

Abstract
Immunotherapy holds great promise for Alzheimer’s disease (AD) and other conformational disorders but certain adverse reactions need to be overcome. The meningoencephalitis observed in the first AD vaccination trial was likely related to excessive cell-mediated immunity caused by the immunogen, amyloid-β (Aβ) 1–42, and the adjuvant, QS–21. To avoid this toxicity, we have been using Aβ derivatives in alum adjuvant that promotes humoral immunity. Other potential side effects of immunotherapy are increased vascular amyloid and associated microhemorrhages that may be related to rapid clearance of parenchymal amyloid. Here, we determined if our immunization strategy was associated with this form of toxicity, and if the therapeutic effect was age-dependent. Tg2576 mice and wild-type littermates were immunized from 11 or 19 months and their behaviour evaluated prior to killing at 24 months. Subsequently, plaque- and vascular-Aβ burden, Aβ levels and associated pathology was assessed. The therapy started at the cusp of amyloidosis reduced cortical Aβ deposit burden by 31% and Aβ levels by 30–37%, which was associated with cognitive improvements. In contrast, treatment from 19 months, when pathology is well established, was not immunogenic and therefore did not reduce Aβ burden or improve cognition. Significantly, the immunotherapy in the 11–24 months treatment group, that reduced Aβ burden, did not increase cerebral bleeding or vascular Aβ deposits in contrast to several Aβ antibody studies. These findings indicate that our approach age-dependently improves cognition and reduces Aβ burden when used with an adjuvant suitable for humans, without increasing vascular Aβ deposits or microhemorrhages.
Keywords: amyloid-β, bleeding, cognition, immunotherapy, microglia
Abbreviations: Aβ, amyloid-β; AD, Alzheimer’s disease; GFAP, glial fibrillary acidic protein
Introduction
Extensive evidence suggests that amyloid β (Aβ) accumulation in the brain is central to the pathogenesis of Alzheimer’s disease (AD). Immunotherapy for AD targeting Aβ remains a promising approach although the initial clinical trial was halted because of encephalitis in a subset of patients. Following the first mouse studies on vaccination with Aβ1–42 (Schenk et al., 1999), several laboratories have confirmed and extended these findings, showing that this approach can prevent Aβ deposition and improves cognition in mouse models (Lemere et al., 2003; Gelinas et al., 2004; Weksler et al., 2005; Sigurdsson, 2006). More limited data from the aborted trial suggest that some patients may have benefited cognitively (Hock et al., 2003; Gilman et al., 2005) and autopsies on participants indicate that parenchymal Aβ deposits were reduced (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005). The meningoencephalitis seen in 6% of the patients was likely caused by the immunogen (Aβ1–42) and excessive cell-mediated immunity. We have proposed to prevent this reaction by using nonfibrillogenic, nontoxic Aβ homologous peptides (Sigurdsson et al., 2001; Sigurdsson et al., 2004). Adjuvants also influence the immune response and most of the prior animal studies used Freund’s adjuvant, which promotes a cytotoxic Th1 response. The adjuvant QS–21, which also promotes Th1 response was used in the Alzheimer’s trial and may have contributed to the encephalitis. Prior to these adverse effects we advocated the use of adjuvants that promote a humoral response such as alum, which is approved for human use (Sigurdsson et al., 2001; Scholtzova et al., 2002). One important aim of our current study was to establish if alum could be used as an effective adjuvant in conjunction with our Aβ derivative immunogen.
Another potential side effect of immunotherapy targeting Aβ is cerebral bleeding, which may be related to rapid clearance of parenchymal Aβ, leading to increased vascular Aβ deposition and vessel friability. Three studies in AD mouse models have observed increased brain microhemorrhages following passive immunization with different Aβ antibodies (Pfeifer et al., 2002; Wilcock et al., 2004; Racke et al., 2005). In the three autopsy cases from the clinical trial, extensive vascular Aβ remained despite parenchymal Aβ clearance, and one of these individuals had numerous brain microhemorrhages (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005). These observations suggest that increased vascular Aβ and hemorrhage may also be an issue with active immunization in humans. In the present study, we determined if our active immunization approach would be associated with this form of toxicity. Furthermore, to determine the prophylactic vs. the therapeutic effect of Aβ derivatives, we immunized Tg2576 mice (Hsiao et al., 1996) with K6Aβ1–30 in alum starting at 11 or 19 months of age with treatment continuing until 24 months. We previously observed an 81–89% reduction in brain amyloid burden in this model using this immunogen with Freund’s adjuvant (Sigurdsson et al., 2001). Towards the end of the study, extensive behavioural testing was performed with subsequent brain analysis, which included assessment of parenchymal- and vascular-Aβ deposits, Aβ levels, as well as associated gliosis and microhemorrhages.
Materials and methods
Peptides and other chemicals
K6Aβ1-30-NH2 was synthesized at the Keck Foundation at Yale University, as described previously (Sigurdsson et al., 2001). This Aβ derivative maintains the two major immunogenic sites of the Aβ peptide, which are residues 1–11 and 22–28 of Aβ1–42 based on an antigenic index (Jameson & Wolf, 1988). The peptide is amidated on the C terminus to maintain the immunogenicity of that epitope. The six lysyl residues on the N terminus were added to enhance immunogenicity and further reduce β-sheet content. All other chemicals were from Sigma (St. Louis, MO) unless otherwise stated.
Animals
The vaccination was performed in the heterozygous Tg2576 amyloid precursor protein (APP) mouse model (Hsiao et al., 1996). These mice show rapid increases in Aβ levels at approximately 6 months of age with Aβ deposition developing in the following months although extensive amyloid burden is usually not observed until the animals are well into their second year (Kawarabayashi et al., 2001). The animals were maintained on a 12-h light : 12-h dark cycle, and animal care was in accordance with institutional guidelines in AAALAC approved facilities. The animal experiments were approved by the IACUC committee at NYU School of Medicine and were carried out in accordance with NIH Guidelines.
Vaccine administration
K6Aβ1-30-NH2 was administered as described previously (Sigurdsson et al., 2001). The peptide was mixed with Adju-Phos adjuvant (Brenntag Biosector, Denmark) at a concentration of 1 mg / mL and the solution was rotated overnight at 4 °C prior to administration to allow the peptide to adsorb onto the aluminium phosphate particles. The mice received a subcutaneous injection of 100 μL followed by a second injection 2 weeks later and monthly thereafter. Vaccination started at 11–12 months and 19–20 months of age and continued until the animals were 23–24 months of age at which time the animals were perfused and their organs collected for analysis. The mice went through a battery of behavioural tests in the two months prior to killing. In the younger treatment group, 19 mice received K6Aβ1–30 and 21 mice received the adjuvant alone. Fifteen wild-type littermates served as additional controls. At the end of the study, 12 and 16 Tg animals remained in the treatment and control groups, respectively. In the older treatment group, 13 mice were immunized with K6Aβ1–30 and 12 mice received adjuvant alone. At the end of the study, ten and eight mice remained in the treatment and control groups, respectively. For gender split and number of mice in the behavioural tests, see Table 1.
Table 1.
Numbers of male and female animals used in the study
| Beginning of study
|
Radial arm maze
|
Closed field symmetrical maze
|
Sensorimotor tasks
|
End of study (brains analysed)
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Males | Females | Males | Females | Males | Females | Males | Females | Males | Females | |
| 11–24 months | ||||||||||
| Tg Control | 4 | 17 | 3 | 16 | 2 | 14 | 2 | 14 | 2 | 14 |
| Tg K6Aβ1–30 | 7 | 12 | 4 | 10 | 2 | 9 | 2 | 10 | 2 | 10 |
| Wild-type | 4 | 11 | 4 | 11 | 4 | 11 | 4 | 11 | 4 | 11 |
| 19–24 months | ||||||||||
| Tg Control | 3 | 9 | 0 | 8 | 0 | 7 | 1 | 7 | 1 | 7 |
| Tg K6Aβ1–30 | 4 | 9 | 4 | 7 | 3 | 7 | 3 | 8 | 3 | 7 |
Behaviour
Cognitive tests
Prior to testing, the mice were adapted to the room with lights on for 15 min.
Radial arm maze
Spatial learning was evaluated using an eight-arm radial maze with a water well at the end of each arm. Clear Plexiglas guillotine doors, operated by a remote pulley system, controlled access to the arms from a central area from which the animals entered and exited the apparatus. After 3–4 days of adaptation, water-restricted mice (2 h daily access to water) were given one training session per day for ten consecutive days. For each session, all arms were baited with saccharine flavored water, and animals were permitted to enter all arms until the eight rewards had been consumed. The number of errors (entries to previously visited arms) and time to complete each session were recorded.
Closed field symmetrical maze
This apparatus is a rectangular field 30 cm square with 9 cm high walls divided into 36, 9.5 cm squares and covered by a clear Plexiglas top. Endboxes, each 11 × 16 × 9 cm, are situated at diagonal corners of the field. The symmetrical maze (Davenport et al., 1970) is a modification of the Hebb–Williams (Hebb & Williams, 1946) and Rabinovitch–Rosvold (Rabinovitch & Rosvold, 1951) type of tests. Briefly, the main difference is that each end-compartment functions as both a startbox and a goalbox, and the mice run in opposite direction on alternate trials, thereby eliminating intertrial handling. The barriers are placed in the field in symmetrical patterns, so that mice face the same turns going in either direction within a given problem. Prior to testing, the mice were adapted to a water restriction schedule (2 h daily access to water). The mice were given two adaptation sessions prior to the beginning of testing. In the first session, all animals were given saccharine flavored water in the goal box for 10 min. In session 2, they were placed in the start chamber and permitted to explore the field and enter the goal box where water reward (0.05 mL) was available. When the mice were running reliably from the start chamber to the goal box, they were given three practice sessions on simple problems where one or two barriers were placed in different positions in the field so as to obstruct direct access to the goal box. Formal testing consisted of the presentation of seven problems graded in difficulty. One problem was presented per day and the mice were given five trials on each problem with an intertrial interval of 2 min. Performance was scored manually by the same observer in terms of errors (i.e. entries and reentries into designated error zones) and time to complete each trial.
Sensorimotor tests
Prior to testing, the mice were adapted to the room with lights on for 15 min. The main objective of performing these sensorimotor tasks was to verify that any treatment related effects observed in the cognitive tasks could not be explained by differences in sensorimotor abilities.
Locomotor activity
A Hamilton–Kinder Smart-frame Photobeam System was used to make a computerized recording of animal activity over a designated period of time. Exploratory locomotor activity is recorded in a circular open field activity chamber measuring 70 × 70 cm. A video camera mounted above the chamber automatically recorded horizontal movements in the open field in each dimension (i.e. x, y, and two z planes). Total distance was measured in centimeters (cm) travelled and is defined as sequential movement interruptions of the animal measured relative to the background. The duration of the behaviour was timed for 15 min. Results are reported based on distance travelled (cm), mean resting time, and velocity (average and maximum) of the animal.
Traverse beam
This task tests balance and general motor coordination and function integration. Mice were assessed by measuring their ability to traverse a graded narrow wooden beam to reach a goal box [modification of (Torres et al., 1994)] which specifically examines hindlimb function. The mice were placed on a 1.1-cm wide beam 50.8-cm long suspended 30 cm above a padded surface by two identical columns. Attached at each end of the beam was a shaded goal box. Mice were placed on the beam in a perpendicular orientation to habituate, and were then monitored for a maximum of 60 s. The number of foot slips each mouse has before falling or reaching the goal box was recorded for each of three successive trials. The average foot slips for all four trials was calculated and recorded. Errors are defined as footslips and recorded numerically. To prevent injury from falling, a soft foam cushion was always kept underneath the beam. Animals that fell off were placed back in their position prior to the fall.
Rotarod
The animal was placed onto the rod (diameter 3.6 cm) apparatus to measure forelimb and hindlimb motor coordination and balance (Rotarod 7650 accelerating model; Ugo Basile, Biological Research Apparatus, Varese, Italy; Carter et al., 1999; Hyde et al., 2001). This procedure was designed to assess motor behaviour without a practice confound. The animals were habituated to the apparatus by receiving training sessions of two trials, sufficient to reach a baseline level of performance. Then the mice were tested a further three times, with increasing speed of the rotating rod. During habituation, the rotor rod was set at 1.0 r.p.m., which was gradually raised every 30 s, and it was also wiped clean with 30% ethanol solution after each session. A soft foam cushion was placed beneath the apparatus to prevent potential injury from falling. Each animal was tested for three sessions, with each session separated by 15 min, and measures were taken for latency to fall or invert (by clinging) from the top of the rotating barrel.
Antibody levels
Antibody levels were determined by 1 : 200 and 1 : 1000 dilutions of plasma using ELISA as described previously (Sigurdsson et al., 2001), in which Aβ or its derivative is coated onto microtiter wells. The antibodies were detected by a goat anti-mouse IgG linked to a horseradish peroxidase (Amersham Biosciences, Piscataway, NJ) or a goat anti-mouse IgM peroxidase conjugate (Sigma; A8786), and tetramethyl benzidine (TMB; Pierce, Rockford, IL) was the substrate.
Histology
Mice were anaesthetized with sodium pentobarbital (150 mg / kg, i.p.), perfused transaortically with phosphate buffer, and the brains processed as described previously (Sigurdsson et al., 1996). The right hemisphere was immersion-fixed in periodate-lysine-paraformaldehyde, whereas the left hemisphere was snap-frozen for measurements of Aβ levels. Serial coronal sections (40 μm) were cut, and every fifth section (30–40 sections in total) was stained with 6E10, a monoclonal antibody that recognizes Aβ and stains both preamyloid and Aβ plaques (Kim et al., 1990). Staining was performed as described previously (Sadowski et al., 2004; Sigurdsson et al., 2004). Every tenth section (15–20 sections in total) was stained with tomato lectin (Vector Laboratories, Burlingame, CA), polyclonal interleukin-1β antibody (Biosource International, Camarillo, CA), polyclonal anti-glial fibrillary acidic protein (GFAP; Dako, Glostrup, Denmark), or with Perl’s iron stain. Tomato lectin binds to poly N-acetyl lactosamine residues and in neural tissue it has specific affinity for microglial cells (Acarin et al., 1994). Interleukin-1β is a cytokine that is primarily found in microglia. GFAP is a component of the glial intermediate filaments that form part of the cytoskeleton and is found predominantly in astrocytes. Both microglia and astrocytes are associated with Aβ deposits.
Immunohistochemistry
Immunostaining was performed as described previously (Sigurdsson et al., 2001; Sigurdsson, 2005). Briefly, sections were incubated in 6E10 at a 1 : 1000 dilution for 3 h. A mouse-on-mouse immunodetection kit (Vector Laboratories, Burlingame, CA) was used, with the biotinylated anti-mouse IgG secondary antibody reacted for 1 h at a 1 : 2000 dilution. The avidin-peroxidase complex was subsequently reacted for 30 min at the same dilution. GFAP- (1 : 500; 3 h) and interleukin-1β-staining (1 : 250, overnight) was performed as described (Sigurdsson et al., 1996) with the diluent for the primary antibody composed of 2.0% Triton X-100, 0.1% sodium azide, 0.01% bacitracin, 2% bovine serum albumin, and 10% normal goat serum in PBS, and the secondary biotinylated anti-rabbit antibody (Vectastain, ABC Elite kit, Vector) was reacted for 1 h at a 1 : 1300 dilution followed by avidin-peroxidase reaction for 30 min at the same dilution. Tomato lectin staining was performed as described (Sigurdsson et al., 2001) with a 2 h incubation (biotinylated tomato lectin, 10 μg / mL PBS; Vector) followed by 1 h reaction in avidin-horseradish peroxidase (Vector). The sections were reacted in 3,3-diaminobenzidine tetrahydrochloride with nickel ammonium sulphate (Ni; Mallinckrodt, Paris, KY) intensification.
Iron staining
Perl’s iron stain was performed to detect cerebral bleeding by placing defatted and hydrated sections in a solution containing 5% potassium ferrocyanide and 10% hydrochloric acid for 30 min. The slides were then rinsed in distilled water, and the sections were dehydrated, cleared in Hemo-De, and coverslipped. This same method was used in the three previous reports that showed that passive immunization against Aβ increased the frequency of microhemorrhages in AD model mice (Pfeifer et al., 2002; Wilcock et al., 2004; Racke et al., 2005). Diamino benzidine intensification of the iron staining, which is useful for detecting low levels of iron in Aβ plaques, did not appear to improve sensitivity for detecting the microhemorrhages and was therefore not employed. To verify that our methodology would allow us to detect increases in hemorrhages, positive controls were used. These mice had brain hemorrhages that were caused by intracerebral cannula placement. These hemorrhages were more extensive than the microhemorrhages observed in the Tg mice.
Image analysis
Immunohistochemistry of tissue sections was quantified with a Bioquant image analysis system (BIOQUANT Image Analysis Corporation, Nashville, TN), and unbiased sampling was used (West, 1999). All procedures were performed by an individual blinded to the experimental condition of the study. The cortical area analysed was dorsomedial from the cingulate cortex and extended ventrolaterally to the rhinal fissure within the right hemisphere. The area of the grid was 800 μm2 ×800 μm2, and Aβ deposit load was measured in 20 cortical frames per mouse (640 ×480 μm2 each) chosen randomly. For the initial Aβ burden measurements, no distinction was made between vascular and parenchymal Aβ deposits. Subsequently, vascular Aβ deposits were specifically measured in the 11–24 months old group in an additional 20 randomly selected cortical frames per mouse. The Aβ burden is defined as the percentage of area in the measurement field occupied by reaction product.
Rating of microgliosis
The assessment of the tomato lectin (microglia) stained sections was based on a semiquantitative analysis of the extent of microgliosis associated with the Aβ deposits (0, a few resting microglia; +, a few ramified and / or phagocytic microglia; ++, moderate number of ramified / phagocytic microglia; +++, numerous ramified / phagocytic microglia)
Tissue homogenization and sandwich ELISA assay for soluble Aβ levels
Extraction of Aβ from brain tissue was performed as described in detail (Schmidt et al., 2005a; Schmidt et al., 2005b) with the following modification. Instead of adding three protease inhibitors (leupeptin, antipain, and pepstatin A; 1 / 1000 volume of 5 mg / mL stock solution) to the homogenization buffer, a protease inhibitor cocktail (Complete, Roche Diagnostics, Indianapolis, IN) was used according to the manufacturer’s description as well as pepstatin A at the concentration listed above. Brains were weighed and homogenized (10% w / v) in homogenization buffer [(20 mμ Tris base, 250 mμ sucrose, 1 mμ EDTA, 1 mμ EGTA) with 100 mμ phenylmethylsulphonyl fluoride in ethanol and the protease inhibitors added immediately before homogenization].
Total Aβ levels
Homogenate (200 μL) was added to 440 μL cold formic acid and sonicated for one minute on ice. Subsequently, 400 μL of this solution was spun down at 100 000 × g for one hour at 4 °C. Then, 210 μL of the supernatant was diluted into 4 mL of formic acid neutralization solution (1 μ Tris base, 0.5 μ Na2HPO4, 0.05% NaN3), aliquotted, flash frozen on dry ice and stored at −80 °C until used for Aβ measurements with ELISA. For measurements of Aβ1–40 vs. Aβ1–42, the aliquots were diluted 1000–2500-fold vs. 100–500-fold, respectively, prior to loading onto ELISA plates. The ELISA procedure was performed as described by the ELISA kit manufacturer (Biosource International).
Soluble Aβ levels
Homogenate (200 μL) was mixed with equal volume of cold 0.4% diethylamine containing 100 mμ NaCl, and subsequently centrifuged at 100 000 ×g for 1 h at 4 °C. Then, the supernatant was neutralized with 1 / 10 volume of 0.5 μ Tris pH 6.8, aliquotted, flash frozen on dry ice and stored at −80 °C until used for Aβ measurement. For measurements of Aβ1–40 vs. Aβ1–42, the aliquots were diluted 100–1000-fold vs. 20–50-fold, respectively, prior to loading onto ELISA plates (Biosource International).
Data analysis
Aβ deposit burden and the levels of total and soluble Aβ within the brain were analysed by a Student’s t-test, one-tailed (Graph Pad Prism 4.0). The data from the radial arm maze, traverse beam and rotarod were analysed by two-way ANOVA repeated measures and the Neuman–Keuls posthoc test (Statistica 6.1). The data from the closed field symmetrical maze was analysed by regular two-way ANOVA with the Neuman–Keuls posthoc test (Statistica 6.1). The data from the locomotor activity and brain microhemorrhage analysis was analysed by one-way ANOVA followed by the Neuman–Keuls posthoc test (11–24 months) or a t-test (19–24 months). When the data analysed with one-way ANOVA failed the Bartlett’s test for equal variance, the Kruskal–Wallis test was performed with the Dunn’s multiple comparison posthoc test. Correlation was determined by calculating the Pearson r correlation coefficient.
Results
In the Tg2576 mouse model, Aβ deposition is usually modest around one year of age with numerous parenchymal plaques and extensive vascular amyloid observed towards the end of their second year (Hsiao et al., 1996). To compare a prophylactic vs. a therapeutic effect of Aβ-targeting immunization in an adjuvant that is approved for human use (alum), mice were immunized from (i) 11 to 24 months and (ii) 19–24 months of age. Antibody response towards the vaccine was assessed periodically, and at the end of the study the animals went through extensive behavioural testing with subsequent brain analyses of levels of Aβ and its deposition, as well as associated gliosis and cerebral bleeding. The behavioural analyses consisted of both a cognitive assessment as well as measurements of sensorimotor performance. The latter tests were run primarily to verify that cognitive performance was not influenced by abnormalities in sensory and motor systems and to assess if the vaccine affected these regions. The brain analyses were designed to determine efficacy and possible toxicity of immunization with our Aβ derivative under these conditions.
Antibody levels
Treatment from 11 to 24 months
The K6Aβ1–30 immunized mice developed IgG antibodies that recognized the immunogen and Aβ1–40 (Fig. 1A). As expected, the antibody levels subsided over time but remained overall higher than levels of autoantibodies in vehicle-treated control animals. Because the animals were bled two weeks following immunization, IgM levels were similar to baseline values.
Fig. 1.
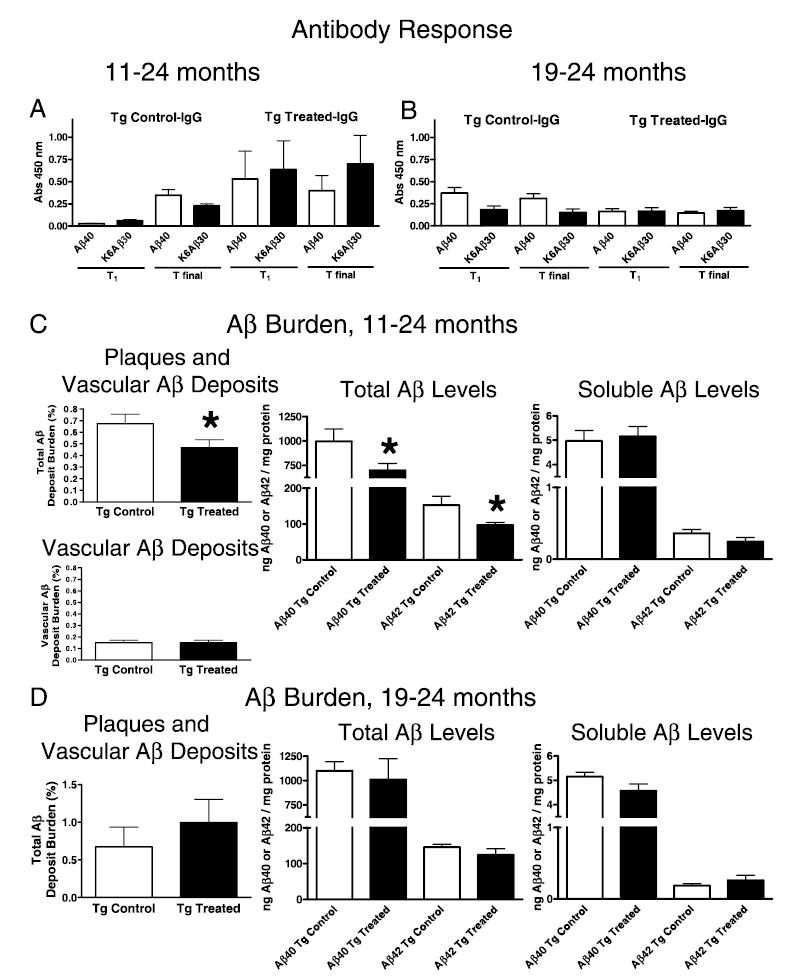
Antibody levels and brain amyloid burden. Groups were treated from 11 to 24 months (A and C) and from 19 to 24 months (B and D). (A) 11–24 months. K6Aβ1–30 in alum adjuvant elicits a good antibody response that is maintained (T1, 13 months; Tfinal, 24 months). The antibodies recognize both the immunogen and Aβ1–40. Vehicle-treated controls have some autoantibodies at old age. Treatment and antibody type measured (IgG) is indicated in the title. The x-axis depicts which peptide (coated on ELISA plate) the antibodies are recognizing. The y-axis depicts the absorbance at 450 nm. (B) 19–24 months. K6Aβ1–30 in alum adjuvant is not immunogenic when treatment is started at old age (19 months). (C) 11–24 months. Cortical amyloid burden (parenchymal and vascular) was reduced by 31% (P < 0.05) in Tg2576 mice immunized with K6Aβ1–30 in alum adjuvant from 11 to 24 months of age, compared to control mice. When analysed separately, vascular amyloid burden was not significantly altered by the immunotherapy. Total brain Aβ levels (Aβ1–40, 30% reduction, P = 0.03; Aβ1–42, 37% reduction, P = 0.02) were reduced to a similar extent as total amyloid burden. Levels of soluble Aβ were not significantly affected although soluble Aβ1–42 was reduced by 32% in the immunized mice (P = 0.08). (D) 19–24 months. No significant difference in cortical amyloid burden was observed between the immunized and nonimmunized Tg mice (P = 0.206). The error bars are standard error of the mean that applies also to all subsequent figures.
Treatment from 19 to 24 month
In these older animals, the immunization did not elicit measurable antibody response (Fig. 1B).
Amyloid-β burden
Treatment from 11 to 24 months
Quantitative analysis of brain Aβ deposition (plaques and vascular Aβ) revealed that the volume of total cortical Aβ deposits was reduced by 31% (P < 0.05) in mice immunized with K6Aβ1–30 in alum adjuvant from 11 to 24 months of age, compared to vehicle-treated animals (Fig. 1C). The amount of vascular Aβ deposits was near identical in the two groups and was 22% and 32% of total Aβ burden in Tg controls vs. Tg K6Aβ1–30-treated animals, respectively (Fig. 1C). Similar treatment effect was observed in total brain Aβ levels (Aβ1–40, 30% reduction, P = 0.03; Aβ1–42, 37% reduction, P = 0.02), but soluble Aβ levels were not significantly affected although there was a strong trend for lower levels of soluble Aβ1–42 in the vaccinated mice (32% reduction, P = 0.08). A comparison of the percentages of total Aβ burden (plaques and Aβ in vessels) and Aβ burden in the vasculature indicates that the vaccination preferentially removes parenchymal- rather than vascular-Aβ deposits.
Histological observation in the 11–24-month-old group indicated that immunized mice appeared to have fewer large plaques and fewer diffuse plaques of all sizes compared to control mice (Fig. 2A and B), which fits with the quantitative analysis. The measurements of total Aβ deposit burden and total Aβ levels correlated well and indicated a similar percentage reduction in the vaccinated mice. The total Aβ deposit burden correlated better with total Aβ levels (Aβ1–40, P < 0.0001; Aβ1–42, P < 0.01) than with soluble Aβ levels (Aβ1–40, NS; Aβ1–42, P < 0.05). Likewise, soluble Aβ1–40 levels did not correlate with total Aβ1–40 levels but there was a strong trend for a correlation between total and soluble Aβ1–42 levels (Aβ1–40, NS; Aβ1–42, P = 0.07). No significant gender differences were observed in plaque burden or Aβ1–40/ 42 levels but only a few males were in the study (see Table 1).
Fig. 2.
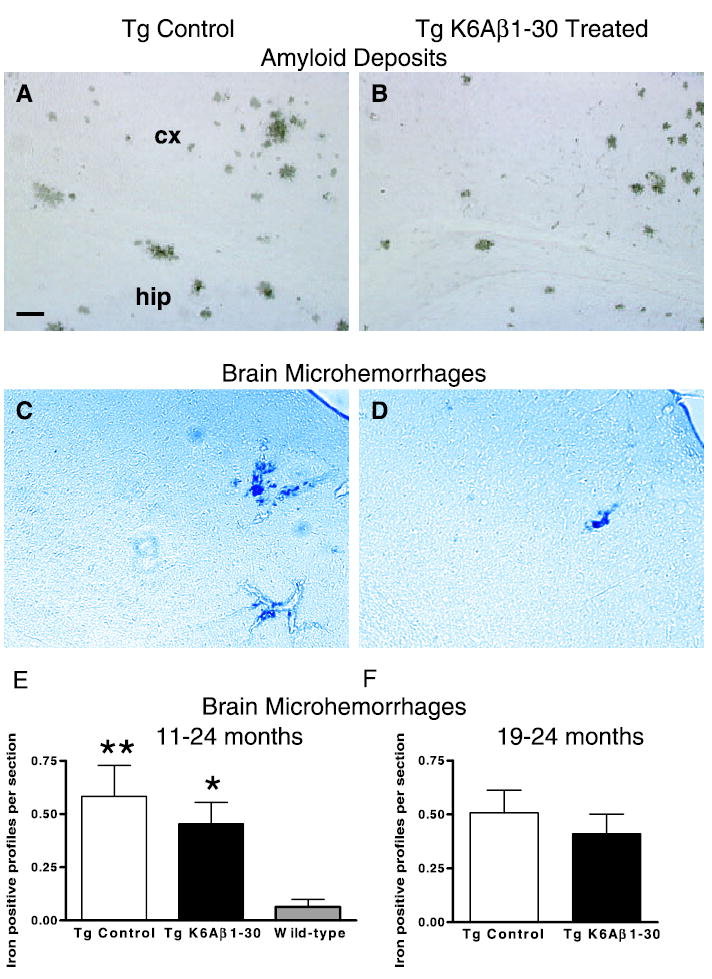
Histology of plaques and microhemorrhages. (A and B) Immunohistochemistry (6E10) of Aβ plaques revealed that compared to controls (A), immunized mice (B) had fewer large plaques and fewer diffuse plaques of all sizes. This observation fits with the 31% reduction in cortical amyloid burden (see Fig. 1A). A and B are representative coronal sections (original magnification, ×50) through the retrosplenial cortex (cx) and the hippocampus (hip). (C and D) Cerebral microhemorrhages were often observed in Tg2576 mouse brain sections stained with Perls’ stain for ferric iron in haemosiderin (blue), both in controls (C) and immunized mice (D). The immunotherapy did not increase the extent of the bleeding as quantified in E and F. C and D are representative coronal sections (original magnification, ×50) through the parietal cortex of Tg2576 mice. (E) 11–24 months. Immunization with K6Aβ1–30 did not increase brain microhemorrhages in Tg2576 mice but the Tg animals had significantly more iron positive profiles per brain section compared to wild-type (Wt) littermates (Tg control vs. Wt, P < 0.01; Tg K6Aβ1–30 vs. Wt, P < 0.05). (F) 19–24 months. No significant difference was seen in brain microhemorrhages between immunized and control Tg mice.
Treatment from 19 to 24 months
No significant difference was observed between the groups in cortical amyloid deposits or Aβ levels (Fig. 1D).
Cerebral bleeding
Treatment from 11 to 24 months
Immunization with K6Aβ1–30 in alum adjuvant did not increase the extent of brain microhemorrhages (Fig. 2C–E). However, Tg2576 mice had more iron-positive profiles per section than wild-type animals, which is likely related to vascular Aβ deposition in the transgenic mice (Kruskal–Wallis P < 0.01; Dunn’s posthoc test Tg control vs. wild-type, P < 0.01; Tg K6Aβ1–30 vs. wild-type P < 0.05). To verify that our staining method would allow us to detect increases in cerebral bleeding we used positive controls. These mice had brain hemorrhages caused by intracerebral cannula placement. The bleeding in these animals was associated with the cannula track and was substantially more extensive than the microhemorrhages that we observed in the Tg mice (data not shown).
Treatment from 19 to 24 months
As in the other treatment group (11–24 months), immunization with K6Aβ1–30 in alum adjuvant did not increase the extent of brain microhemorrhages compared to control Tg mice (Fig. 2F).
Microglia and astrocytes
Treatment from 11 to 24 months
Semiquantitative analysis of microglial staining with lectin indicated more microglial involvement with Aβ plaques in the immunized animals compared to their Tg controls but the differences were not robust (Mann–Whitney, P < 0.05; see Table 2 and Fig. 3). Subsequent staining for IL-1β that is primarily found in microglia and for GFAP, a marker of astrocytes, did not reveal obvious differences between the treatment groups.
Table 2.
Semiquantitative analysis of microglial staining with tomatolectin
| Numbers of animals
|
||
|---|---|---|
| Microgliosis | Tg control | Tg-treated |
| +++ | 3 | 4 |
| ++ | 5 | 5 |
| + | 4 | 2 |
| 0 | 4 | 0 |
Extent of microgliosis associated with the Aβ deposits: 0, a few resting microglia; +, a few ramified and / or phagocytic microglia; ++, moderate number of ramified / phagocytic microglia; +++, numerous ramified / phagocytic microglia
Fig. 3.
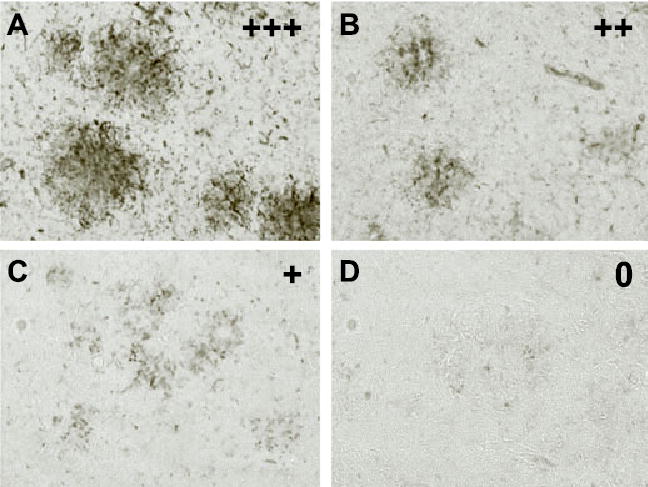
Microgliosis. The semiquantitative analysis of microglial staining with tomatolectin (Table 2) indicated more extensive microgliosis associated with Aβ plaques in the immunized animals in the 11–24 months group compared to transgenic controls (P < 0.05). No apparent differences were observed between the groups in the 19–24-month study (data not shown). (A–D) Representative images of the microglia quantified in Table 2. Original magnification ×200. In sections with +++ rating, ramified and phagocytic microglia were prominent throughout the Aβ deposit (A). Microglial involvement with the Aβ deposits was less in sections rated as ++ (B) and + (C) and virtually absent in sections rated as 0 (D).
Treatment from 19 to 24 months
No apparent differences were observed between the groups in microglial or astroglial activation.
Behaviour
The immunized mice and their vehicle controls were assessed on two cognitive tests in addition to various sensorimotor tasks. We and others have used these tests extensively in AD model mice except for the closed-field symmetrical maze, which consists of mazes of varying difficulties and was included to confirm the cognitive effect of the vaccine.
Cognitive tests
Radial arm maze
Treatment from 11 to 24 months
The Tg mice immunized with K6Aβ1–30 (n = 14) navigated the maze much better than the vehicle-treated Tg mice (n = 19), and their performance was similar to that of their wild-type littermates [(n = 15), Fig. 4A, ANOVA, repeated measures; group effect, P < 0.001; days effect, P = 0.198, interaction (days × treatment), P = 0.024]. Posthoc analysis revealed a treatment effect (Tg C vs. Tg K6Aβ1–30, Newman–Keuls P = 0.01). Also, wild-type mice differed from Tg controls (P = 0.001) but not from immunized Tg mice. Wild-type littermates were vaccinated with the peptide, received adjuvant alone or no injection. No difference was observed between these groups, which were then combined for subsequent analysis. The groups did not differ significantly in their time taken to run the maze (data not shown).
Fig. 4.
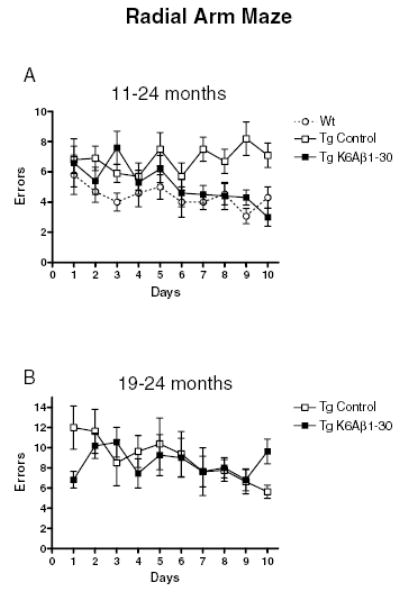
Radial arm maze. (A) 11–24 months. The Tg mice immunized with K6Aβ1–30 navigated the radial arm maze with fewer errors than control Tg mice, and performed as well as their wild-type (Wt) littermates (group effect P < 0.001; Tg control vs. Tg K6Aβ1–30 P = 0.01; Tg control vs. Wt mice P = 0.001). (B) 19–24 months. The Tg mice immunized with K6Aβ1–30 did not differ significantly from their control Tg mice in their performance in the radial arm maze.
Treatment from 19 to 24 months
No difference was observed between the K6Aβ1–30 immunized mice (n = 11) and their transgenic controls (n = 8) in their performance in the radial arm maze (Fig. 4B).
Closed field symmetrical maze
Treatment from 11 to 24 months
The overall performance (number of errors) of the mice differed between groups (P < 0.0001) and mazes (P < 0.0001) as analysed by two-way ANOVA. There was also an interaction (P < 0.0001) between these two factors, which indicates that group performance depended on the maze that the animals were navigating. Posthoc analysis between the groups indicated that the Tg controls (n = 16) differed significantly from the Tg K6Aβ1–30 treated mice (n = 11, P < 0.001) and the wild-type mice (n = 15, P < 0.0001), whereas the TgK6Aβ1–30 mice did not differ significantly from the wild-type animals. Significant differences between individual groups were observed in Mazes 3 and 7 (Fig. 5C and G). In Maze 3, Tg controls performed worse than wild-type mice (P < 0.05), and in Maze 7, Tg controls had more errors than both wild-type mice and immunized Tg mice (P < 0.0001) but those latter two groups showed a more modest difference (P < 0.01). Of all the mazes, #7 had the most complex navigation pattern, followed by #3, as shown by the total number of errors committed. A strong trend for significance, with treated Tg mice performing similarly to wild-type littermates, was observed in #1 and 6 that are of medium difficulty (Fig. 5A and F). In the three simplest mazes (#2, 4 and 5), as judged by the number of errors committed, the three groups performed at a similar level (Fig. 5B, D and E). A similar pattern was observed when maze and group differences in the time taken to run the maze were analysed instead of the number of errors (data not shown).
Fig. 5.
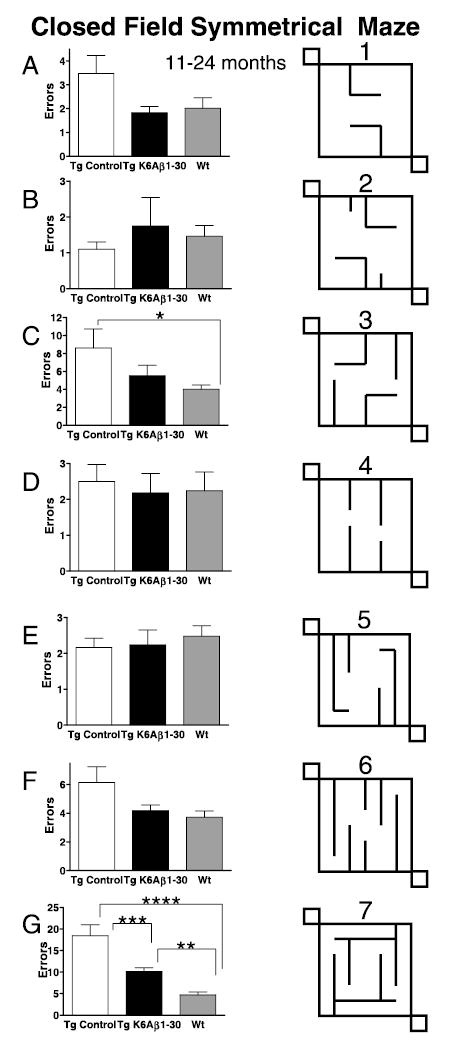
Closed field symmetrical maze. (A–G) 11–24 months. Overall, the Tg controls performed significantly worse than the immunized Tg mice (P < 0.001) and the wild-type mice (P < 0.0001), whereas the immunized Tg animals performed as well as their wild-type (Wt) littermates. Significant differences in the number of errors were observed in Maze 3 (C, Tg control vs. Wt, P < 0.05) and Maze 7 (G, Tg control vs. Tg K6Aβ1–30, P < 0.001; Tg control vs. Wt, P < 0.0001; Tg K6Aβ1–30 vs. Wt, P < 0.01). Maze 7 had the most difficult navigation pattern and Maze 3 was the second most difficult maze, as shown by the total number of errors committed (see y-axis). A strong trend for significance was observed in two other relatively complex mazes (Mazes 1 and 6) and in these mazes the Tg K6Aβ1–30 mice had similar number of errors as Wt mice. The Tg mice immunized from 19 to 24 months did not differ significantly from their Tg controls in any of the mazes (data not shown).
Treatment from 19 to 24 months
Group differences were not observed in any of the seven mazes (data not shown).
Sensorimotor tests
Locomotor activity
Treatment from 11 to 24 months
The transgenic mice were more active than their wild-type littermates (n = 15) but significant differences were not observed between the K6Aβ1–30 treated (n = 12) and control Tg mice (n = 16) in any of the parameters measured (Fig. 6A). Group differences were observed in: (i) distance travelled (ANOVA, P < 0.01); (ii) average speed (ANOVA, P < 0.01) and (iii) rest time (ANOVA, P < 0.001). Maximum speed obtained did not differ between the groups. Posthoc analysis revealed that the wild-type mice travelled less distance than Tg controls (P < 0.01) or immunized Tg mice (P < 0.05). They also moved at a slower speed than both Tg groups (P < 0.01–0.05), and rested longer as well (P < 0.001–0.05).
Fig. 6.

Locomotor activity. (A) 11–24 months. Both the Tg groups were more active than their wild-type (Wt) littermates but the immunized and control Tg groups did not differ significantly in any of the parameters measured. Distance travelled (P < 0.01, Wt vs. Tg control, P < 0.01; Wt vs. Tg K6Aβ1–30, P < 0.05). Maximum speed (Vmax) obtained did not differ between the groups. Average speed (Vmean, P < 0.01, Wt vs. Tg control, P < 0.01; Wt vs. Tg K6Aβ1–30, P < 0.05). Rest time (P < 0.001, Wt vs. Tg control, P < 0.001; Wt vs. Tg K6Aβ1–30, P < 0.05). (B) 19–24 months. The Tg groups did not differ in any of the locomotor parameters measured.
Treatment from 19 to 24 months
The treated (n = 11) and control Tg mice (n = 8) performed in a similar manner in all of the measured parameters (Fig. 6B).
All the animals that went through the locomotor activity test were assessed in the traverse beam and rotarod (see also Table 1).
Traverse beam and rotarod
Treatment from 11 to 24 months
Group differences were observed in the animals’ performance on the traverse beam (Fig. 7A; ANOVA repeated measures, P < 0.05). Although the vaccinated Tg mice had more foot slips than wild-type mice (P = 0.03), they did not differ from their Tg controls. This indicates that the slips are probably related to the increased locomotor activity observed in both groups of Tg mice, compared to wild-type controls. The animals performed better on subsequent trials as expected (ANOVA, P < 0.0001) and the improvement in performance was group dependent (ANOVA, treatment × trials, P = 0.0003). On the rotarod, the groups performed equally well and the animals did better on subsequent trials as expected (Fig. 7B; ANOVA repeated measures, P = 0.0003).
Fig. 7.
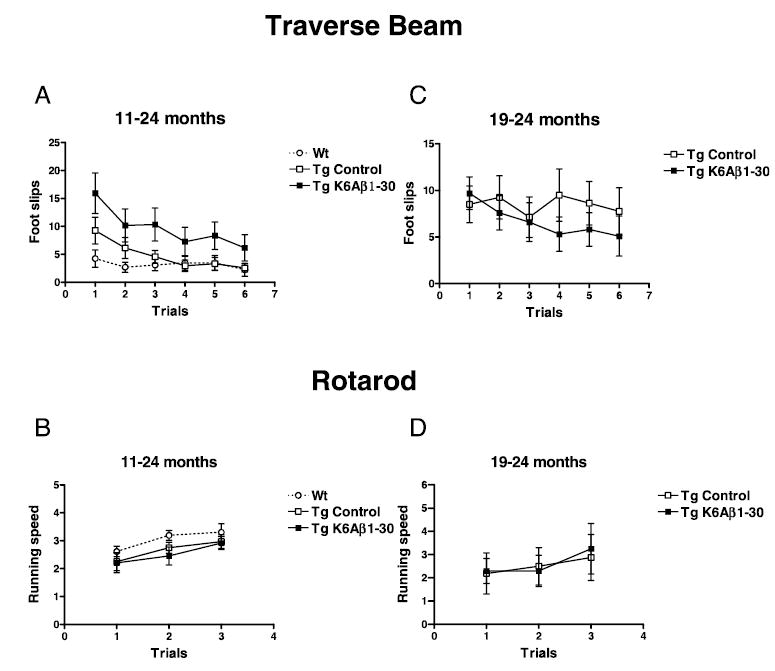
Traverse beam and rotarod. (A) 11–24 months. The animals differed in their performance on the traverse beam (P < 0.05, Tg K6Aβ1–30 vs. Wt, P = 0.03). As expected, the mice had fewer foot slips with more trials (P < 0.0001) and the improvement was group dependent (treatment × trials P = 0.0003). (B) 11–24 months. No group differences were observed but the animals’ performance on the rotarod improved over time as expected (P = 0.0003). (C) 19–24 months. No differences were observed between the Tg groups in their performance on the traverse beam but there was a strong overall trend for significantly fewer foot slips with more trials (P = 0.06). (D) 19–24 months. No group differences were observed on the rotarod but the animals’ performance improved over time as expected (P = 0.0023).
Treatment from 19 to 24 months
No statistical difference was observed between the groups in their performance on the traverse beam, and there was a strong overall trend for fewer foot slips with more trials (Fig. 7C; ANOVA repeated measures, P = 0.06). On the rotarod, the immunized and control Tg mice performed equally well and the mice performed better on subsequent trials as expected (Fig. 7D; ANOVA repeated measures, P = 0.0023).
Discussion
Whether immunotherapy for AD will go into clinical practice depends on minimizing potential side-effects. In the aborted clinical trial, toxicity was linked to excessive cell-mediated immunity while a therapeutic response was thought to be associated with humoral immunity (Bennett & Holtzman, 2005; Gilman et al., 2005; Sigurdsson, 2006). Hence, passive immunization appears feasible. However, as AD is a chronic disease, multiple injections of monotypic antibodies or Fv fragments would likely be needed, which could result in an anti-idiotypic response with subsequent complications. Furthermore, passive immunization can lead to increased cerebral hemorrhages in animal models (Pfeifer et al., 2002; Wilcock et al., 2004; Racke et al., 2005) and has been associated with encephalitis (Lee et al., 2005). Therefore, if toxicity can be reduced, active immunization may prove simpler and more effective.
Prior to the encephalitis in the clinical trial of AD vaccination, we promoted the use of an immunogen and adjuvant that favours a Th2 instead of a Th1 response (Sigurdsson et al., 2001). We have shown previously that our nontoxic, nonfibrillogenic Aβ derivatives with altered sequences to minimize Th1 response, reduce the Aβ deposit burden when used with strong adjuvants such as Freund’s (Sigurdsson et al., 2001; Sigurdsson et al., 2004). In the present study we extend those observations by demonstrating that one of our homologs reduces the Aβ burden when used in conjunction with alum adjuvant, which is commonly used in humans and primarily induces a Th2 response (Gupta, 1998). This contrasts to Freund’s adjuvant, which was used in most prior AD model vaccination protocols and QS-21, which was employed in the clinical trial. Alum has not been previously tested in AD mouse models. Our use of it is supported by our preliminary findings (Scholtzova et al., 2002), and a recent study, in which wild-type mice were immunized with truncated Aβ peptides (Agadjanyan et al., 2005). In that study, high titres of Aβ antibodies were generated but splenocytes were not reactive with Aβ.
We show a robust reduction of 31% in Aβ deposit burden (parenchymal and vascular) in the group treated from 11 to 24 months but vascular Aβ specifically was not altered. These findings indicate that our approach, in contrast to prior passive immunization studies does not increase vascular Aβ deposition (Wilcock et al., 2004). The reduction in deposited Aβ and total Aβ levels (30–37% reduction) correlated very well, which strengthens the findings. Soluble Aβ levels measured approximately 200- and 800-fold less than total Aβ1–40 and Aβ1–42, respectively. These substantially lower levels were not altered by the therapy, which can be explained by their small percentage of total Aβ.
While our results show a group correlation between cognitive improvements and Aβ burden, no relationship was observed when an individual animal’s Aβ deposition or Aβ levels were correlated with its performance on the two cognitive tests (radial arm maze and symmetrical maze). This fits with prior reports by us and others, and it may be that behaviour will correlate more with levels of oligomeric Aβ (Janus et al., 2000; Morgan et al., 2000; Dodart et al., 2002; Kotilinek et al., 2002; Sigurdsson et al., 2004; Jensen et al., 2005). However, although plaque deposition and subsequent encapsulation by glial cells is likely to be an attempt to isolate excess Aβ that cannot be cleared, this pathological process can eventually affect neuronal connectivity, and chemicals released by reactive glial cells can be neurotoxic. Hence, any immunotherapy should at least slow the progression of plaque deposition.
Cognitive improvements were observed in the radial arm maze and the symmetrical maze. The radial arm maze has consistently allowed us to detect improvements in spatial / working memory in vaccinated mice (Sigurdsson et al., 2004). We have now confirmed these findings in another cognitive measure, the closed-field symmetrical maze, which has not been previously used for AD model mice. This test consists of several mazes of varying difficulties, that have permitted comparisons of spatial learning across species (Shore et al., 2001), and its virtual form can easily be applied to humans. Hence in future studies, the potential cognitive effects of therapy for AD could be compared using the same test in mouse models and AD patients.
To verify that the apparent enhanced cognitive abilities of the vaccinated mice were not due to unrelated behavioural effects, the mice went through a battery of sensorimotor tests. Although the transgenic mice were more active than wild-type littermates in a locomotor activity test, no differences were observed between the treated and control transgenic groups. The rotarod test did not reveal any differences between the groups and although the vaccinated transgenic mice had more foot slips than wild-type mice on the traverse beam, they did not differ from their transgenic controls, indicating that this effect is probably related to their increased locomotor activity. Also, the impaired motor balance while crossing a beam cannot explain why the vaccinated mice performed better on the cognitive tests. Overall, the results from the sensorimotor tests strengthen the findings of cognitive improvements in the radial arm maze and the closed field symmetrical maze.
We did not observe changes in Aβ burden or cognition in mice immunized from 19 to 24 months. This can likely be explained by the lack of an antibody response in these older mice, which in turn may be due to immunosenescence observed with aging and the relatively mild adjuvant employed. These findings are in agreement with a prior study showing that Aβ1–42 immunization in Freund’s adjuvant at different ages reduced Aβ levels and / or Aβ deposition more effectively when treatment was initiated prior to extensive plaque development (Das et al., 2001). This has also been found to be the case with passive immunization approaches using a variety of Aβ antibodies and mouse AD models (Levites et al., 2006). However, because cognitive improvements have often been observed following immunotherapy in different AD model mice without an obvious relationship to various Aβ measurements (Janus et al., 2000; Morgan et al., 2000; Dodart et al., 2002; Kotilinek et al., 2002; Sigurdsson et al., 2004; Jensen et al., 2005), it remained to be determined if immunotherapy at a late stage of Aβ deposition could improve cognition even though plaques were not cleared.
Besides an excessive Th1 response, another potential side-effect of Aβ immunotherapy is cerebral bleeding. Several reports have shown an increase in microhemorrhages in different AD mouse models following passive intraperitoneal immunization with different monoclonal antibodies with high affinity for Aβ plaques and congophilic angiopathy (Pfeifer et al., 2002; Wilcock et al., 2004; Racke et al., 2005). This effect was not observed with yet another Aβ antibody following a single intracerebroventricular injection in the Tg2576 model (Chauhan & Siegel, 2003), or with a monoclonal Aβ antibody which binds soluble Aβ and does not recognize amyloid deposits (Racke et al., 2005). Also, in a recent study using several different Aβ antibodies, microhemorrhages were not observed even in control animals (Levites et al., 2006). However, those AD model mice were much younger than the mice in the current study or those used in the previous reports discussed above. There are no prior reports of microhemorrhage assessment following active immunization in animal models; however, the clinical trial data from the few autopsies suggest that vascular Aβ deposits were not being cleared and hemorrhage may have been increased (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005). In the present study we did not observe increased cerebral bleeding in the vaccinated mice, indicating that active immunization with an immunogen and adjuvant favouring a Th2 response, may be a safer approach than passive immunization. Our findings need to be interpreted with caution because we did not include a group that received passive immunization, but the extent of microhemorrhages in our Tg2576 control mice is similar to a previous report in the same model at a similar age (Wilcock et al., 2004). Also, our positive control sections indicated that our staining method was of a sufficient sensitivity to detect increases in vascular bleeding. The microhemorrhages are likely caused by rapid clearance of parenchymal Aβ deposits and subsequent vascular deposition by high doses of high affinity antibodies, as well as a direct immune response to Aβ laden vessels. In active immunization paradigms, a polyclonal response with antibodies of different affinities and against various epitopes is likely to have a less acute effect on Aβ clearance, which may provide for compensatory changes to minimize the detrimental effects related to rapid removal of Aβ. Because the vaccine was not immunogenic in the old treatment group (19–24 months), the present data does not clarify if active immunization such as ours would result in microhemorrhages if animals generate Aβ antibodies once extensive vascular amyloid is in place. The passive immunization studies resulting in microhemorrhages were conducted in old mice that very likely had severe congophilic amyloid angiopathy (CAA) at the onset of the treatment whereas CAA in our 11-month-old group was presumably modest.
Our results extend our previous findings showing that our Aβ derivatives can reduce Aβ burden, and lead to cognitive improvement, when used with an adjuvant suitable for humans, without any associated increase in vascular Aβ burden or microhemorrhages. As in other studies, we also found that immunization has to be initiated early in the course of pathology to be effective. This highlights the need to develop methods to detect Aβ deposits prior to the development of clinical symptoms of dementia, at which point the Aβ burden may be quite high (Wisniewski & Frangione, 2005). It can be envisioned that because a self-antigen is being targeted and individual immune responses vary, that future immunotherapy may have to be individually tailored. The aim would be to modestly shift the balance between brain Aβ accumulation and clearance. For example, an Aβ-derived immunogen may be selected based on haplotype screening to provide first a T cell-independent IgM response, which will generate antibodies that recognize different epitopes and conformations of Aβ, which may be more efficacious than targeting a single entity. If this relatively modest immune response would prove ineffective, vaccination could proceed with other Aβ derivatives predicted to elicit a slightly stronger immune response. This process could be continued until cognitive measures would improve and / or Aβ burden would be reduced as assessed by imaging techniques that are currently under development (Wadghiri et al., 2003; Klunk et al., 2004; Sigurdsson et al., 2006).
Acknowledgments
Supported by NIH grants AG020197, AG020245 and AG005891, and the Alzheimer’s Association. We thank Julie Eng and Ashlyn Lines for technical assistance.
References
- Acarin L, Vela JM, Gonzalez B, Castellano B. Demonstration of poly--acetyl lactosamine residues in ameboid and ramified microglial cells in rat brain by tomato lectin binding. J Histochem Cytochem. 1994;42:1033–1041. doi: 10.1177/42.8.8027523. [DOI] [PubMed] [Google Scholar]
- Agadjanyan MG, Ghochikyan A, Petrushina I, Vasilevko V, Movsesyan N, Mkrtichyan M, Saing T, Cribbs DH. Prototype Alzheimer’s disease vaccine using the immunodominant B cell epitope from β-amyloid and promiscuous T cell epitope pan HLA DR-binding peptide. J Immunol. 2005;174:1580–1586. doi: 10.4049/jimmunol.174.3.1580. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Holtzman DM. Immunization therapy for Alzheimer disease? Neurology. 2005;64:10–12. doi: 10.1212/01.WNL.0000150527.24596.29. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ. Intracerebroventricular passive immunization with anti-Aβ antibody in Tg2576. J Neurosci Res. 2003;74:142–147. doi: 10.1002/jnr.10721. [DOI] [PubMed] [Google Scholar]
- Das P, Murphy MP, Younkin LH, Younkin SG, Golde TE. Reduced effectiveness of Aβ1–42 immunization in APP transgenic mice with significant amyloid deposition. Neurobiol Aging. 2001;22:721–727. doi: 10.1016/s0197-4580(01)00245-7. [DOI] [PubMed] [Google Scholar]
- Davenport JW, Hagquist WW, Rankin GR. Symmetrical Maze –An Automated Closed-Field Test Series for Rats. Behav Res Meth Instrumentation. 1970;2:112–118. [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer’s disease model. Nature Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Boada RM, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-β immunization in Alzheimer’s disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas DS, DaSilva K, Fenili D, George-Hyslop P, McLaurin J. Immunotherapy for Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101:14657–14662. doi: 10.1073/pnas.0404866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Boada RM, Forette F, Orgogozo JM. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Gupta RK. Aluminum compounds as vaccine. Adv Drug Deliv Rev. 1998;32 :155–172. doi: 10.1016/s0169-409x(98)00008-8. [DOI] [PubMed] [Google Scholar]
- Hebb DO, Williams KA. A method of rating animal intelligence. J Gen Psychol. 1946;34:59–65. doi: 10.1080/00221309.1946.10544520. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against β-amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hyde LA, Crnic LS, Pollock A, Bickford PC. Motor learning in Ts65Dn mice, a model for Down syndrome. Dev Psychobiol. 2001;38:33–45. doi: 10.1002/1098-2302(2001)38:1<33::aid-dev3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Jameson BA, Wolf H. The antigenic index: a novel algorithm for predicting antigenic determinants. Comput Appl Biosci. 1988;4:181–186. doi: 10.1093/bioinformatics/4.1.181. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, George-Hyslop P, Westaway D. Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Jensen MT, Mottin MD, Cracchiolo JR, Leighty RE, Arendash GW. Lifelong immunization with human β-amyloid (1–42) protects Alzheimer’s transgenic mice against cognitive impairment throughout aging. Neuroscience. 2005;130:667–684. doi: 10.1016/j.neuroscience.2004.09.055. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Wen GY, Bancher C, Chen CMJ, Sapienza V, Hong H, Wisniewski HM. Detection and quantification of amyloid β-peptide with 2 monoclonal antibodies. Neurosci Res Comm. 1990;7:113–122. [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH. Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J Neurosci. 2002;22:6331–6335. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Leng LZ, Lee VM, Trojanowski JQ. Meningoencephalitis associated with passive immunization of a transgenic murine model of Alzheimer’s amyloidosis. FEBS Lett. 2005;579:2564–2568. doi: 10.1016/j.febslet.2005.03.070. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Spooner ET, Leverone JF, Mori C, Iglesias M, Bloom JK, Seabrook TJ. Amyloid-β immunization in Alzheimer’s disease transgenic mouse models and wildtype mice. Neurochem Res. 2003;28:1017–1027. doi: 10.1023/a:1023203122036. [DOI] [PubMed] [Google Scholar]
- Levites Y, Das P, Price RW, Rochette MJ, Kostura LA, McGowan EM, Murphy MP, Golde TE. Anti-Aβ (42)- and anti-Aβ(40)- specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J Clin Invest. 2006;116:193–201. doi: 10.1172/JCI25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Aβ vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64 :129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nature Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Aβ immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Rabinovitch MS, Rosvold HE. A closed-field intelligence test for rats. Can J Psychol. 1951;5:122–128. doi: 10.1037/h0083542. [DOI] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li Y, Schmidt SD, Mathews PM, Fryer JD, Holtzman DM, Sigurdsson EM, Wisniewski T. A synthetic peptide blocking the apolipoprotein E / β-amyloid binding mitigates β-amyloid toxicity and fibril formation in vitro and reduces β-amyloid plaques in transgenic mice. Am J Pathol. 2004;165:937–948. doi: 10.1016/s0002-9440(10)63355-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Schmidt SD, Jiang Y, Nixon RA, Mathews PM. Tissue processing prior to protein analysis and amyloid-β quantitation. [In E.M. Sigurdsson, (Ed.), Amyloid Proteins: Methods and Protocols, Humana Press, Totowa] . Meth Mol Biol. 2005a;299:267–278. doi: 10.1385/1-59259-874-9:267. [DOI] [PubMed] [Google Scholar]
- Schmidt SD, Nixon RA, Mathews PM. ELISA method for measurement of amyloid-β levels. [In E.M. Sigurdsson, (Ed.), Amyloid Proteins: Methods and Protocols, Humana Press, Totowa] . Meth Mol Biol. 2005b;299:279–298. doi: 10.1385/1-59259-874-9:279. [DOI] [PubMed] [Google Scholar]
- Scholtzova H, Wisniewski T, Ahlawat S, Watanabe M, Quartermain D, Frangione B, Sigurdsson EM. Safety of potential vaccines for Alzheimer’s disease. Soc Neurosci Abstr. 2002:227.1. [Google Scholar]
- Shore DI, Stanford L, MacInnes WJ, Klein RM, Brown RE. Of mice and men: virtual Hebb–Williams mazes permit comparison of spatial learning across species. Cogn Affect Behav Neurosci. 2001;1:83–89. doi: 10.3758/cabn.1.1.83. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM. Histological staining of amyloid-β in mouse brains. [In E.M. Sigurdsson, (Ed.), Amyloid Proteins: Methods and Protocols, Humana Press, Totowa] . Meth Mol Biol. 2005;299:299–308. doi: 10.1385/1-59259-874-9:299. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM. Immunotherapy for conformational diseases. Curr Pharmaceut Design. 2006;12:2569–2585. doi: 10.2174/138161206777698837. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Wadghiri YZ, Mosconi L, Blind JA, Li Y, Knudsen E, Asuni AA, Tsui WH, Sadowski M, Turnbull DH, de Leon MJ, Wisniewski T. A non-toxic ligand for voxel-based MRI analysis of plaques in AD transgenic mice. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.12.018. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Knudsen E, Asuni A, Fitzer-Attas C, Sage D, Quartermain D, Goni F, Frangione B, Wisniewski T. An attenuated immune response is sufficient to enhance cognition in an Alzheimer’s disease mouse model immunized with amyloid-β derivatives. J Neurosci. 2004;24:6277–6282. doi: 10.1523/JNEUROSCI.1344-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Lorens SA, Hejna MJ, Dong XW, Lee JM. Local and distant histopathological effects of unilateral amyloid-β 25–35 injections into the amygdala of young F344 rats. Neurobiol Aging. 1996;17:893–901. doi: 10.1016/s0197-4580(96)00169-8. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T. Immunization with a non-toxic / non-fibrillar amyloid-β homologous peptide reduces Alzheimer’s disease associated pathology in transgenic mice. Am J Pathol. 2001;159:439–447. doi: 10.1016/s0002-9440(10)61715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Perry TA, Blockland A, Wilkinson LS, Wiley RG, Lappi DA, Dunnet SB. Behavioural, histochemical and biochemical consequences of selective immunolesions in discrete regions of the basal forebrain cholinergic system. Neuroscience. 1994;63:95–122. doi: 10.1016/0306-4522(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Wadghiri YZ, Sigurdsson EM, Sadowski M, Elliott JI, Li Y, Scholtzova H, Tang CY, Aguinaldo G, Pappolla M, Duff K, Wisniewski T, Turnbull DH. Detection of Alzheimer’s amyloid in transgenic mice using magnetic resonance microimaging. Magn Reson Med. 2003;50:293–302. doi: 10.1002/mrm.10529. [DOI] [PubMed] [Google Scholar]
- Weksler ME, Gouras G, Relkin NR, Szabo P. The immune system, amyloid-β peptide, and Alzheimer’s disease. Immunol Rev. 2005;205:244–256. doi: 10.1111/j.0105-2896.2005.00264.x. [DOI] [PubMed] [Google Scholar]
- West MJ. Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. TINS. 1999;22:51–61. doi: 10.1016/s0166-2236(98)01362-9. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Aβ in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Frangione B. Immunological and anti-chaperone therapeutic approaches for Alzheimer disease. Brain Pathol. 2005;15:72–77. doi: 10.1111/j.1750-3639.2005.tb00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]