforti et al. 2003; Robbins et al. 2005). Kv1.3 channels are expressed in T cells, where they participate in the T cell activation process, thus regulating proliferation and cytokine production/release (Cahalan et al. 2001).
T cell activation results from the concerted function of various membrane channels and signalling molecules (Cahalan et al. 2001). Binding of the antigen to the T cell receptor (TCR) complex leads to the coordinated activation of protein tyrosine kinases, which causes Ca2+ efflux from the endoplasmatic reticulum through the activation of phospholipase Cγ. Depletion of the intracellular calcium stores activates the calcium release activated calcium (CRAC) channels, resulting in Ca2+ influx into the lymphocyte. The electrochemical driving force necessary for Ca2+ influx is maintained by the voltage-dependent Kv1.3 and the Ca2+-activated KCa3.1 channels (Cahalan et al. 2001; Lewis, 2001). Expression of these channels depends on the T cell activation state and Kv1.3 channels constitute the dominant K+ conductance of resting T lymphocytes (Chandy et al. 2004). Inhibition of Kv1.3 channels induces membrane depolarization and suppresses the activation response of human primary T cells (Cahalan et al. 2001). The importance of Kv1.3 channels in T cell activation is emphasized by the recent effort to design specific Kv1.3 channel blockers to use as immunosuppressive agents (Chandy et al. 2004). Still, the early ionic membrane events triggered upon receptor binding constitute one of the least well understood steps in the overall TCR activation process (Winslow & Crabtree, 2005). Even more limited information is available on the ionic mechanisms underlying altered T lymphocyte behaviour in hypoxia. Our laboratory has shown that hypoxia inhibits both Kv1.3 channel expression and function in human T cells and that this inhibition correlates with a decrease in TCR-mediated Ca2+ influx and proliferation (Conforti et al. 2003; Robbins et al. 2005). However, the mechanisms that mediate the O2 sensitivity of Kv1.3 channels in human T cells are yet to be understood.
Various signalling pathways, including protein tyrosine kinases (PTK) as well as serine/threonine kinases, can modulate Kv1.3 channel activity (Payet & Dupuis, 1992; Holmes et al. 1996a; Szabo et al. 1996, 1997; Chung & Schlichter, 1997; Fadool et al. 1997; Gulbins et al. 1997; Holmes et al. 1997; Fadool, 1998; Fadool & Levitan, 1998; Dellis et al. 1999; Cook & Fadool, 2002). Cayabyab et al. (2000) showed that inhibition of Kv1.3 current by anoxia/glucose deprivation in rat microglia is mediated by src-family PTKs. The main src-PTKs expressed in T cells are p56lck (Lck) and p59fyn (Fyn) (Palacios & Weiss, 2004). In human T cells, Lck has been reported to exist in physical association with Kv1.3 channels via the PDZ-containing Drosophila discs large (hDlg) protein which is able to bind both Lck and the Kv1.3 alpha subunit (Hanada et al. 1997). Furthermore, Lck has been implicated in mediating the inhibition of Kv1.3 current in Jurkat T cells upon Fas receptor ligation, ceramide treatment, and HIV-1 exposure (Szabo et al. 1996; Gulbins et al. 1997; Dellis et al. 1999). Although these latter studies demonstrated that Lck is necessary to mediate Kv1.3 current inhibition, they did not conclusively showed that Lck is by itself sufficient to down-regulate Kv1.3 channel activity.
In this paper we have investigated the role of Lck in mediating Kv1.3 channel response to hypoxia in primary human T cells. Furthermore, we have directly examined the functional correlation between active Lck and Kv1.3 channel activity.
The data presented here indicate that Lck is required for Kv1.3 modulation by hypoxia, but that Lck itself has no direct inhibitory effect on Kv1.3 channels in human T lymphocytes.
Methods
Cells
CD4+ and CD3+ lymphocytes were obtained from the blood of healthy donors by E-rosetting (StemCell Tech., Vancouver, Canada) and Ficoll density gradient separation (Amersham Biosciences, Uppsala, Sweden) as previously described (Conforti et al. 2003). The purity of the CD4+ population was confirmed by fluorescence-activated cell sorting (FACS) analysis: 98% of the cells isolated were CD4+ (n = 8 donors). T lymphocytes were maintained in RPMI medium supplemented with 10% pooled male human AB serum (Sigma, St. Louis, MO), 200 units ml−1 penicillin, 200 μg ml−1 streptomycin, 1 mm Hepes, in a humidified incubator at 37°C with 5% CO2. L929 cells stably expressing mKv1.3 (a kind gift of G. Chandy, University of California, Irvine) (Grissmer et al. 1994) were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal calf serum, 4 mml-glutamine, 1 mm sodium pyruvate and 500 μg ml−1 G418. Jurkat (E6-1; ATCC, Manassas, VA, USA) and JCaM1.6 cells (kind gift of Dr A. Weiss, University of California, San Francisco) were maintained in the same RPMI medium used for fresh T cells with FBS substituted for human serum. All the experiments with primary T lymphocytes were done exclusively on resting T cells to minimize the possible contribution of KCa3.1 channels to the overall response to hypoxia (Chandy et al. 2004).
Electrophysiology
Patch-clamp experiments were performed either in the cell-attached or in the whole cell configuration as previously described (Conforti & Millhorn, 1997; Conforti et al. 2003). Kv1.3 whole-cell currents were recorded with an external solution of the following composition (mm): 150 NaCl, 5 KCl, 2.5 CaCl2, 1.0 MgCl2, 10 glucose and 10 Hepes, pH 7.4. The pipette solution was composed of (mm): 134 KCl, 1 CaCl2, 10 EGTA, 2 MgCl2, 5 ATP-sodium and 10 Hepes, pH 7.4, with an estimated free Ca2+ concentration of 10 nm (Szabo et al. 1997). Single channel experiments were performed in cell-attached configuration using 5 MΩ fire-polished and Sylgard-coated electrodes (WPI, Sarasota, FL). The external and pipette solution compositions were (in mM): 110 K-gluconate, 20 KCl, 2.0 MgCl2, 10 Hepes, 1.0 CaCl2, 5 glucose, 11 EGTA (pH 7.4) and 140 NMG-gluconate, 2.8 KCl, 2.0 MgCl2, 10 Hepes, and 2.0 CaCl2 (pH 7.4), respectively. Single channel conductance is reported as slope factor of the fitted single-channel I–V relationship measured by ramp depolarization from −80 mV to +40 mV (900 ms duration). The number of active channels in a patch (N) was determined at +50 mV, by observing the maximum number of simultaneous current steps that appear over a long recording period. The open state probability (NPo) was calculated by dividing the time spent in the open state at each current level by the total duration of the recording. Experiments were performed using an Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA). The digitized signals were stored and analysed using pCLAMP 8 software (Axon Instruments). Experiments were conducted at room temperature (22°C). To study the effect of hypoxia, cells were placed in a perfusion chamber where the oxygen tension (PO2) of the medium could be controlled. This chamber allowed an immediate exchange of solution and equilibration with the perfusate saturated with the desired gas composition (Conforti et al. 2003). The effect of hypoxia was studied by switching from a perfusion medium bubbled with air (21% O2, PO2ca 150 mmHg) to a medium equilibrated with 100% N2 (PO2 in the perfusion chamber ca 20 mmHg). The PO2 in the chamber is monitored by a polarographic O2 electrode (WPI, Sarasota, FL, USA). Throughout the whole paper, when referring to hypoxia we refer to a PO2 of 20 mmHg, unless otherwise indicated.
Membrane potential measurements
The membrane potential of CD4+ cells was measured by flow cytometry as previously described (Robbins et al. 2005). Briefly, ca 1 × 106 cells ml−1 were suspended in a solution of the following composition (mm): 150 NaCl, 5 KCl, 1.8 CaCl2, 1 MgCl2, 10 Hepes, 10 glucose (pH 7.4). T cells were then exposed to either normoxia or hypoxia for 15 min at room temperature. To study the effect of hypoxia on membrane potential, cells were maintained in a modular incubator chamber (Billups-Rothenberg, Inc., Del Mar, CA, USA) in an atmosphere saturated with 1% O2, 5% CO2, and 94% N2. The 75 mm potassium solution was made by altering the KCl to 75 mm and adjusting the NaCl concentration to a total monovalent cation concentration of 155 mm. To measure the membrane potential, 300 nm bis-(1,3-dibutylbarbituric acid)trimethine oxonol (DiBAC4(3)) dye (Molecular Probes, Eugene, OR, USA) was added to the cells, and 5 min later the fluorescence intensity of the T cell population was analysed with a FACSCalibur flow cytometer (Becton-Dickinson, San Jose, CA, USA); 488 nm wavelength excitation. DiBAC4(3) was prepared in DMSO according to the manufacturer's instructions. All flow cytometric analyses were accomplished using CellQuest software (Becton-Dickinson).
Immunoprecipitation
Freshly isolated human CD3+ cells were maintained for 24 h in normoxia (150 mmHg), followed by incubation for the indicated time in either normoxia or hypoxia (1% O2, ca 8 mmHg) in a cell incubator with controlled PO2 (ThermoForma, Marietta, OH, USA). After this time, cells were lysed with a lysis buffer of the following composition: 10 mm Tris, 1 mm EDTA, 1 mm PMSF, 1% Triton X-100, 2 μg ml−1 aprotinin, 2 μg ml−1 leupeptin, 1 mm sodium orthovanadate. Cell lysates were sonicated for 30 s, and centrifuged at 15 000 g for 10 min at 4°C. Supernatant was precleared with protein G agarose beads (Upstate Biotech., Lake Placid, NY, USA) for 1 h at 4°C. Beads were removed by centrifugation at 15 000 g for 10 min at 4°C and protein content was measured using the Pierce BSA Protein Assay. Five hundred micrograms of precleared protein lysate in 500 μl lysis buffer was combined with 5 μl anti-phosphotyrosine antibody (Cell Signalling Technology, Beverly, MA, USA) as the precipitating antibody and the sample was incubated at 4°C for 2 h on a rocking platform. Protein G agarose beads and 1.25 μl horse anti-mouse antibody (Pierce, Rockford, IL, USA) were then added and incubated overnight at 4°C. Beads were then sedimented, washed with lysis buffer and the bound proteins eluted from the beads by boiling in SDS sample buffer. Proteins were separated by SDS-PAGE and immunoblotted with either polyclonal anti-Lck antibody (Upstate Biotech.) or anti-Kv1.3 antibody (Alomone Laboratories, Jerusalem, Israel). After blotting, the membrane was washed and treated with horseradish peroxidase-conjugated secondary antibody. The immunoreactive bands were visualized using enhanced chemiluminescence reagents (Pierce). The levels of tyrosine phosphorylated proteins were determined by densitometric analysis of the immunoprecipitated bands normalized to the corresponding values in the crude protein blots.
Near-infrared cytoblot assay
Freshly isolated CD3+ cells were plated on poly l-lysine coated coverslips and then perfused with either normoxic (150 mmHg) or hypoxic (20 mmHg) perfusate as described in the electrophysiological experiments. The cells were then fixed with 3.7% formaldehyde for 20 min and permeabilized with 0.1% Triton X-100 in PBS. Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE, USA) was then added and the cells were incubated for 1.5 h at room temperature with gentle rocking. Blocking buffer was discarded prior to antibody staining. Tyrosine phosphorylation of Lck was detected by either anti-pY505Lck (pY505Lck) or anti-phospho-src family Y416 (pY416) polyclonal antibodies (Cell Signalling Technology). The latter antibody recognizes active src-PTKs and was used to detect the active Lck form, phosphorylated at the Y394, since, to our knowledge, an anti-pY394 specific antibody is not commercially available. To normalize the phosphorylation signal against total target protein, each of the phospho-specific antibodies was diluted in blocking buffer with the pan monoclonal anti-Lck antibody (Upstate Biotech.) and the cells were incubated overnight with the primary antibody mixtures at 4°C. After five washes with 0.1% Tween 20, cells were then incubated with the appropriate labelled secondary antibodies, anti-rabbit Alexa Fluor 680 (Invitrogen, Carlsbad, CA, USA) and anti-mouse IRDye 800CW (Rockland Immunochemicals, Gilbertsville, PA, USA) for 60 min at room temperature. The cells were imaged by scanning simultaneously at 700 and 800 nm with 169 μm resolution using an Odyssey Infrared Imaging System (LI-COR Biosci.) (Chen et al. 2005).
Overexpression of constitutively active p56Lck
Constitutively active Lck (LckY505F), obtained by replacing the Y505 with F, was kindly provided by C. T. Baldari (University of Siena, Italy) (Di Somma et al. 1995). The LckY505F construct (subcloned into the green fluorescent protein (GFP)-expressing vector pIRES2-eGFP; Clontech, Mountain View, CA, USA) was transiently transfected into L929 cells expressing recombinant Kv1.3 channels using the lipofectamine method according to the manufacturer's protocol (Invitrogen). After 24 h, cells were plated on coverslips and later assayed for GFP and LckY505F expression. Fluorescence was normally observed 48 h later. Cells transfected with pIRES2-eGFP empty vector served as the control. Overexpression of Lck was determined by immunocytochemistry as previously described (Conforti et al. 2000). Briefly, transfected L929 cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. Lck was labelled with rabbit polyclonal anti-Lck antibody (Upstate Biotech.) and visualized with an Alexa-Fluor 568 conjugated secondary antibody (Invitrogen).
Data analysis
All data are presented as means ± s.e.m., unless otherwise indicated. Statistical analyses were performed using Student's t test (paired or unpaired); P ≤ 0.05 was defined as significant. Wilcoxon's test was used as a non-parametric test. For each experiment with fresh T cells, the number of experiments (n) and the number of donors are reported.
Chemicals
PP2 and PP3 were purchased from Calbiochem (San Diego, CA, USA). Active Lck was purchased from Upstate Biotechnology, Inc. Src activator peptide was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Chemicals were purchased from Sigma (St Louis, MO, USA), unless indicated otherwise.
Results
Src-PTKs mediate the hypoxic inhibition of Kv1.3 channels in human primary T lymphocytes
Cell-attached patch-clamp experiments were first performed to study the effect of hypoxia on Kv1.3 channels in resting CD4+ T lymphocytes (Fig. 1). This recording configuration maintains the cell's integrity and allows one to fully appreciate the membrane bound and cytosolic signalling molecules that may be involved in the inhibitory effects of hypoxia on Kv1.3 channels (Conforti et al. 2003). Kv1.3 single channel currents were recorded in normoxia and after 3 min exposure to hypoxia (PO2 in the chamber ca 20 mmHg). At this time a complete change of PO2 in the recording chamber and a complete and steady-state inhibition of Kv1.3 current were achieved (Conforti et al. 2003). We found that exposure to hypoxia reduced the amplitude of the peak ensemble average current by 22.4 ± 9.7% (n = 11 individual cells from 4 different donors, P = 0.044) of the current measured during normoxia (Fig. 1A). The inhibitory effect of hypoxia on Kv1.3 current was the result of a decrease in Kv1.3 channel open probability (NPo, Fig. 1B) and it occurred with no change in single channel conductance (Fig. 1C).
Figure 1. Hypoxia decreases the open probability of Kv1.3 channels in human primary T lymphocytes.
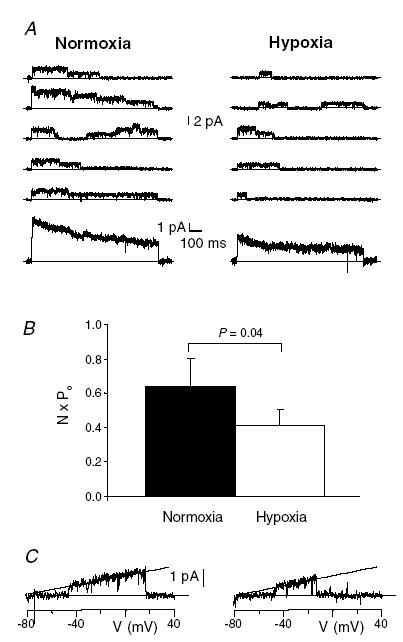
A, the upper part of the panel shows representative current traces recorded under cell-attached patch-clamp condition during 900 ms long step depolarizing pulses from −80 mV holding potential (HP) to +50 mV in normoxia and after 3 min hypoxia. Leak and capacitative currents were subtracted from the record. Upward current deflections from the zero line correspond to the opening of the channel. The corresponding ensemble average traces (from 50 consecutive traces) are shown below the representative traces. Dashed lines represent the zero current. B, average NPo values in normoxia and hypoxia (n = 11). C, hypoxia did not change the single channel conductance of Kv1.3 channels. The i–V curves were obtained with a 900 ms ramp pulse from −80 mV HP to +40 mV and the recordings were fitted with a straight line which had a slope factor of 10.9 ± 0.12 pS in normoxia (left panel) and 10.8 ± 0.18 pS in hypoxia (right panel; n = 4, P = 0.6).
The Kv1.3 response to hypoxia was completely abolished by the src-PTK inhibitor PP2 (50 nm, Fig. 2). PP2 is a protein tyrosine kinase inhibitor with selective inhibitory potential for Lck and Fyn at the concentration used (Hanke et al. 1996). Cell-attached patch-clamp experiments showed that the hypoxia-induced inhibition of Kv1.3 current was prevented by pre-exposure to PP2, but not by its inactive analogue PP3 (Fig. 2). In the presence of PP3, hypoxia significantly decreased NPo (Fig. 2) and the ensemble average current (from 2.26 ± 0.46 pA in normoxia to 1.43 ± 0.37 in hypoxia, n = 6, 4 donors, P = 0.023). Neither PP2 nor PP3 treatment affected the NPo and ensemble average current amplitudes in normoxia. The ensemble average current amplitudes were: 1.57 ± 0.32 pA (n = 11) for control (non-treated cells, Fig. 1), 2.26 ± 0.46 pA (n = 6) for PP3 (P = 0.23 versus control) and 2.28 ± 0.29 pA (n = 13) for PP2 (P = 0.11 versus control and P = 0.97 versus PP3). The NPo values in normoxia were: 0.64 ± 0.17 (n = 11) for control, 0.71 ± 0.20 (n = 6) for PP3 (P = 0.77 versus control) and 0.78 ± 0.15 (n = 12) for PP2 (P = 0.53 versus control and P = 0.81 versus PP3).
Figure 2. The src-PTK inhibitor PP2 abolishes the hypoxic inhibition of Kv1.3 channels in primary T lymphocytes.
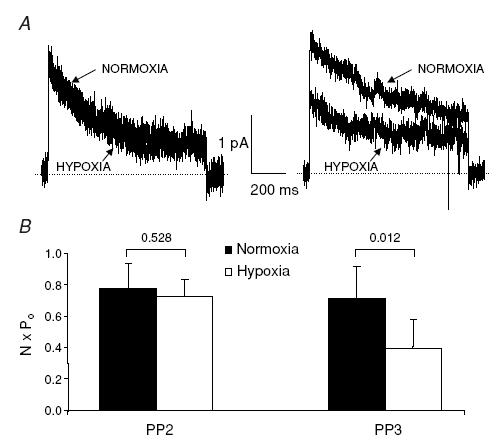
A, representative Kv1.3 ensemble average currents (from 50 consecutive traces) recorded in cell-attached patch-clamp experiments during 900 ms long step depolarizing pulses from −80 mV HP to +50 mV in normoxia and after 3 min hypoxia in the presence of either 50 nm PP2 (left panel) or PP3 (right panel). CD4+ lymphocytes were preincubated with PP2/PP3 for 20 min before electrophysiological experiments and PP2/PP3 was maintained thereafter throughout the entire experiment. Leak and capacitative currents were subtracted from the record. Dashed lines represent the zero current. B, the corresponding average NPo values in normoxia and hypoxia in the presence of PP2 (n = 13) and PP3 (n = 6).
We have previously shown that Kv1.3 channel inhibition during hypoxia induces membrane depolarization, thereby decreasing the driving force for Ca2+ influx upon stimulation of the T cell receptor (Robbins et al. 2005). We thus expect that inhibition of src-PTKs and disruption of the communication between hypoxia and Kv1.3 channel interferes with the early functional responses of T cells in hypoxia. To test whether the membrane depolarization that occurs in hypoxia is inhibited by src-PTK blockade, we exposed T cells to hypoxia in the presence or absence of either PP2 or PP3. Hypoxia-induced membrane depolarization, indicated by a shift in fluorescence similar to that observed in high [K+]o, is not affected by PP3 treatment (Fig. 3A and C). Instead, 50 nm PP2 decreased the shift in fluorescence induced by hypoxia by 55 ± 4% (n = 3 donors, P = 0.008; Fig. 3B and C). The mean channel fluorescence (MCF) in normoxia in control cells and PP3- and PP2-treated cells was not statistically different: 61 ± 10 in control (n = 5), 96 ± 24 with PP2 (50 nm, n = 3, P = 0.09 versus control) and 66 ± 19 with PP3 (1 μm, n = 2).
Figure 3. Src-protein tyrosine kinase inhibition reduces hypoxia-induced membrane depolarization in T cells.
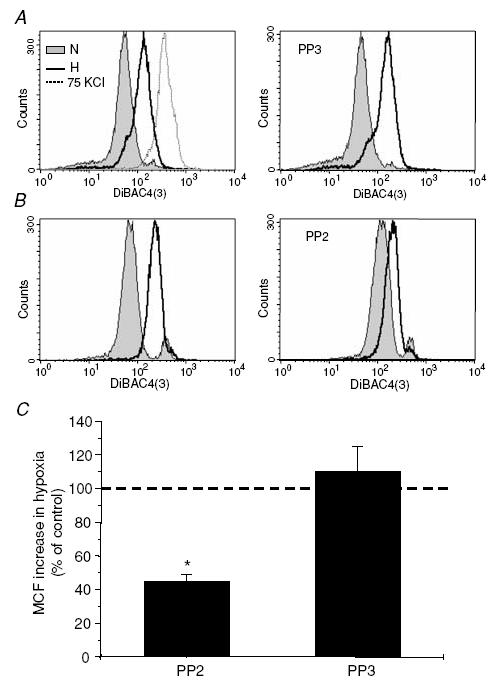
A, hypoxia-induced membrane depolarization in CD4+ lymphocytes is not affected by PP3 treatment. Hypoxia induces membrane depolarization in control (non-treated) cells, as indicated by the shift in DiBAC4(3) fluorescence in the same direction as that induced by 75 mm KCl. Similar shifts in fluorescence were induced by hypoxia in cells pre-exposed to PP3 (1 μm for 15 min, right panel). The data shown are from the same donor. B, PP2 inhibits hypoxia-induced membrane depolarization. Pre-exposure to 50 nm PP2 for 15 min reduced the shift in fluorescence produced by hypoxia (right panel) as compared to the corresponding values in control cells (left panel) in T cells from the same donor. C, average values of mean channel fluorescence (MCF) shift produced by hypoxia and compared to the shift produced in control (untreated cells) set as 100 (dashed line). n = 3 donors for PP2 and n = 2 donors for PP3. *P = 0.008.
These results indicate that src-kinases are necessary for the Kv1.3 channel response to hypoxia and the consequent membrane depolarization of freshly isolated human T lymphocytes. Still, it is not clear if these signalling molecules are directly inhibiting Kv1.3 channels.
Activation of endogenous src-PTKs does not inhibit Kv1.3 channels in T lymphocytes
The direct inhibitory effect of src-PTKs on Kv1.3 channels was tested in CD4+ lymphocytes by application of the src kinase activating peptide EPQYEEIPIYL. This peptide binds to the SH2 domain of the src-kinase and activates it (Songyang et al. 1993; Pawson, 1995). We delivered the EPQYEEIPIYL peptide into CD4+ lymphocytes through the patch pipette in whole-cell voltage-clamp experiments (Cayabyab et al. 2000; Conforti et al. 2000) and recorded the effect of src-PTK activation on the Kv1.3 current (Fig. 4). Evoked whole-cell Kv1.3 current, after an initial steady phase, continuously decreased in control cells and at 14 min it was 69 ± 4% (n = 5) of the maximum current, while in the presence of the src activator at this time point the amplitude of Kv1.3 current was 84 ± 5% of the maximum current (n = 5; P = 0.046). The continuous decrease in the Kv1.3 current amplitude upon repetitive stimulation in control cells could be explained by cumulative inactivation, exacerbated by the dilution of intracellular signalling molecules (Chung & Schlichter, 1997; Cayabyab et al. 2000). These results indicate that src-PTKs do not directly down-regulate wild-type Kv1.3 channel activity in human primary T cells, but instead they appear to be necessary to stabilize the Kv1.3 current. It still remains to be determined whether Lck or Fyn is the kinase in question.
Figure 4. Effect of src-PTK activation on Kv1.3 current in freshly isolated human T lymphocytes.
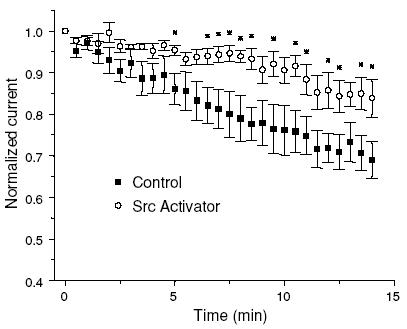
Kv1.3 currents were elicited with 900 ms long depolarizing pulses to +50 mV (−80 mV HP) every 30 s either in the present (n = 5) or absence (control, n = 5) of the src activator peptide EPQYEEIPIYL (1 mm) in the patch pipette. The peak current amplitude values (normalized for the maximum current observed after establishing a whole cell configuration) were plotted against time from the time point of maximum current recording (set as 0). *P < 0.05
Lck is the necessary mediator of O2 sensitivity of Kv1.3 channels in T cells
To further dissect the role of src-PTKs we narrowed our investigation to Lck, known to exist in T cells in physical association with Kv1.3 channels (Hanada et al. 1997). The response of Kv1.3 channels to hypoxia was therefore studied in the JCaM1.6 cell line. This is a mutant derivative of the Jurkat cell line lacking Lck while still retaining Fyn expression (Straus & Weiss, 1992). Exposure of JCaM1.6 cells to different degrees of hypoxia (20–0 mmHg) did not inhibit the Kv1.3 current (Fig. 5), while we have previously shown that a PO2 of 20 mmHg inhibits Kv1.3 current in the parent Jurkat T cell line by 20% (Conforti et al. 2003). The amplitude of Kv1.3 current in JCaM1.6 cells in hypoxia (20 mmHg) was 102 ± 3% of the normoxic value (n = 9, P = 0.40). Thus, these data indicate that Kv1.3 channels in JCaM1.6 cells lack O2 sensitivity, although they have been shown to display pharmacological and electrophysiological properties similar to Kv1.3 channels in the parent Jurkat T cells (Dellis et al. 1999). Interestingly, the current density (measured normalizing the peak Kv1.3 current to the membrane capacitance) was significantly lower in JCaM1.6 cells than in Jurkat cells (Fig. 5C). The membrane capacitances measured in Jurkat and JCaM1.6 cells were 4.2 ± 0.3 pF (n = 10) and 7.3 ± 1.0 pF (n = 9; P = 0.015), respectively. Delivery of recombinant full length Lck (1 U ml−1) into the JCaM1.6 cells through the patch pipette in whole-cell voltage-clamp experiments produced a significant increase in Kv1.3 current density, although this amount of Lck was not sufficient to restore the Kv1.3 channel density of Jurkat cells (P = 0.0002). Moreover, this amount of Lck did not restore the hypoxic response of Jurkat cells (the current amplitude in hypoxia of Lck-dialysed JCaM1.6 cells was 104 ± 4% of the normoxic value, n = 9, P = 0.35), although a lower concentration of c-srcp60 (0.5 U ml−1) was proven to be sufficient to modulate Kv1.3 current in olfactory bulb neurons (Fadool, 1998). These data are consistent with a role of Lck as a necessary mediator of the O2 sensitivity of Kv1.3 channels in T cells, although this kinase has no direct inhibitory effect on Kv1.3 channels. Additionally, they suggest that other mechanisms which might be also compromised in this mutant cell line are necessary for the channel response to hypoxia.
Figure 5. Lack of hypoxia sensitivity of Kv1.3 current in JCaM1.6 cells.
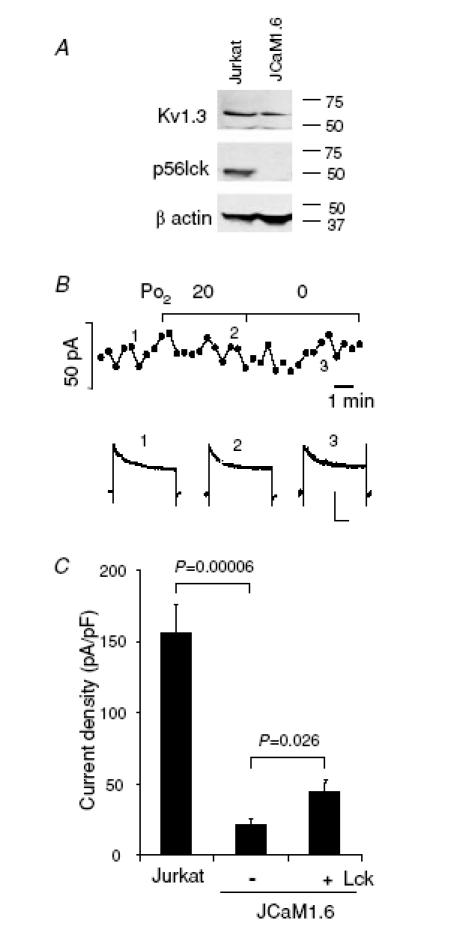
A, lack of Lck expression in JCaM1.6 cells. Immunoblot analysis of Kv1.3α subunit and Lck proteins in Jurkat (lane 1) and JCaM1.6 cells (lane 2). β-Actin was used as a constitutive protein in these samples. B, time course of Kv1.3 current amplitude measured in normoxia and during exposure to progressively more severe hypoxia (top). The continuous lines indicate the time of exposure to hypoxia. The Kv1.3 current recordings corresponding to the time-points marked by numbers are shown in the bottom panel. Scale bars correspond to 200 ms and 100 pA. Kv1.3 currents were elicited by step depolarizing steps from −80 mV HP to +50 mV, every 30 s. C, introduction of Lck into JCaM1.6 cells increases the Kv1.3 current amplitude. Kv1.3 current density, measured under whole cell patch-clamp configuration, is significantly lower in JCAM1.6 cells than in Jurkat cells (n = 10 in Jurkat and n = 9 in JCaM1.6). Kv1.3 currents were elicited as described in panel B. Introducing 1 U ml−1 active Lck into the cells by dialysing through the patch pipette significantly increases the current density in JCaM1.6 cells (n = 11).
p56Lck confers sensitivity to hypoxia on otherwise O2-insensitive recombinant Kv1.3 channels, but it displays no direct inhibitory effect
The effect of Lck on the O2 sensitivity of Kv1.3 channels was thus further investigated in recombinant Kv1.3 channels expressed in L929 cells. These recombinant channels have electrophysiological properties identical to those of the Kv1.3 channels in T cells (Grissmer et al. 1994). Recombinant Kv1.3 channels are not sensitive to hypoxia in this setting (Fig. 6A). We assessed the expression level of Lck in L929 cells by Western blotting and found that L929 cells express negligible levels of Lck as compared to T lymphocytes (data not shown). The intracellular levels of Lck were thus increased by delivering full length human recombinant Lck (0.5 U ml−1) through the patch pipette during whole cell patch clamp measurements. Hypoxia did not inhibit the recombinant Kv1.3 current in control cells (the Kv1.3 current amplitude was 2.9 ± 0.4 nA in normoxia and 2.8 ± 0.4 nA in hypoxia, n = 15, P = 0.716; Fig. 6A and C). On the other hand, after introduction of Lck by the patch pipette, hypoxia decreased the amplitude of the Kv1.3 current from 3.1 ± 0.3 nA to 2.7 ± 0.3 nA (n = 12 and n = 15, respectively, P = 0.0006; Fig. 6B and C). The Kv1.3 current densities in these two experimental conditions were similar (Fig. 6D). Kv1.3 current densities were compared at the time point before introduction of hypoxia, 10–15 min after entering into whole cell configuration. This time has been shown to be sufficient for successful delivery of proteins into cells via the patch pipette (Conforti et al. 2000). Overall, recombinant Lck did not change the amplitude of the whole cell Kv1.3 current, but conferred hypoxia sensitivity on heterologous Kv1.3 channels.
Figure 6. Delivery of active Lck into L929 cells stably expressing Kv1.3 channels does not inhibit Kv1.3 currents, but restores their hypoxia sensitivity.
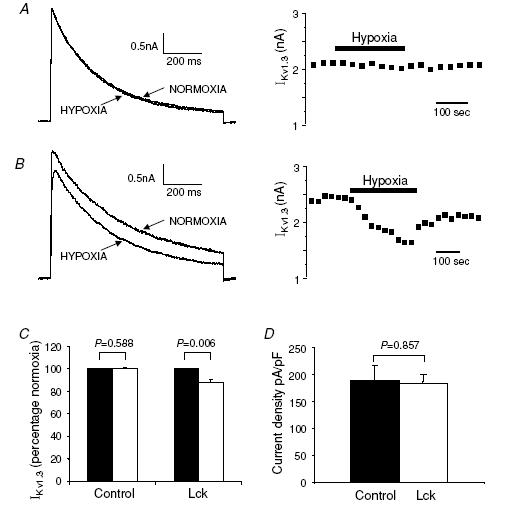
A, representative Kv1.3 current traces recorded in whole cell patch-clamp configuration were elicited with 900 ms long depolarizing pulses from −80 mV HP to +50 mV. The currents were recorded using regular pipette solution. Hypoxia has no effect on the current. The corresponding time course is shown on the right. B, Kv1.3 currents were recorded in experiments in which 0.5 U ml−1 active Lck was dialysed into the cell through the patch pipette during recording. Hypoxia inhibits the current only in the presence of active Lck. The corresponding time course is shown on the right. C, effect of hypoxia on Kv1.3 current in the presence and absence of Lck. Data are expressed as a percentage of control (normoxia, set as 100, black bars). D, introducing active Lck into the cells does not change the Kv1.3 current density (n = 12 in the presence of Lck and n = 15 in control).
There is a possibility that the lack of a direct effect of Lck on Kv1.3 channels in the above experiments results from the rapid inactivation of Lck once in the cytoplasm. Lck exists in active and inactive forms linked to the phosphorylation state of the key tyrosines Y394 and Y505 (Hermiston et al. 2002). Phosphorylation of Y394, located in the kinase domain, potentiates the kinase activity while phosphorylation of Y505 at the carboxy-terminal domain negatively regulates kinase activity. Although it is known that the full length recombinant Lck is able to activate by autophosphorylation (Jullien et al. 1994), we cannot exclude the possibility that in the above whole-cell experiments the recombinant Lck was quickly converted into its inactive analogue upon delivery into the cell. To overcome these limitations, we transiently transfected L929 cells with the constitutively active form of Lck (LckY505F). In this construct the Y505 is mutated to phenylalanine (F) to prevent phosphorylation at this site, thus abrogating the inactivation and making the Lck constitutively active (Di Somma et al. 1995). The successful overexpression of LckY505F was confirmed by immunostaining with anti-Lck antibody (Fig. 7A). The overexpression of LckY505F was confirmed by comparing the ratio of the red fluorescence intensity of Lck in the transfected cells and the surrounding non-transfected cells in mock and LckY505F-transfected cells. The Lck red fluorescence ratio was 10-fold higher in LckY505F overexpressing cells as compared to the mock transfected cells (10.5 ± 2.1 and 0.9 ± 0.1, respectively, n = 13, P = 0.0006). L929 cells overexpressing LckY505F and visualized by the GFP fluorescence were subjected to patch-clamp experiments. Overexpression of LckY505F conferred O2 sensitivity on the recombinant Kv1.3 channels (Fig. 7). Hypoxia did not change the Kv1.3 current amplitude in mock transfected cells (2.3 ± 0.7 nA in normoxia and 2.3 ± 0.7 nA in hypoxia, n = 4, P = 0.352), while in cells overexpressing LckY505F it decreased Kv1.3 current from 2.5 ± 0.3 nA to 2.2 ± 0.2 nA (n = 8, P = 0.032). LckY505F and mock transfected cells displayed similar membrane capacitances: 16.5 ± 1.4 pF (n = 11) and 12.7 ± 1.5 pF (n = 15; P = 0.08), respectively. LckY505F showed no direct inhibitory effect on Kv1.3 currents, as demonstrated by the insignificant increase in Kv1.3 current density in LckY505F compared to mock transfected cells (Fig. 7C). These results are consistent with the conclusion that Lck is required for Kv1.3 channel O2 sensitivity, but that it does not directly inhibit Kv1.3 channels.
Figure 7. Effect of constitutively active Lck on recombinant Kv1.3 current amplitude and O2 sensitivity.
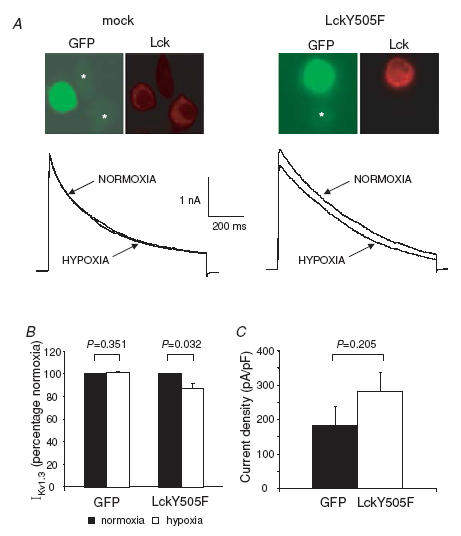
A, top panels show representative fluorescence images of rat L929 cells transiently transfected with pIRES2-EGFP empty vector (mock) and with pIRES2-EGFP vector containing constitutively active Lck (LckY505F). Non-transfected cells are marked with an asterisk in the GFP panels. In the mock experiments the intensity of red fluorescence, indicative of Lck levels, is identical in transfected and non-transfected cells. A 10-fold increase in Lck levels was instead observed in the LckY505F transfected cells. Representative Kv1.3 current traces recorded in LckY505F and mock transfected L929 cells in normoxia and after 3 min in hypoxia are shown in the bottom panels. Currents were elicited as described in the legend of Fig. 6. B, averaged effect of hypoxia on Kv1.3 currents in LckY505F and mock transfected cells. Data are expressed as a percentage of control (normoxia). C, current density in LckY505F (n = 15) and mock (n = 11) cells.
Lck activity and Kv1.3 channel tyrosine phosphorylation state in T cells during hypoxia
T lymphocytes express higher levels of endogenous Lck than L929 cells, but it is still not clear if the Lck basal activity of resting T cells is sufficient to confer O2 sensitivity on the Kv1.3 channels, or if Lck needs to be activated by hypoxia for the channel to become O2 sensitive. Biochemical studies were performed to determine whether hypoxia activates Lck. T cells were maintained in either normoxia or hypoxia (ca 8 mmHg) for 1 h, 6 h and 24 h. The level of tyrosine phosphorylated Lck was determined by immunoprecipitation (Fig. 8A). The amount of tyrosine phosphorylated Lck did not change after 1 h exposure to hypoxia (105 ± 15% of the normoxic cells, P = 0.739). The effect of shorter exposure to hypoxia (15 min) was tested in two donors and no increase in Lck tyrosine phosphorylation was detected at this time point either (95 ± 7%versus normoxia). However, after prolonged hypoxia (6–24 h) a 60% increase in Lck tyrosine phosphorylation occurred (P = 0.004). Consistent with these findings, we observed that prolonged exposure to hypoxia (24 h, ca 8 mmHg) increased tyrosine phosphorylation of Kv1.3 channels in Jurkat T cells as determined by immunoprecipitation (Fig. 8B). This exposure to hypoxia produced a 40% decrease in total Kv1.3 protein levels as previously reported (Conforti et al. 2003). Interestingly, the amount of tyrosine phosphorylated Kv1.3 protein in the immunoprecipitate in hypoxia was threefold higher than in normoxia (Fig. 8B). Overall prolonged hypoxia increased the Kv1.3 channel protein tyrosine phosphorylation level. In contrast, a shorter exposure to hypoxia (15 min) did not result in any detectable increase in tyrosine phosphorylated Kv1.3 protein (Fig. 8C). Our data are consistent with the finding that prolonged hypoxia (3 h or more) increases the tyrosine phosphorylation of Lck in MCF-7 and MDA-MB-231 human breast cancer cells (Mahabeleshwar & Kundu, 2003). They further indicate that the level of tyrosine phosphorylation of Kv1.3 channels also increases with prolonged exposure to hypoxia.
Figure 8. Activation of Lck by prolonged hypoxia and tyrosine phosphorylation of Kv1.3 channels.
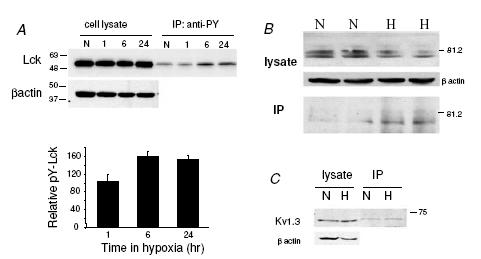
A, T lymphocytes were exposed to normoxia and hypoxia (8 mmHg) for 1, 6 and 24 h. Cell lysates containing equal amount of total proteins were immunoprecipitated with rabbit polyclonal anti-phosphotyrosine antibody and the immunoprecipitated samples were analysed by Western blotting using rabbit polyclonal anti-Lck antibody. β-Actin was used as loading control. Hypoxia induced an increase in tyrosine phosphorylated Lck level after 6 and 24 h exposure. The lower panel shows the average densitometric values of tyrosine phosphorylated Lck after 1, 6 and 24 h exposure to hypoxia. Densitometric values of the immunoprecipitated bands were normalized to the corresponding values in the crude protein blots and were expressed as a percentage of the normoxic value. n = 6 donors for 1 h, and n = 2 donors for 6 and 24 h. B, Jurkat cells were exposed to normoxia and hypoxia (PO2 of 8 mmHg) for 24 h. Cell lysates containing equal amount of total proteins were immunoprecipitated with rabbit polyclonal anti-phosphotyrosine antibody. The cell lysate and the immunoprecipitated samples (IP) were analysed by Western blotting using rabbit polyclonal anti-Kv1.3 antibody. β-Actin was used as loading control. Hypoxia induced an increase in tyrosine phosphorylated Kv1.3 level. C, experiments similar to those described in A were conducted after maintaining Jurkat cells in either normoxia or hypoxia (8 mmHg) for 15 min. This blot is representative of 2 separate experiments.
Still, in patch-clamp experiments, the observed changes in the activity of Kv1.3 channels reflect a very quick response to hypoxia, within minutes. To our knowledge, such short-term changes in kinase activity during hypoxia have never been investigated. During the immunoprecipitation experiments large numbers of cells were exposed to hypoxia in a hypoxic incubator. These experiments require a long equilibration time and therefore do not allow study of the effect of acute changes in O2 pressure. To evaluate the effect of acute hypoxia on Lck activity we used the near-infrared Cytoblot Assay which allowed us to study the phosphorylation state of Lck under controlled acute hypoxic conditions (identical to those used in the electrophysiological experiments). We studied the response to acute hypoxia of primary T cells from 11 healthy donors (Fig. 9). On average, acute (3 min) exposure to hypoxia did not change the proportion of either the inactive phosphorylated Lck (Y505Lck) or its active tyrosine phosphorylated form, while 15 min hydrogen peroxide treatment caused a significant increase in the proportion of Y505Lck and active tyrosine phosphorylated Lck (Hardwick & Sefton, 1997). On closer inspection, individual items of data indicated that cells from 8/11 donors underwent a decrease in Y505 phosphorylated Lck during hypoxia while the proportion of the active form of phosphorylated Lck was unchanged, suggesting a variability in the response to hypoxia among individuals. These data raise the possibility that Lck activation might also occur during acute hypoxia within the heterogeneous population of lymphocytes in some normal individuals.
Figure 9. Effect of acute hypoxia on Lck activity.
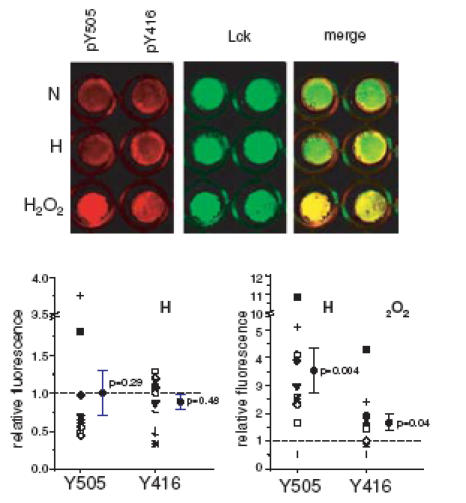
Near-infrared cytoblot assay of phosphorylated Y505 Lck and active Lck (pY396Lck, detected using anti-phosho-src family pY416 antibodies) in normoxia (N), after 3 min exposure to hypoxia (H, 20 mmHg) and after 15 min treatment with 2 mm H2O2. Red pseudocolour images represent the 700 nm wavelength signal of pY505 and pY416 phosphospecific antibodies, while the green pseudocolour images represent the 800 nm wavelength signal of anti-Lck antibody. The right images show the merged picture of red and green images. The lower panels show the averages (with the corresponding P-values) and the individual levels of pY505 and pY416 after 3 min hypoxia (H, left) and after 15 min treatment with H2O2 (right) in 11 donors. Note the different Y axes for the two graphs. The actual values were obtained by normalizing the phosphospecific signals to the signal of Lck in double labelled cells. The values are expressed as relative to normoxia (set as 1 and marked by a dashed line). The symbols indicating each donor are the same in the two graphs.
Discussion
In this paper we have presented evidence indicating that the src-protein tyrosine kinase Lck is required for acute O2 sensing in human T lymphocytes, as it confers O2 sensitivity on Kv1.3 channels. To our knowledge, this is the first report of a signalling pathway involved in mediating the response to acute hypoxia of Kv1.3 channels in primary human T cells.
We have previously shown that Kv1.3 channels expressed in human T lymphocytes are inhibited by hypoxia and their inhibition accounts for hypoxia-induced membrane depolarization and decrease in cell proliferation (Conforti et al. 2003; Robbins et al. 2005). Detailed single-channel patch-clamp studies presented here indicate that hypoxia inhibits Kv1.3 channel activity because it regulates channel open probability without changing the single-channel conductance. This is a common feature shared by other O2-sensitive K (KO2) channels, such as those expressed in chemosensitive cells (Lopez-Barneo et al. 2001). KO2 channels are effector proteins, able to respond quickly to hypoxia and to translate changes in O2 tension into the appropriate functional response. As such, KO2 channels play a key role in the early events of the O2-sensing process. Despite the functional importance of these channels, very little is known about the mechanisms that mediate their O2 sensitivity. Evidence from a variety of studies indicates that KO2 channels lack intrinsic O2 sensitivity and auxiliary subunit(s) and/or signalling molecule(s), as yet unknown, are necessary for making Kv channels O2 sensitive (Lopez-Barneo et al. 2001). Our studies provide evidence indicating that the src-PTK Lck is necessary to confer O2 sensitivity on Kv1.3 channels. We have shown that the hypoxic inhibition of native Kv1.3 channels expressed in human T cells is lost upon src-PTK inhibition and is not observed in Lck-deficient JCaM1.6 cells. Moreover, blockade of src-PTKs inhibits hypoxia-induced membrane depolarization known to be a direct consequence of Kv1.3 channel inhibition (Robbins et al. 2005). Further evidence of the involvement of Lck in the Kv1.3 channel response to hypoxia can be found in the experiments conducted with recombinant Kv1.3 channels. Heterologous Kv1.3 channels expressed in L929 cells (which have minimal levels of Lck) are insensitive to changes in O2 tension, whereas overexpression of Lck in L929 cells reinstates sensitivity to hypoxia. Yet, this was not the case for JCaM1.6 cells: reintroduction of Lck into these cells failed to restore the Kv1.3 channel response to hypoxia. It is, however, possible that in this mutated cell line the expression and function of other signalling molecules necessary for the Kv1.3 channel response to hypoxia might be altered due to the absence of Lck. Lck is known to regulate the activity of other kinases such as PKC (Palacios & Weiss, 2004). This finding raises the possibility that Lck, although necessary, is not sufficient to mediate the Kv1.3 channel response to hypoxia and that other signalling molecules may be involved.
Previous publications have concluded that Lck is necessary for Kv1.3 channel modulation. The inhibitory effect of Fas stimulation and ceramide treatment on Kv1.3 channels in Jurkat cells requires Lck. This finding, associated with the fact that exogenous Lck expression increases Kv1.3 phosphorylation, suggested that Lck-mediated phosphorylation of Kv1.3 channels determines channel inhibition (Szabo et al. 1996; Gulbins et al. 1997). However, no clear evidence of a direct inhibitory effect of Lck on Kv1.3 channel activity has yet been presented. On the contrary, Dellis et al. (1999) have shown that Lck is necessary, but not sufficient, to mediate inhibition of Kv1.3 channels during HIV exposure of Jurkat T cells. Indeed, it seems unlikely that this specific PTK, which plays such an important role in the T cell activation process, would inhibit Kv1.3 channels. Activation of Lck is the earliest signalling event triggered upon ligation of the TCR by peptide antigens and is necessary for the activation process to occur (Palacios & Weiss, 2004). Lck-mediated inhibition of Kv1.3 channels would result in membrane depolarization and immediate termination of the activation process. Instead it has been shown that T cell activation is followed by an initial hyperpolarization and DeCoursey et al. (1984, 1985) have provided evidence for increased Kv1.3 channel activity immediately after mitogen stimuli. In this study we show that Lck does not directly inhibit Kv1.3 channel activity: neither overexpression of constitutively active Lck nor the delivery of full length Lck through the patch pipette inhibited recombinant Kv1.3 currents in L929 cells. On the contrary, experiments in human T cells indicated that Lck increases Kv1.3 currents. In fact, increasing the activity of Lck with the general src kinase activator peptide stabilized Kv1.3 current in freshly isolated human CD4+ cells. Furthermore, introduction of Lck into JCaM1.6 cells increased Kv1.3 current density. Overall, our data indicate that while Lck is necessary to confer O2 sensitivity on Kv1.3 channels, this src-PTK does not directly inhibit Kv1.3 channels.
A question that still remains unanswered is whether hypoxia effects occur through tyrosine phosphorylation. It is generally believed that the effect of Lck, and src-PTKs in general, on Kv1.3 channels occurs via phosphorylation of specific tyrosine residues on the channel protein (Holmes et al. 1996b; Szabo et al. 1996; Fadool et al. 1997; Gulbins et al. 1997; Fadool, 1998; Cook & Fadool, 2002). Kv1.3 channels have several consensus sites for tyrosine phosphorylation and it has been shown that the functional outcome depends on the site as well as on the degree of phosphorylation. The immunoprecipitation studies we have performed showed that 6–24 h exposure to hypoxia increases Lck tyrosine phosphorylation (Mahabeleshwar & Kundu, 2003). Concurrently, the Kv1.3 channel protein is up-phosphorylated. These data indicate that a phosphorylative mechanism is important for the T cell response to chronic hypoxia and it might mediate the down-regulation of Kv1.3 channel expression that occurs in T cells after long exposure to hypoxia (Conforti et al. 2003). Instead, the response to acute hypoxia appeared to be different. The studies we performed indicated that short exposures to hypoxia (3 min to 1 h) do not change the tyrosine phosphorylation of either Lck or Kv1.3. However, when we looked closely at the effect of acute hypoxia on Lck tyrosine phosphorylated active (pY416) and inactive (pY505) forms we saw that some degree of Lck activation might occur in some normal individuals. Heterogeneity in T cell response to hypoxia was also seen in our previous study and it may be related to the activation state of individual cells (Robbins et al. 2005). Accordingly, although short-term hypoxia did not change the tyrosine phosphorylation state of Kv1.3 channels in the immortalized Jurkat T cell line, we cannot exclude that some degree of Kv1.3 channel tyrosine phosphorylation might be observed in primary T cells. Unfortunately, our attempts to determine if acute hypoxia changes the phosphorylation state of Kv1.3 channels in primary T cells were not successful possibly because of the low expression of Kv1.3 channel proteins and the limited sample available.
Altogether, the results we present herein raise the possibility that the function of Lck in human T lymphocytes is to maintain the Kv1.3 channel tyrosine phosphorylated state necessary for its proper function and to allow the function of other kinases involved in the process of O2 sensing. Alternatively, it is also possible that Lck plays a structural role and maintains the channel in the proper scaffolding to allow contact with other signalling molecules activated during hypoxia. Kv1.3 channels have been shown to exist in association with Lck and hDlg1, a PDZ protein involved in channel localization and membrane stability (Hanada et al. 1997). This scaffold structure might also maintain a close proximity between Kv1.3 channels and other signalling molecules activated during hypoxia and responsible for Kv1.3 channel inhibition. It is known, for example, that Kv1.3 channels are modulated by serine and threonine phosphorylation (Payet & Dupuis, 1992; Chung & Schlichter, 1997). Further experiments are underway to determine the additional mechanism(s) involved in Kv1.3 channel inhibition during hypoxia in human T cells.
In conclusion, the findings presented here answer two fundamental questions regarding the role of Lck in the O2 sensing process of human T cells and the functional relationship between Lck and Kv1.3 channels. The latter finding could have important implications in the T cell activation process and based on this, we propose that an additional role of Lck during T cell activation may be to set the conditions for proper Kv1.3 channel function and to prepare Kv1.3 channels for further modulation either by other signalling molecules activated later on during the T cell activation process or by exogenous factors such as hypoxia.
Acknowledgments
We thank Drs G. K. Chandy and C. T. Baldari for providing the L929 cells stably transfected with Kv1.3 and the constitutively active Lck construct, respectively. We also thank Dr A. Weiss for the JCaM1.6 cells. This work was supported by NIH grant no. CA95286 to L.C. no. HL074111 to K.T., and AI056927 to CC.
References
- Cahalan MD, Wulff H, Chandy KG. Molecular properties and physiological roles of ion channels in the immune system. J Clin Immunol. 2001;21:235–252. doi: 10.1023/a:1010958907271. [DOI] [PubMed] [Google Scholar]
- Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG, Sitkovsky MV. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol. 2001;167:6140–6149. doi: 10.4049/jimmunol.167.11.6140. [DOI] [PubMed] [Google Scholar]
- Cayabyab FS, Khanna R, Jones OT, Schlichter LC. Suppression of the rat microglia Kv1.3 current by src-family tyrosine kinases and oxygen/glucose deprivation. Eur J Neurosci. 2000;12:1949–1960. doi: 10.1046/j.1460-9568.2000.00083.x. [DOI] [PubMed] [Google Scholar]
- Chandy GK, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci. 2004;25:280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Kovar J, Sissons S, Cox K, Matter W, Chadwell F, Luan P, Vlahos CJ, Schutz-Geschwender A, Olive DM. A cell-based immunocytochemical assay for monitoring kinase signaling pathways and drug efficacy. Anal Biochem. 2005;338:136–142. doi: 10.1016/j.ab.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Chung I, Schlichter LC. Native Kv1.3 channels are upregulated by protein kinase C. J Membr Biol. 1997;156:73–85. doi: 10.1007/s002329900189. [DOI] [PubMed] [Google Scholar]
- Conforti L, Bodi I, Nisbet JW, Millhorn DE. O2-sensitive K+ channels: role of the Kv1.2-subunit in mediating the hypoxic response. J Physiol. 2000;524:783–793. doi: 10.1111/j.1469-7793.2000.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Millhorn DE. Selective inhibition of a slow-inactivating voltage-dependent K+ channel in rat PC12 cells by hypoxia. J Physiol. 1997;502:293–305. doi: 10.1111/j.1469-7793.1997.293bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Petrovic M, Mohammad D, Lee S, Ma Q, Barone S, Filipovich AH. Hypoxia regulates expression and activity of Kv1.3 channels in T lymphocytes: a possible role in T cell proliferation. J Immunol. 2003;170:695–702. doi: 10.4049/jimmunol.170.2.695. [DOI] [PubMed] [Google Scholar]
- Cook KK, Fadool DA. Two adaptor proteins differentially modulate the phosphorylation and biophysics of Kv1.3 ion channel by SRC kinase. J Biol Chem. 2002;277:13268–13280. doi: 10.1074/jbc.M108898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 1984;307:465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage-dependent ion channels in T-lymphocytes. J Neuroimmunol. 1985;10:71–95. doi: 10.1016/0165-5728(85)90035-9. [DOI] [PubMed] [Google Scholar]
- Dellis O, Bouteau F, Guenounou M, Rona JP. HIV-1 gp160 decreases the K+ voltage-gated current from Jurkat E6.1 T cells by up-phosphorylation. FEBS Lett. 1999;443:187–191. doi: 10.1016/s0014-5793(98)01691-3. [DOI] [PubMed] [Google Scholar]
- Di Somma MM, Nuti S, Telford JL, Baldari CT. p56lck plays a key role in transducing apoptotic signals in T cells. FEBS Lett. 1995;363:101–104. doi: 10.1016/0014-5793(95)00292-h. [DOI] [PubMed] [Google Scholar]
- Fadool DA. Tyrosine phosphorylation downregulates a potassium current in rat olfactory bulb neurons and a cloned Kv1.3 channel. Ann N Y Acad Sci. 1998;855:529–532. doi: 10.1111/j.1749-6632.1998.tb10621.x. [DOI] [PubMed] [Google Scholar]
- Fadool DA, Holmes TC, Berman K, Dagan D, Levitan IB. Tyrosine phosphorylation modulates current amplitude and kinetics of a neuronal voltage-gated potassium channel. J Neurophysiol. 1997;78:1563–1573. doi: 10.1152/jn.1997.78.3.1563. [DOI] [PubMed] [Google Scholar]
- Fadool DA, Levitan IB. Modulation of olfactory bulb neuron potassium current by tyrosine phosphorylation. J Neurosci. 1998;18:6126–6137. doi: 10.1523/JNEUROSCI.18-16-06126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissmer S, Nguyen A, Aiyar J, Hanson D, Mather R, Gutman G, Karmilowicz M, Auperin D, Chandy K. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- Gulbins E, Szabo I, Baltzer K, Lang F. Ceramide-induced inhibition of T lymphocyte voltage-gated potassium channel is mediated by tyrosine kinases. Proc Natl Acad Sci U S A. 1997;94:7661–7666. doi: 10.1073/pnas.94.14.7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Lin L, Chandy KG, Oh SS, Chishti AH. Human homologue of the Drosophila discs large tumor suppressor binds to p56lck tyrosine kinase and Shaker type Kv1.3 potassium channel in T lymphocytes. J Biol Chem. 1997;272:26899–26904. doi: 10.1074/jbc.272.43.26899. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Hardwick JS, Sefton BM. The activated form of the Lck tyrosine protein kinase in cells exposed to hydrogen peroxide is phosphorylated at both Tyr-394 and Tyr-505. J Biol Chem. 1997;272:25429–25432. doi: 10.1074/jbc.272.41.25429. [DOI] [PubMed] [Google Scholar]
- Hermiston ML, Xu Z, Majeti R, Weiss A. Reciprocal regulation of lymphocyte activation by tyrosine kinases and phosphatases. J Clin Invest. 2002;109:9–14. doi: 10.1172/JCI14794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes TC, Berman K, Swartz JE, Dagan D, Levitan IB. Expression of voltage-gated potassium channels decreases cellular protein tyrosine phosphorylation. J Neurosci. 1997;17:8964–8974. doi: 10.1523/JNEUROSCI.17-23-08964.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes TC, Fadool DA, Levitan IB. Tyrosine phosphorylation of the Kv1.3 potassium channel. J Neurosci. 1996a;16:1581–1590. doi: 10.1523/JNEUROSCI.16-05-01581.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes TC, Fadool DA, Ren R, Levitan IB. Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain. Science. 1996b;274:2089–2091. doi: 10.1126/science.274.5295.2089. [DOI] [PubMed] [Google Scholar]
- Jullien P, Bougeret C, Camoin L, Bodeus M, Durand H, Disanto JP, Fischer S, Benarous R. Tyr394 and Tyr505 are autophosphorylated in recombinant Lck protein-tyrosine kinase expressed in Escherichia coli. Eur J Biochem. 1994;224:589–596. doi: 10.1111/j.1432-1033.1994.00589.x. [DOI] [PubMed] [Google Scholar]
- Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- Loeffler DA, Juneau PL, Heppner GH. Natural killer-cell activity under conditions reflective of tumor micro-environment. Int J Cancer. 1991;48:895–899. doi: 10.1002/ijc.2910480617. [DOI] [PubMed] [Google Scholar]
- Loeffler DA, Juneau PL, Masserant S. Influence of tumour physico-chemical conditions on interleukin-2-stimulated lymphocyte proliferation. Br J Cancer. 1992;66:619–622. doi: 10.1038/bjc.1992.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler DA, Keng PC, Baggs RB, Lord EM. Lymphocytic infiltration and cytotoxicity under hypoxic conditions in the EMT6 mouse mammary tumor. Int J Cancer. 1990;45:462–467. doi: 10.1002/ijc.2910450315. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Ortega-Saenz P. Cellular mechanism of oxygen sensing. Annu Rev Physiol. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- Mahabeleshwar GH, Kundu GC. Tyrosine kinase p56lck regulates cell motility and nuclear factor κB-mediated secretion of urokinase type plasminogen activator through tyrosine phosphorylation of IκBα following hypoxia/reoxygenation. J Biol Chem. 2003;278:52598–52612. doi: 10.1074/jbc.M308941200. [DOI] [PubMed] [Google Scholar]
- Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000;74:181–273. doi: 10.1016/s0065-2776(08)60911-6. [DOI] [PubMed] [Google Scholar]
- Naldini A, Carraro F. Hypoxia modulates cyclin and cytokine expression and inhibits peripheral mononuclear cell proliferation. J Cell Physiol. 1999;181:448–454. doi: 10.1002/(SICI)1097-4652(199912)181:3<448::AID-JCP8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23:7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- Payet MD, Dupuis G. Dual regulation of the N type K+ channel in Jurkat T lymphocytes by protein kinases A and C. J Biol Chem. 1992;267:18270–18273. [PubMed] [Google Scholar]
- Robbins JR, Lee SM, Filipovich AH, Szigligeti P, Neumeier L, Petrovic M, Conforti L. Hypoxia modulates early events in T cell receptor-mediated activation in human T lymphocytes via Kv1.3 channels. J Physiol. 2005;564:131–143. doi: 10.1113/jphysiol.2004.081893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- Straus DB, Weiss A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell. 1992;70:585–593. doi: 10.1016/0092-8674(92)90428-f. [DOI] [PubMed] [Google Scholar]
- Szabo I, Gulbins E, Apfel H, Zhang X, Barth P, Busch AE, Schlottmann K, Pongs O, Lang F. Tyrosine phosphorylation-dependent suppression of a voltage-gated K+ channel in T lymphocytes upon Fas stimulation. J Biol Chem. 1996;271:20465–20469. doi: 10.1074/jbc.271.34.20465. [DOI] [PubMed] [Google Scholar]
- Szabo I, Nilius B, Zhang X, Busch AE, Gulbins E, Suessbrich H, Lang F. Inhibitory effects of oxidants on n-type K+ channels in T lymphocytes and Xenopus oocytes. Pflugers Arch. 1997;433:626–632. doi: 10.1007/s004240050323. [DOI] [PubMed] [Google Scholar]
- Whiteside TL. Immune cells in the tumor microenvironment. Mechanisms responsible for functional and signaling defects. Adv Exp Med Biol. 1998;451:167–171. [PubMed] [Google Scholar]
- Winslow MM, Crabtree GR. Immunology. Decoding calcium signaling. Science. 2005;307:56–57. doi: 10.1126/science.1108163. [DOI] [PubMed] [Google Scholar]
- Zuckerberg AL, Goldberg LI, Lederman HM. Effects of hypoxia on interleukin-2 mRNA expression by T lymphocytes. Crit Care Med. 1994;22:197–203. doi: 10.1097/00003246-199402000-00008. [DOI] [PubMed] [Google Scholar]