

Abstract
Prion diseases are transmissible and invariably fatal neurodegenerative disorders associated with a conformational transformation of the cellular prion protein (PrPC) into a self-replicating and proteinase K (PK)-resistant conformer, scrapie PrP (PrPSc). Humoral immunity may significantly prolong the incubation period and even prevent disease in murine models of prionoses. However, the mechanism(s) of action of anti-PrP monoclonal antibodies (Mabs) remain(s) obscure. The murine neuroblastoma N2a cell line, infected with the 22L mouse-adapted scrapie strain, was used to screen a large library of Mabs with similar binding affinities to PrP, to identify those antibodies which could clear established infection and/or prevent infection de novo. Three Mabs were found capable of complete and persistent clearing of already-infected N2a cells of PrPSc. These antibodies were 6D11 (generated to PK-resistant PrPSc and detecting PrP residues 93–109), and 7H6 and 7A12, which were raised against recombinant PrP and react with neighbouring epitopes of PrP residues 130–140 and 143–155, respectively. Mabs were found to interact with PrPSc formation both on the cell surface and after internalization in the cytosol. Treatment with Mabs was not associated with toxicity nor did it result in decreased expression of PrPC. Both preincubation of N2a cells with Mabs prior to exposure to 22L inoculum and preincubation of the inoculum with Mabs prior to infecting N2a cells resulted in a significant reduction in PrPSc levels. Information provided in these studies is important for the rational design of humoral immune therapy for prion infection in animals and eventually in humans.
Keywords: conformational disorder, monoclonal antibodies, N2a cell line, scrapie, treatment
Abbreviations: ATCC, American Type Culture Collection; BSE, bovine spongiform encephalopathy; Mab, monoclonal antibody; MEM, minimal essential medium; MTT, 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide; N2a/22L cells, N2a cells infected with the 22L mouse-adapted scrapie strain; PK, proteinase K; PrPC, cellular prion protein; PrPSc, scrapie prion protein; recPrP, recombinant PrP; vCJD, variant Creutzfeldt – Jakob disease
Introduction
Currently no effective treatment for prion diseases exists. As a result of the bovine spongiform encephalopathy (BSE) epidemic large populations have been exposed to this animal prion disease with the potential for human transmission. In the United Kingdom alone, the total number of definite or probable cases of variant Creutzfeldt–Jakob disease (vCJD) has currently reached 160 patients, who contracted the infection from prion-contaminated beef products (UK Department of Health & Monthly CJD Statistics, 2006). Due to the very long incubation period of prionoses, it has been suggested that a much larger number of people could be asymptomatic carriers of this disease (Balter, 2002; Hilton et al., 2002). This cohort is at risk of developing disease in the future and also constitutes a reservoir of infectivity. Transmissibility of prions through blood products has been experimentally shown with the induction of scrapie in sheep by blood transfusion from BSE cattle (Houston et al., 2000). Furthermore, scrapie prion protein (PrPSc) was detected in the lymphatic system of a patient who had received blood products donated by an asymptomatic prion carrier (Peden et al., 2004). Spread of infection from asymptomatic carriers is possible because of the accumulation of PrPSc in the lymphatic system long before the appearance of clinical disease (Wadsworth et al., 2001; Hilton et al., 2004a,b). The presence of PrPSc may remain limited to the extraneural tissues for a prolonged period (Eklund et al., 1967; Rubenstein et al., 1991; Yasuhara et al., 1993; Herzog et al., 2004); therefore, PrPSc formation can be targeted in the early stages of infection by treatment approaches which do not have to be capable of penetrating the blood–brain barrier.
Reports from several laboratories have demonstrated that immunization can significantly prolong the incubation period of prionoses or even prevent disease symptoms (Goni et al., 2005; Sigurdsson et al., 2002a; Schwarz et al., 2003). The effectiveness of these approaches appears to be associated chiefly with humoral immunity (Heppner et al., 2001; Sigurdsson et al., 2003; White et al., 2003). However, the exact mechanism by which anti-PrP antibodies interfere with PrPSc formation remains obscure. Analysis of in vitro and in vivo treatment studies indicated that only some anti-PrP monoclonal antibodies (Mabs) demonstrate therapeutic efficacy (Peretz et al., 2001; Sigurdsson et al., 2003; Perrier et al., 2004; Feraudet et al., 2005). Therefore, understanding how Mabs interfere with PrPSc formation, and selecting those Mabs which are therapeutically active, is crucial for the rational design of the humoral immune therapy for prion infection. Hypotheses for the mechanisms of action of Mabs include perturbation of PrPC trafficking by Mabs (Feraudet et al., 2005), lowering PrPC content (Perrier et al., 2004), interference with the interaction of PrPC and PrPSc (Peretz et al., 2001) and sequestration of PrPC and/or PrPSc on the cell surface by Mabs (Enari et al., 2001a; Kim et al., 2004). In this study we used N2a murine neuroblastoma cell lines susceptible to infection with the 22L mouse-adapted scrapie strain (Bosque & Prusiner, 2000; Nishida et al., 2000; Perrier et al., 2004) to test the effectiveness of a number of Mabs for their ability both to prevent and to clear established infection. We attempted to map PrPSc epitopes which are the most crucial for the propagation of prion infectivity.
Materials and methods
Cell lines were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). Animals were obtained from Taconic (Germantown, NY, USA). All reagents and antibodies used in this study were purchased from Sigma (St Louis, MO, USA) unless specified otherwise. Animal studies were approved by the NYU School of Medicine Institutional Animal Care and Use Committee and were consistent with the recommendations of the American Veterinary Association.
Infection of N2a cells with 22L mouse-adapted prion strain
N2a mouse neuroblastoma cells (ATCC line CCL 131) were maintained in minimal essential medium (MEM) supplemented with heat-inactivated 10% fetal bovine serum, penicillin (100 units/mL) and streptomycin (100 μg/mL) at 37 °C in 5% CO2. Brains of terminally sick CD-1 mice infected with mouse-adapted 22L prion strain were homogenized by sonication (10% weight/volume) in cold phosphate-buffered saline and 5% dextrose in sterile conditions. For infection the brain homogenate was further diluted to 2% in Opti-MEM and added to subconfluent six-well plates (Corning, Acton, MA, USA), 1 mL per 10-cm2 well. After 5 h, 1 mL of regular MEM was added and the cells were incubated in the presence of infectious brain homogenate for an additional 12 h. After the medium was replaced with standard MEM, N2a cells were grown until confluence and then they were split into 1 : 2 dilutions and transferred to 25-cm2 flasks (Corning). Cells grown in one of the flasks were split 1 : 2 every 4–5 days to give rise to subsequent passages, whereas cells grown in the other flask were harvested and homogenized to monitor the level of PrPSc (see below). As a control experiment N2a cells were exposed to other mouse-adapted scrapie strains including 87V, 139A and ME7 in experimental conditions analogous to these described above. Previously published data indicate that wild-type N2a cells are resistant to infection with these strains (Bosque & Prusiner, 2000; Nishida et al., 2000).
Detection and quantification of PrPSc in infected N2a cells (N2a/22L)
N2a cells infected with the 22L mouse-adapted scrapie strain (N2a/22L) were harvested using ice-cold lysis buffer [NaCl, 150 mM; triton X-100, 0.5%; sodium deoxycholate, 0.5%; and Tris-HCL, 50 mM, pH 7.5; with a protease inhibitor cocktail (Roche, Indianapolis, IN, USA)]. The lysates were centrifuged for 3 min at 10 000 g to remove cell debris and the total protein concentration was measured in the supernatant using the bicinchoninic acid assay (BCA; Pierce, Rockford, IL, USA). Aliquots containing 200 μg of total protein were titrated by adding buffer to achieve a final protein concentration of 1 μg/μL. Samples were digested with proteinase K (PK; Roche) for 30 min at 37 °C. The enzyme-to-protein weight ratio was 1 : 50 (Perrier et al., 2004). PK activity was quenched by adding phenylmethanesulphonyl fluoride to achieve a final concentration of 3 mM. Samples were then centrifuged at 20 000 g for 45 min at 4° C. Pellets were resuspended in 15 μL Tris-buffered saline and 15 μL sample buffer, boiled for 5 min and then subjected to electrophoresis on 12.5% SDS-polyacrylamide Tris-tricine gels (Jimenez-Huete et al., 1998). In each experiment at least three samples of noninfected N2a cells were subjected to PK digestion to assure that PK-sensitive PrPC was completely digested. In samples not digested with PK, which were used to estimate the absolute level of PrP in the cell lysate, the total amount of protein loaded onto the SDS-polyacrylamide was 10 μg. Following overnight electrophoresis the protein was transferred onto nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ, USA) for 1 h at 400 mA using CAPS buffer (3-cyclohexylamino-1-propanesulphonic acid) containing 10% methanol. The membranes were blocked with 5% nonfat milk in TBST (Tris, 10 mM; NaCl, 150 mM; Tween 20, 0.1%, pH 7.5) for 1 h at room temperature and then incubated with Mab 6D11 diluted 1 : 3000. Following extensive washing in TBST the membranes were incubated with a horseradish peroxidase-conjugated sheep antimouse antibody (Amersham) and then developed using an enhanced chemiluminescent substrate (SuperSignal; Pierce). Membranes were apposed to autoradiography film (X-Omat Blue XB-1; Kodak, New Haven, CT, USA). The exposure time was kept standard for all experiments at 30 s. Developed films were converted into eight-bit grayscale digital files using a Epson Perfection 4990 scanner (Epson America, Long Beach, CA, USA) and Adobe Photoshop software 7.01 (Adobe Systems, San Jose, CA, USA) and saved in a TIF format with a resolution of 600 dpi. Quantification of PrPSc was performed by densitometric analysis using NIH Image J software v. 1.34. Areas under the curves for three PrP bands representing non-, mono- and diglycosylated isoforms of the protein were summarized from each sample to calculate the total PrP level.
Mabs
Production and characterization of anti-PrP Mabs 8B4, 11G5, 7H6, 7A12, 2C2, 8H4, 8F9 and 9H7 (Table 1) have been described previously (Zanusso et al., 1998; Liu et al., 2001; Pan et al., 2002; 2004; 2005; Wong et al., 2003). This set of Mabs reacts with the major structural domains of PrP. All are capable of binding both PrPC and PrPSc with varying levels of affinity (Pan et al., 2004; Wong et al., 2003). 7D9 is a Mab raised against recombinant PrP (recPrP) and which recognizes a nonlinear epitope on PrPSc and PrPC (Adler et al., 2003; Sadowski et al., 2003). 6D11 is a novel Mab raised against a nondenaturated PK-resistant fragment of PrPSc which has been purified from brains of CD-1 mice infected with the 139A mouse-adapted scrapie agent according to previously published protocols (Kascsak et al., 1986; Carp et al., 2000). Immunization of Prnp0/0 mice, isolation of splenocytes and their immortalization by fusion to myeloma cells, and identifications of clones by ELISA were performed using routine protocols (Kascsak et al., 1987; Zanusso et al., 1998). Binding of 6D11 to PrPC, recPrP, PK-resistant PrPSc fragments from brain homogenates and N2a/22L cell lysates was tested using Western blot as described above. Characterization of the 6D11 binding epitope was performed using a dot–blot assay. Thirty-mer peptides spanning the sequence of the PK-resistant PrPSc fragment (PrP residues 93–229) were synthesized at the W. M. Keck Foundation Facility (Yale University, New Haven, CT, USA) by solid-phase technique and purified by reverse-phase liquid chromatography, and their mass and purity were assessed by mass spectrometry (Sigurdsson et al., 2001; Sigurdsson et al., 2002a; Sadowski et al., 2004a). The peptides (0.3, 0.6, 1.2 and 1.8 μg/well) were blotted onto Immobilon PVDF membrane (Millipore Corporation, Billerica, MA, USA). Full-length recPrP (manufactured as described previously; Wong et al., 2003) and unrelated peptides Aβ1–40 and Aβ12–28 (Sadowski et al., 2004a) were used as positive and negative controls, respectively. Blocking, immunoblotting and development of the dot–blot were performed as described above.
Table 1.
Characterization of anti-PrP Mabs
| Antibody | Antigen localization† | KD |
|---|---|---|
| 7D9 | Unknown, nonlinear | 8.2 ± 0.19 × 10−10M |
| 8B4 | 35–45 | 2.4 ± 0.60 × 10−10M |
| 6D11 | 93–109 | 8.5 ± 0.18 × 10−11M |
| 11G5 | 115–130 (1st β-strand) | 5.4 ± 0.17 × 10−10M |
| 7H6 | 130–140 | 2.2 ± 0.04 × 10−10M |
| 7A12 | 143–155 (α-helix A) | 1.9 ± 0.05 × 10−10M |
| 2C2 | 153–165 (2nd β-strand) | 3.4 ± 0.15 × 10−9M |
| 8H4 | 175–185 (α-helix B) | 3.8 ± 0.11 × 10−10M |
| 9H7 | 143–231 | 10.3 ± 0.43 × 10−10M |
| 8F9 | 220–231 | 10.6 ± 0.50 × 10−9M |
Numbers correspond to residues of murine PrP. KD values are given as mean ± SD from at least three independent experiments.
The binding affinities of Mabs were compared using a solid-phase binding assay. ELISA plates (Immulon 2 HB; Canadawide Scientific, Ottawa, ON, USA) were coated overnight with recPrP (50 ng/well) and then blocked with Superblock (Pierce) for 2 h prior to adding Mabs. Binding with the primary antibody using dilutions ranging from 12 × 10−12 to 12 × 10−9M was performed at room temperature for 2 h, and was followed by adding rabbit antimouse IgG horseradish peroxidase-conjugated secondary antibody 1 : 3000 (Amersham). The color reaction was developed using 3,3′,5,5′-tetramethylbenzidine and stopped by adding 2 M sulphuric acid. The absorbance was measured at wavelength 450 nM. Experiments were run in triplicate. Data were fitted into a one-site binding curve, and the KD was calculated using GraphPad Prism Software v4.03 (GraphPad Prism Software, Inc., San Diego, CA, USA).
Treatment of N2a/22L cells with Mabs
N2a/22L cells (from the fifth passage after infection and higher) were plated in six-well plates and cultured until they reached 70–80% confluence. The therapeutic efficacy of Mabs was assessed by culturing N2a/22L cells in the presence of antibodies at a concentration 10 μg/mL for 96 h. The level of PK-resistant PrPSc was measured in Western blots as described above. Each Mab was tested in three independent experiments using independently infected cell lines. Each experiment included both a positive control (nontreated N2a/22L cells) and a negative control (N2a cells); both controls were subjected to PK digestion. The levels of PrPSc were expressed as percentages of the average value from a positive control (nontreated N2a/22L cells), whereas the optic density of the background was taken from negative control lanes (N2a cells). The effect of various Mabs was analysed using one-way ANOVA followed by a Dunnett post hoc test (Graph Pad Prism Software, v4.03).
Fifty per cent of maximal inhibitory concentration (IC50) was established by growing N2a/22L cells in the presence of increasing concentrations of Mabs for 96 h. The PrPSc level was then measured and data were fitted in a sigmoidal dose–response curve using Graph Pad Prism Software (v4.03). Experiments were performed in triplicate.
In experiments designed to check whether treatment with Mabs resulted in a persistent abrogation of PrPSc in treated cells, N2a/22L cells were cultured in the presence of Mabs (10 μg/mL) for 8 days, changing the medium every other day. Cells were then cultured in the absence of Mabs for an additional 14 days, harvested and lysed, and the level of PrPSc was measured in cell lysates as described above.
Total PrP levels (PrPC + PrPSc) and levels of β-actin and Thy-1 in N2a/22L cells treated with Mabs were measured in PK-nontreated samples. Following gel electrophoresis and Western blotting, membranes were probed with either 6D11, mouse antiβ-actin Mab (1 : 1000; Abcam, Cambridge, MA, USA) or AS02 Mab (1 : 1000; Calbiochem, CA, USA) recognizing mouse Thy-1 protein. For detection of Thy-1 protein the electrophoresis was performed under nonreducing conditions, according to the manufacturer’s recommendations. Densitometric measurement was performed as described above and the optic densities are expressed as percentages of the average protein level of nontreated N2a/22L cells. Experiments were performed in triplicate. Values were compared using one-way ANOVA followed by Dunnett’s post hoc test. The β-actin is a structural 43-kDa protein (Shashidhar et al., 2005), unrelated to PrPSc pathological biology, and was used as a marker of general protein expression in treated cells (Korth et al., 2001; Perrier et al., 2004). Thy-1 is a 31-kDa glycosylphosphatidylinositol-anchored protein, abundantly expressed by neurons, which coexists with PrPC on the external plasma membrane surface and shares similarities in trafficking and metabolism (Tiveron et al., 1994; Madore et al., 1999; Sunyach et al., 2003).
The cytotoxicity of anti-PrP Mabs was tested by culturing N2a and N2a/22L cells in 96-well microtiter plates in the presence of 10 μg/mL Mabs for 96 h. Following completion of treatment, the viability of cells was assessed using a colorimetric 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide (MTT) assay (Roche) performed according to the manufacturer’s instructions (Sadowski et al., 2004a; Sigurdsson et al., 2001). The viability of cells treated with various Mabs, murine IgG and nontreated control cells were compared using one-way ANOVA followed by a Dunnett post hoc test.
In vivo PrP labeling was performed in N2a and N2a/22L cells using Mab 6D11 conjugated with Cy3 fluorescent dye (6D11/Cy3). 6D11 was conjugated with Cy3 using a Pierce antibody labeling kit applied according to the manufacturer’s instructions. Cells were cultured on coverslips placed in 10-cm2 wells in the presence of 10 μg/mL 6D11/Cy3 for 12, 24, 48 or 72 h, fixed with 20% ice-cold methanol, counterstained with DAPI (4′,6-diamidino-2-phenylindole dihydro-chloride) and analysed under a deconvolution fluorescence microscope Zeiss Axioskop 40 (Carl Zeiss AG, Gottingen, Germany) or a Bio-Rad (Hercules, CA, USA) Radiance 2000 confocal system attached to the Olympus BX50WI fluorescence microscope.
Prevention of N2a infection with Mabs
In experiments designed to prevent infection, N2a cells were cultured overnight in six-well plates with 10 μg/mL of Mabs in 2 mL of MEM. After washing with phosphate-buffered saline, cells were infected with 22L brain homogenates as described above. In similar experiments 2% brain homogenates diluted in 1 mL of Opti-MEM was incubated with 20 μg Mabs for 2 h and then used to infect cells. The PrPSc levels were measured in subsequent passages and compared to the level of PrPSc in N2a/22L cells infected simultaneously without preincubation with Mabs. Differences between control and treated groups were compared using repeated-measures ANOVA (Statistica v6.1; StatSoft Inc, Tulsa, OK, USA).
In addition we performed an animal experiment to ascertain whether prophylactic treatment with Mabs administered immediately following prion exposure may delay the onset of neurological symptoms. Two cohorts of 3-month-old male CD-1 mice, 20 animals each, were inoculated by intraperitoneal injection of 100 μL of 10% brain homogenate from terminally sick 22L-infected mice (Sigurdsson et al., 2002a; Sadowski et al., 2003; Sadowski et al., 2004b). An hour after exposure one cohort received intravenous infusion of murine IgG and the other cohort received Mab 7D9. Selection of this particular Mab was dictated by its effectiveness in the prevention of infection in cell culture as well as its availability in large quantities. Starting from the beginning of the 4th month following inoculation mice were tested for the first symptoms of prion infection using an apparatus containing a series of parallel bars (3 mm in diameter) placed 7 mm apart. The earliest detectable clinical symptoms of central nervous system involvement include an impaired activity level and competency when mice attempt to cross a series of parallel bars. An animal was considered clinically symptomatic if it scored positive for disease for 3 weeks in a row by an observer blinded to the animal’s treatment group assignment. Upon receiving the third positive score in a row, an animal was killed with an overdose of pentobarbital (150 mg/kg body weight) and the clinical diagnosis was confirmed by demonstration of PrPSc on Western blot from PK-digested brain homogenate. This method of scoring in prion-infected mice for early detection of neurological symptoms has been validated and widely used by the authors previously (Sadowski et al., 2003; Sigurdsson et al., 2002b; Sigurdsson et al., 2003). Data were analysed using Kaplan–Meier survival curves followed by a log rank test. Analysis was performed using GraphPad Prism Software.
Results
6D11 is a highly sensitive Mab recognizing PrPC, PrPSc and recPrP. Western blot studies demonstrated that 6D11 reacted equally well with both PrPC and PrPSc, recognizing all three isoforms: di-, mono- and nonglycosylated (Fig. 1). 6D11 also recognized recPrP. Solid-phase binding studies showed that the affinity of 6D11 for PrP is higher than that of other Mabs (Table 1). The KD value calculated for 6D11 (8.5 ± 0.18 × 10−11 M) was about one-half that calculated for 7A12 and ~40 and 125 times lower than those calculated for 2C2 and 8F9, respectively. However, the binding affinities of the other Mabs used in this study are in a similar range (Table 1).
Fig. 1.

(A) Mab 6D11 detected di-, mono- and nonglycosylated PrP isoforms prior to and after PK treatment in brain homogenate from 22L-infected CD-1 mouse (brain/22L) and in cell lysate from N2a cells infected with the 22L prion strain (N2a/22L). 6D11 also reacted with PrPC produced by noninfected N2a cells (N2a) and with recombinant PrP (recPrP). Ten micrograms of total protein from brain homogenate and 40 and 10 μg of total protein from cell lysates were loaded onto lanes 1, 3 and 4, respectively. Two hundred micrograms of total protein from brain homogenate or cell lysate were subjected to PK digestion and loaded onto lanes 2 and 5, and 3 μg of recPrP was loaded onto lane 6. PK-, protein not treated with PK; PK+, protein treated with PK. (B) Dot–blot analysis of 6D11 binding epitope. 6D11 reacted with a peptide homologous to PrP residues 93–122 with an affinity similar to that shown for full-length recPrP.
Antigen mapping studies demonstrated that 6D11 reacted with a synthetic peptide homologous to PrP residues 93–122 but showed no reaction with a peptide spanning residues 109–141, which indicates that 6D11 epitope is localized between position 93 and 109. In a dot–blot experiment, 6D11 reacted with similar affinity with PrP 93–122 as it did with full-length recPrP. This was further confirmed by an ELISA experiment, where the KD of 6D11 binding to PrP 93–122 (15.5 ± 0.24 × 10−11 M) was found to be similar to that calculated for its binding to recPrP.
22L mouse-adapted scrapie strain induced PrPSc formation in the N2a cell line
Exposure of N2a murine neuroblastoma cells to brain homogenate from terminally sick CD-1 mice infected with 22L mouse-adapted scrapie strain resulted in stable infection of this cell line. A high level of PK-resistant PrPSc could be demonstrated in subsequent passages (Fig. 2A) and N2a/22L cells divided vigorously for 12–14 passages after infection. A decreased mitotic rate and inefficient growth was routinely observed in passages higher than the 12th.
Fig. 2.
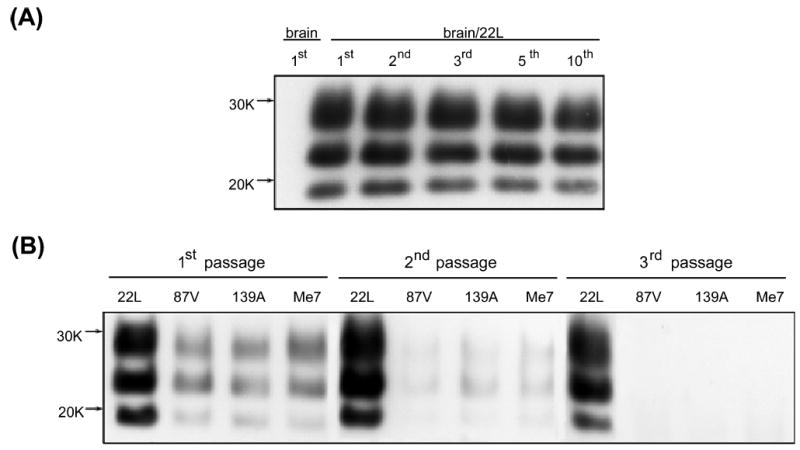
(A) PK-treated cell lysates from N2a murine neuroblastoma cells exposed to homogenate from a control brain (brain) or from a brain of a CD-1 mouse infected with 22L mouse-adapted scrapie strain (brain/22L). No PK-resistant material could be detected in cells cultured in the presence of control brain homogenate. Exposure of N2a cells to 22L mouse-adapted scrapie strain resulted in persistent infection of this line as the stable presence of PK-resistant PrPSc could be demonstrated in subsequent passages. (B) PK-treated cell lysates from N2a cells exposed to different mouse-adapted scrapie strains: 22L, 87V, 139A and ME7. Only exposure to the 22L strain resulted in the stable infection and sustained formation of PK-resistant PrPSc derived from PrPC expressed by the N2a cells. A weak signal can be seen in the first passage; this almost completely fades out in the second passage and is derived either from PrPSc carried over from the inoculum or from limited, unsustained conversion of cellular PrPC to PrPSc. No PrPSc could be detected in the third or higher passages of N2a cells exposed to 87V, 139A or ME7.
As has been described previously, wild-type N2a cells are resistant to infection with many other mouse-adapted scrapie strains including 87V, 139A and ME7 (Bosque & Prusiner, 2000). Infectivity with 139A has only been reported in transfected N2a cells overexpressing PrPC (Nishida et al., 2000). Therefore, we used 87V, 139A and ME7 strains as a negative control for the infection experiment to differentiate between inoculum-derived PrPSc and PrPSc formed de novo from PrPC expressed by N2a cells. As demonstrated in Fig. 2B, only exposure to the 22L strain resulted in the stable infection and sustained formation of PK-resistant PrPSc derived from PrPC expressed by the N2a cells. For comparison, exposure to 87V, 139A or ME7 produced a weak PrPSc signal seen in the first passage, which almost completely faded out in the second passage. No PrPSc could be detected in the third and higher passages.
Treatment of N2a/22L cells with 6D11, 7H6 and 7A12 Mabs abrogated the presence of PrPSc
Culturing of N2a/22L cells in the presence of Mabs produced variable lowering of the PrPSc level (Fig. 3). Three Mabs, 6D11, 7H6 and 7A12, were found capable of complete abrogation of the PrPSc presence in N2a/22L cells. Among these Mabs, 6D11 showed the lowest IC50 (Fig. 4). It was calculated from a sigmoidal dose–responese curve to be 0.07 ± 0.009 μg/mL (0.47 ± 0.06 μM; mean ± SD from at least three independent experiments). Values of IC50 for 7H6 and 7A12 were 0.16 ± 0.02 μg/mL (1.07 ± 0.13 μM) and 0.35 ± 0.05 μg/mL (2.35 ± 0.34 μM), respectively. The treatment effect of these Mabs was persistent as when antibodies were removed from the medium and the cells were grown for another 14 days in antibody-free medium no PrPSc was be detected (Fig. 5).
Fig. 3.
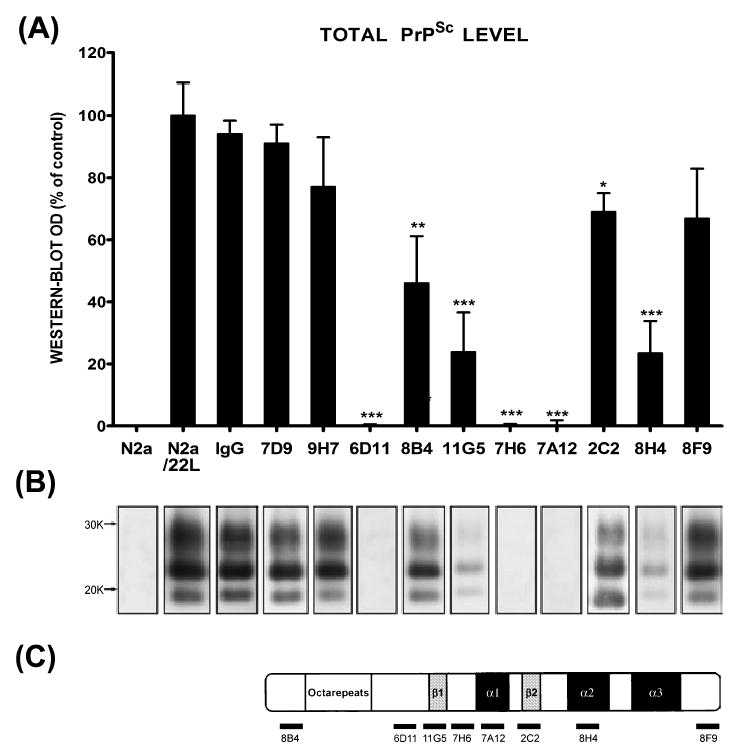
(A) The level of PrPSc in N2a/22L cells grown in the presence of anti-PrP Mabs. Mabs 6D11, 7H6 and 7A12 caused complete abrogation of PrPSc in infected cells. N2a (noninfected N2a cells), N2a/22L (N2a cells infected with the 22L strain) and IgG (N2a/22L cells incubated in the presence of murine IgG) were included as controls. OD, optical density; values are given as a mean + SD from at least three independent experiments. One-way ANOVA (P < 0.0001) was followed by Dunnett’s post hoc test; *P < 0.05, **P < 0.01, ***P < 0.001. (B) PK-resistant PrPSc in cell lysates following treatment with particular Mabs. (C) Schematic representation of epitopes of Mabs used to treat N2a/22L cells.
Fig. 4.
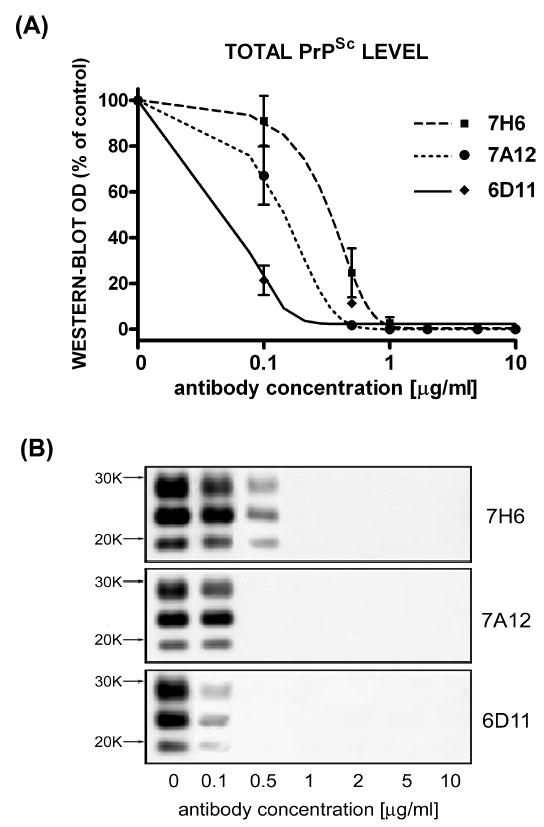
Dose-dependant inhibition of PrPSc formation in N2a/22L cells by Mabs. (A) Densitometric measurements of PrPSc bands detected in the Western blots fitted to sigmoidal dose–response curves. Values are given as mean ± SD from at least three independent experiments. (B) Western blots of PK-treated cell lysates from N2a/22L cells treated for 4 days with different concentrations of Mabs.
Fig. 5.
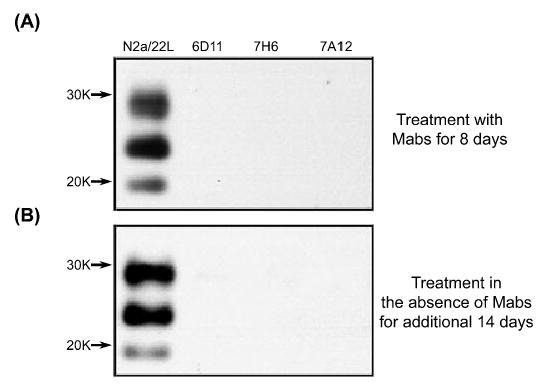
(A) Absence of PK-resistant PrPSc in N2a/22L cells incubated with Mabs 6D11, 7H6 and 7A12 for 8 days. (B) There was no reappearance of PrPSc after the cells were grown for another 14 days in the absence of Mabs. This indicates that the effect of treatment was persistent.
A significant reduction in PrPSc levels was observed with treatment using the following Mabs: 8H4 (reduction in PrPSc level by 76.6%, P < 0.001), 11G5 (76% reduction, P < 0.001), 8B4 (54% reduction, P < 0.01) and 2C2 (33.9% reduction, P < 0.05; Fig. 3). No significant effect on PrPSc levels in N2a/22L cells was observed with treatment using Mabs 7D9, 9H7 or 8F9, or murine IgG. Thus, as all of these Mabs specifically bind PrP with similar affinity, another antibody-specific parameter is required to affect the PrPSc level.
Mabs did not lower the total PrP or β-actin levels but altered the level of Thy-1
To evaluate whether the therapeutic effect of Mabs was mediated by the suppression of PrPC level the total PrP level was measured in N2a/22L cells treated with Mabs. Cell lysates were divided into fractions treated and not treated with PK; these were then subjected to electrophoresis and immunoblotting to measure the levels of PrPSc and total PrP, respectively. While treatment with Mabs such as 2C2 resulted in a partial reduction in PrPSc and treatment with Mabs such as 6D11 and 7H6 completely blocked PrPSc formation, the level of total PrP under treatment with these Mabs was similar to that of nontreated N2a/22L cells (Figs 6 and 7). While comparing the density of immunoblots for PK-treated and -nontreated samples one has to keep in mind that PK-nontreated samples contained 10 μg of protein from cell lysate, whereas 200 μg of protein was subjected to PK digestion prior to loading on the gel to give bands of similar intensity. Therefore, we estimate that PrPSc constituted 5% of the total PrP. We showed that blocking PrPSc formation did not affect the total PrP signal significantly. As the total PrP signal corresponded mainly to PrPC, we suggest that that the therapeutic effect of our Mabs did not appear to be mediated via alteration of the PrPC level.
Fig. 6.
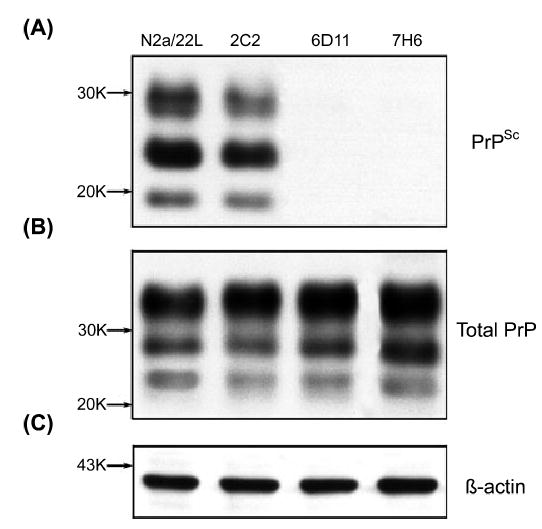
Protein expression in N2a/22L cells treated with Mabs 2C2, 6D11 and 7H6. (A) Level of PrPSc following PK digestion; (B) level of total PrP; (C) level of β-actin.
Fig. 7.
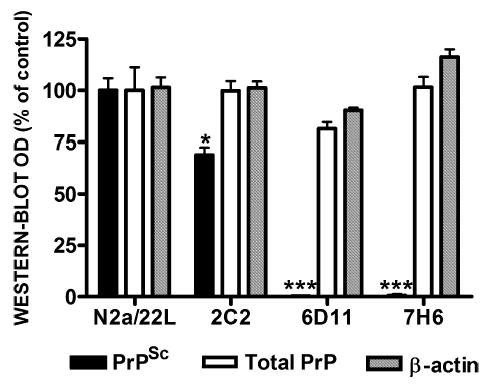
Quantification of protein expression in N2a/22L cells treated with Mabs. While 2C2 resulted in partial reduction in PrPSc and 6D11 and 7H6 caused complete abrogation of PrPSc, treatment with these Mabs did not significantly affect either the total level of PrP (PrPC + PrPSc) or the level of unrelated proteins such as β-actin. Values are given as mean + SD from at least three independent experiments. One-way ANOVA (P < 0.0001) was followed by Dunnett’s post hoc test; *P < 0.05, ***P < 0.001.
To assess whether treatment with Mabs affected expression of other proteins, we quantified the level of β-actin and Thy-1. β-Actin is a 43-kDa structural protein unrelated to PrP metabolism (Shashidhar et al., 2005), and is commonly used as marker of general protein expression in cell culture treatment experiments (Korth et al., 2001; Perrier et al., 2004). Thy-1, however, colocalizes with PrPC on the external plasma membrane surface and to a great extent shares endocytosis and trafficking pathways (Tiveron et al., 1994; Madore et al., 1999; Sunyach et al., 2003). We did not detect differences in the β-actin level between treated and nontreated N2a/22L cells (Figs 6C and 7). However, the level of Thy-1 was increased in infected cells and returned to control levels when infected cells were treated with therapeutically effective Mabs. Mabs which did not have a significant effect on PrPSc formation, such as 7D9 and 8F9, did not alter the level of Thy-1 compared to nontreated N2a/22L cells (Fig. 8A and B). As the level of Thy-1 in N2a/22L cells was over twice that in noninfected cells (P < 0.01), its reduction appears to be associated with a treatment effect rather than indicating cellular toxicity.
Fig. 8.

(A) The level of Thy-1 in N2a/22L cells treated with various Mabs. The level of Thy-1 in N2a/22L cells was more than twice than in N2a cells. Mabs 7D9 and 8F9, which showed no significant therapeutic effect, had no impact on the Thy-1 level. Incubation of N2a/22L cells with therapeutically effective Mabs caused a significant reduction in Thy-1 level. One-way ANOVA (P < 0.001) was followed by a Dunnett post hoc test; *P < 0.05, **P < 0.01. (B) Thy-1 in cell lysates following treatment with specific Mabs.
Mabs were not toxic
Potential toxicity related to treatment with anti-PrP Mabs in cells expressing PrPC and in those actively forming PrPSc was also specifically assessed using the MTT metabolic assay. Both N2a and N2a/22L cells were grown in the presence of anti-PrP Mabs and IgG as a nonspecific control in conditions similar to those used in treatment experiments. A minor trophic effect was observed in N2a cells. Viability of N2a cells was increased by 25–35% of control values (P < 0.01; Fig. 9A). This trophic effect did not appear to be specific as a similar increase in N2a cell viability was observed when these cells were grown in the presence of murine IgG. N2a/22L cells cultured in the presence of anti-PrP Mabs or murine IgG did not show either trophic or toxic effects (Fig. 9B).
Fig. 9.
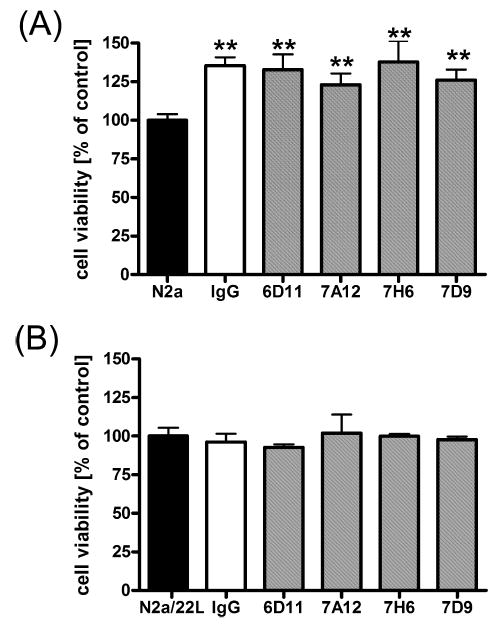
The effect of Mabs on viability of N2a and N2a/22L cells. (A) Murine IgG and anti-PrP Mabs had a nonspecific trophic effect on N2a cells. One-way ANOVA (P < 0.0001) was followed by Dunnett’s post hoc test; **P < 0.01. (B) Anti-PrP Mabs showed neither a trophic effect nor reduced viability of N2a/22L cells. Values are given as a mean + SD from six independent measurements.
Measuring protein expression served also as an indirect test to assess the toxicity of Mabs. In addition to specifically measuring the β-actin level, the total protein amount in cell lysates was always calculated and compared between treated and nontreated cells to monitor for potential toxicity. The total protein amount harvested from a confluent 10-cm2 well containing N2a/22L cells ranged between 350 and 500 μg.
Mab 6D11 was internalized by N2a/22L cells
In order to better understand how anti-PrP Mabs clear infected N2a/22Lcells of PrPSc, N2a and N2a/22L cells were cultured in the presence of Cy3-labeled Mab 6D11. N2a cells showed distinct labeling of the cytoplasmic membrane with only modest intracellular penetration of the dye despite an incubation period extended to 72 h (Fig. 10A and C). Images obtained using confocal microscopy confirmed that the labeling pattern was consistent with binding of 6D11/Cy3 to PrPC expressed primarily on the outer surface of plasma membrane (Fig. 10E, arrowhead). In addition to labeling the cytoplasmic membrane, N2a/22L cells showed internalization of 6D11/Cy3 and their accumulation in the cytoplasm which continued over the time of experiment (Fig. 9B, D and E).
Fig. 10.
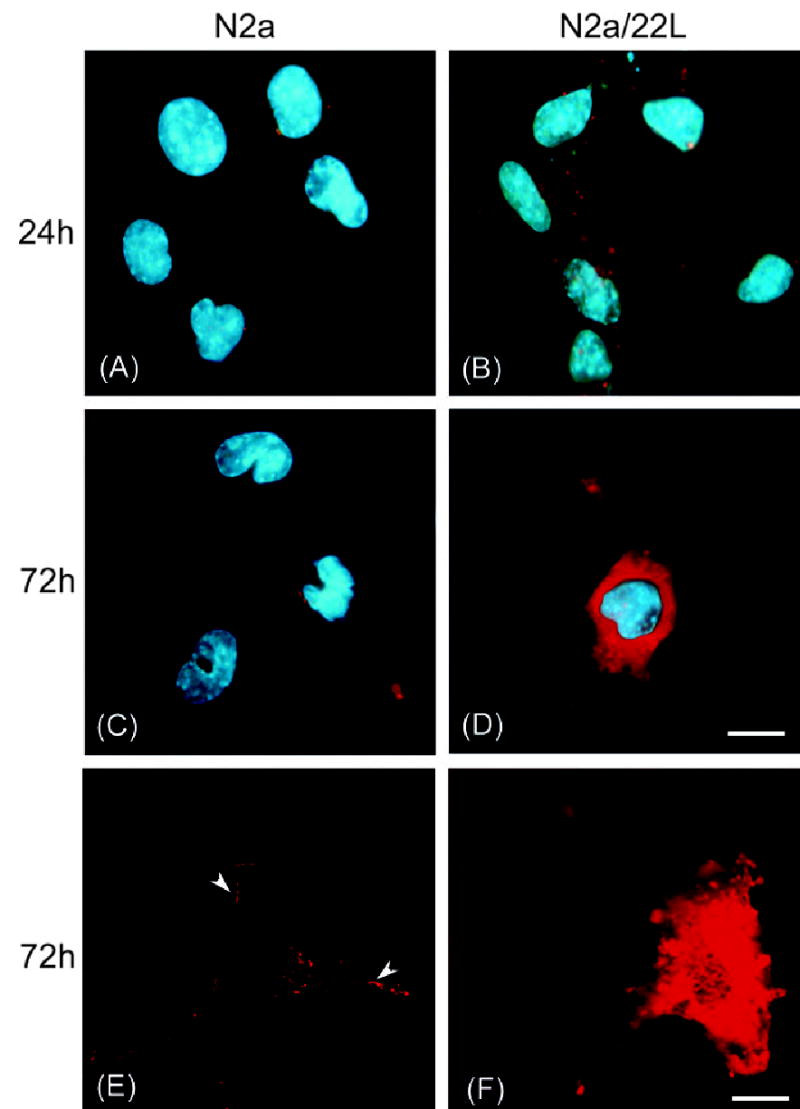
Incubation of N2a and N2a/22L cells in the presence of Mab 6D11 conjugated with Cy3 (6D11/Cy3). Labeling of the plasma membrane in N2a cells grown in the presence of 6D11/Cy3 for (A) 24 h and (C) 72 h. (E) Confocal microscopy images showing binding of Mabs to the outer leaflet of the plasma membrane (arrowheads). (B) In addition to membrane labeling, N2a/22L cells showed internalization of 6D11/Cy3 after 24 h, which (D and F) was more pronounced after 72 h. A–D, deconvolution microscope; E and F, confocal microscope. Red staining in A–F represents Cy3; blue staining in A–D is DAPI nuclear dye. Scale bars, 10 μm (in D for A–D), 20 μm (in F for E and F).
Mabs reduced prion infectivity
Both preincubation of N2a cells prior to inoculation and preincubation of the brain homogenate from 22L-infected mice with anti-PrP Mabs resulted in significantly decreased levels of PrPSc in subsequent passages compared to N2a cells infected in the standard way (Fig. 11). In the third passage, the PrPSc level was reduced by 74% when the inoculum was preincubated with 6D11 Mab, and by 84% when the N2a cells were exposed to 6D11 prior to the infection (Fig. 11A; P < 0.01). The PrPSc level was followed in subsequent passages and it continued to be significantly reduced. The reduction ranged from 4 to 41% of the PrPSc level in control N2a/22L cells in passages 3–7 (Fig. 11B; repeated-measures ANOVA; P < 0.001). Mab 7D9, which did not have therapeutic efficacy in lowering PrPSc levels in already infected N2a/22L cells, showed a surprising and significant preventive effect in that it reduced the PrPSc level by 88.5% (P < 0.01) and by 57% (P < 0.05) when it was incubated with the inoculum and with N2a cells, respectively (Fig. 11A). Mab 7A12, which was capable of completely blocking PrPSc formation in already infected N2a/22L cells, showed the weakest preventive effect. In the third passage, the PrPSc level was reduced by only 41 and 43% (P < 0.05), respectively, when the inoculum and N2a cells were preincubated with 7A12 (Fig. 11A). In the subsequent passages, the PrPSc level in the experiments with 7A12, but not with 6D11 or 7D9, gradually increased and in the sixth passage was not significantly different from that in control N2a/22L cells.
Fig. 11.
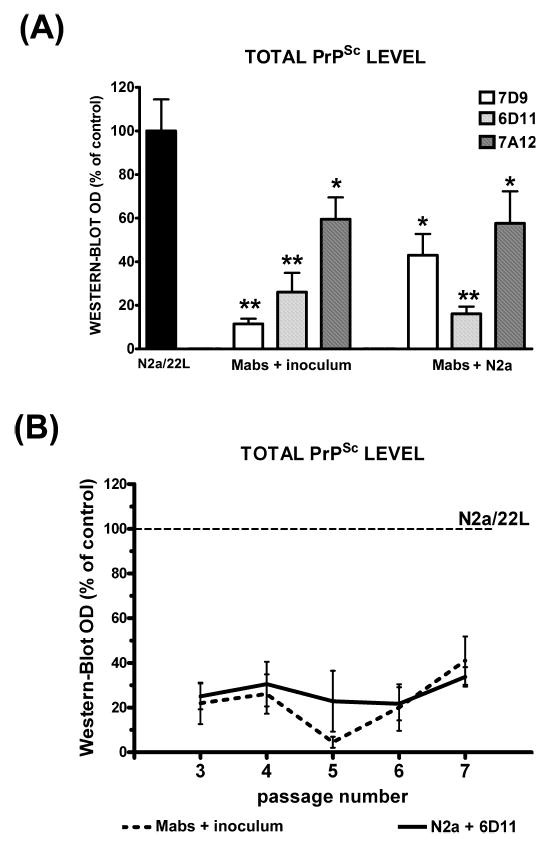
(A) Decreased level of PrPSc in the third passage of N2a/22L cells which were infected with inoculum preincubated with Mabs (Mabs + inoculum) or N2a cells incubated with Mabs before being exposed to the inoculum (Mabs + N2a); results are compared to the level of PrPSc in N2a/22L cells infected in a standard manner. Values are given as mean + SD from at least three independent experiments. One-way ANOVA (P < 0.0001) was followed by Dunnett’s post hoc test; *P < 0.05, **P < 0.01. (B) Persistently decreased level of PrPSc was maintained in subsequent passages of N2a cells infected in the presence of 6D11. Values are given as mean ± SD from at least three independent experiments. Comparison was made to the density of PrPSc bands (designated as 100%) in corresponding passages of N2a/22L cells infected in the standard manner (repeated-measures ANOVA; P < 0.001).
To evaluate whether this preventive effect of Mabs was also relevant in vivo we administered a single dose of 7D9 Mab to CD-1 mice within 1 h following intraperitoneal inoculation with infectious brain homogenate. The selection of 7D9 was based upon the availability of large amounts of this Mab required for the experiment and its effectiveness in tissue culture studies. The Kaplan–Meier survival analysis demonstrated a statistically significant treatment effect. The difference between the median incubation period for groups treated with Mab 7D9 and murine IgG was 28 days (Fig. 12; P < 0.0001, log rank test).
Fig. 12.
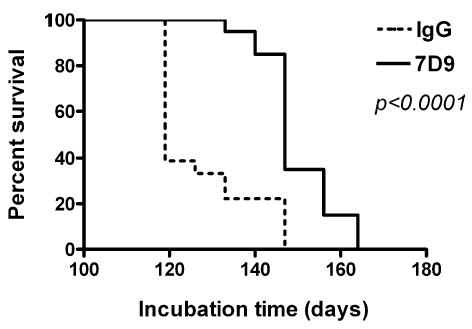
Kaplan–Meier survival analysis of CD-1 mice prophylactically treated with a single dose of Mab 7D9 administered immediately after intraperitoneal exposure to 22L brain homogenate. Animals which received Mab 7D9 showed a statically significant delay in onset of neurological symptoms compared to those which received an equivalent dose of IgG. Log rank test indicates a statistically significant difference between the groups (P < 0.0001).
Discussion
Using a panel of Mabs directed against the major structural domains of PrP we demonstrated that a number of Mabs inhibit PrPSc formation by an antigen-specific disruption of the PrPC–PrPSc interaction. Three Mabs, 6D11, 7H6 and 7A12, were found to be capable of completely eradicating PrPSc from infected N2a cells. Mabs 7H6 and 7A12 bind to neighbouring epitopes corresponding to PrP sequences 130–140 and 143–155, respectively. The importance of the PrP 130–150 sequence, which includes the first α-helical domain (PrP residues 143–155) for the PrPC–PrPSc interaction and the formation of PrPSc, has been suggested by several investigators (Enari et al., 2001a; Heppner et al., 2001; Peretz et al., 2001; White et al., 2003; Perrier et al., 2004). Other reports also identified the 132–156 region of the protein as critical for interspecies transmission of prionoses (Kocisko et al., 1995; Scott et al., 1993; Priola & Chesebro, 1995). Therefore, the PrP sequence 130–150 represents an important target for the development of anti-PrP drugs. Mabs 11G5 and 2C2 reacting with linear epitopes flanking downstream and upstream regions of this sequence had a partial effect in clearing PrPSc. It has been shown that PrP sequences 119–136 and 166–179, partially covered by the epitopes of these antibodies, play an auxiliary role in the PrPC-to-PrPSc conversion (Horiuchi & Caughey, 1999). On the other hand, the effect of these antibodies may be due to steric hindrance. Because of their size, which is several times bigger than PrP itself, they may indirectly block access to the 130–150 domain.
6D11 is a novel Mab, raised against the PK-resistant fragments of PrPSc, which also showed an excellent effect in clearing N2a/22L cells of PrPSc. 6D11 had the highest binding affinity to recPrP among the studied Mabs and also bound robustly to PrPSc. Dot–blot experiments demonstrated that the 6D11 epitope is located between residues 93 and 109, as shown by the finding that the antibody bound with high affinity to a peptide homologous to PrP residues 93–122 and showed no binding to a peptide homologous to sequence 109–141. The localization of the major 6D11 epitope was further narrowed to residues 97–100 by pepscan assay (Pepscan Systems, Lelystad, The Netherlands; data not shown). 6D11 did not recognize the PrP 130–155 sequence as tested by dot–blot, immunoprecipitation and ELISA studies. These observations indicate that the epitope along PrP residues 93–109 is in addition to residues 130–155, another important therapeutic target for the inhibition of PrPSc formation. Neutralization of this antigen by 6D11 may either prevent the PrPC–PrPSc interaction or interfere with binding auxillary molecules essential to prion propagation (Kaneko et al., 1997; Hundt et al., 2001; Leucht et al., 2003).
Among other tested Mabs, a partial effect in lowering the PrPSc level could be demonstrated by using Mabs 8B4, directed against the N-terminus, and 8H4 directed against the second α-helix domain. No effect was observed with Mab 8F9 reacting against a linear epitope on the C-terminus of PrP. These observations are consistent with our in vivo observation where symptoms of prion disease were delayed in mice treated with Mabs 8H4 and 8B4 but not 8F9 (Sigurdsson et al., 2003). No significant effect on PrPSc level in N2a/22L cells was observed following treatment with Mabs 7D9, 9H7 or 8F9, nor with murine IgG. Similarly, Feraudet et al. (2005), who analysed the therapeutic efficacy of 145 Mabs raised against different forms of human, ovine, hamster and murine PrP, demonstrated that a partial treatment response can be achieved with Mabs directed against epitopes located outside the central potion of the PrP sequence. However, the most effective Mab in their study, Sha 31, reacted with an epitope located along residues 145–152 which is similar to the epitope of our 7A12 Mab.
All Mabs used had high binding affinity to the PrP, and this appears to be a prerequisite for therapeutic effectiveness. The KD of binding to PrP for all Mabs used in this study was within the picomolar range as analysed by solid-phase binding assays. The exceptions were Mabs 2C2 and 8F9, which showed slightly higher KD values. As all investigated Mabs specifically bound PrP with closely similar affinity, this feature does not appear to be a primary parameter determining their therapeutic efficacy. Furthermore, 2C2, which had a KD ~40× higher than that of 6D11 and 15× higher than that of 7H6, showed only partial therapeutic effects in N2a/22L cells whereas Mabs 7D9 and 9H7, with binding affinities to PrP ~4× and 3× better, respectively, than that of 2C2 were completely ineffective. Therefore, differences in binding affinity cannot in and of themselves account for their disparate therapeutic effects. Although it is desirable for a therapeutic antibody to have the highest affinity possible, this property appears to be of secondary importance to their targeting of specific critical PrP domains.
The interaction of PrPSc with PrPC, and the replication of PrPSc, take place both on the cell surface and in the cytosol (DeArmond et al., 2002). When N2a/22L cells were incubated with 6D11/Cy3 Mabs, initial labeling of cell membranes was followed by internalization of antibodies and their accumulation in the cytosol. Internalization of Mabs was minimal in noninfected N2a cells. These observations indicate that 6D11 binds PrPSc and is internalized together with PrPSc. Therefore, it can prevent the PrPSc–PrPC interaction not only on the cell membrane surface but also within the cytoplasm, preventing lysosomal and cytoplasmic accumulation and formation of new PrPSc (Ma & Lindquist, 2002; Ma et al., 2002).
Toxicity is a potential adverse effect associated with immunization approaches. Autoimmune meningoencephalitis occurring in 6% of patients resulted in premature termination of a clinical trial of an Alzheimer’s vaccine (Nicoll et al., 2003; Orgogozo et al., 2003). Passive immunization is less likely to initiate an uncontrolled autoimmune reaction (Schenk, 2002; Sadowski & Wisniewski, 2004), but Mabs targeting PrPSc formation in vivo must not exert toxicity toward cells replicating PrPSc and at the same time expressing PrPC. One of the previous studies analysing the effects of anti-PrP Mabs on PrPC metabolism in noninfected cells suggested that the anti-PrP Mabs may shorten the PrPC half-life and thus their therapeutic effect is related to depletion of PrPC (Perrier et al., 2004). However, in a follow-up study the same group reported contradictory results, showing that Mabs do not lower PrPC level in infected cells (Feraudet et al., 2005). In view of our data, anti-PrP Mabs which effectively abrogate PrPSc infection do not appear to lower PrPC level. In addition we investigated the effect of treatment on the level of two proteins, β-actin and Thy-1. Interestingly, we found the level of Thy-1 in N2a/22L cells to be over twice that in noninfected cells. As Thy-1 and PrP coexist in close proximity on the outer surface of the plasma membrane and share similarities in trafficking and metabolism (Madore et al., 1999; Sunyach et al., 2003), an increased level of Thy-1 appears to be related to the infectious status of the N2a cells. Whether this is a result of its overexpression or extended half-life remains yet to be elucidated. Nevertheless in N2a/22L cells cultured in the presence of therapeutically active anti-PrP Mabs, the Thy-1 level is reduced to a level similar to noninfected cells. This appears to be additional evidence demonstrating the therapeutic effect of our Mabs. The level of β-actin, which is a structural protein not involved in the PrPC–PrPSc interaction, is unchanged during treatment. This suggests that our Mabs do not affect general protein expression. The MTT cytotoxicity assay also demonstrated a lack of Mab toxicity in both N2a/22L and N2a cells.
Preincubation of the inoculum with several Mabs resulted in a significant decrease in its infectivity as the Mabs bind and neutralize PrPSc. Likewise, the level of PrPSc formation was effectively decreased when N2a cells were preincubated with several different Mabs. Previous experiments demonstrated that Mabs which preferentially bind PrPC may also successfully clear infected cell cultures by sequestering PrPC on the cell membrane (Enari et al., 2001b; Kim et al., 2004; Feraudet et al., 2005). However, in animal experiments Mabs which equally recognize PrPSc and PrPC appeared to be more effective (White et al., 2003). In experiments with 6D11/Cy3 we were able to directly demonstrate that Mabs bind to PrPC on the membrane surface of noninfected cells. Thus, sequestration of PrPC on the cell surface by Mabs and inhibition of its contact with PrPSc from the inoculum has a strong protective effect against infection. No epitope-specific effect was observed in experiments designed to prevent infection. Mab 7D9, which showed no therapeutic effect in reducing PrPSc level in N2a/22L cells, was more effective than Mab 7A12 which produced persistent abrogation of PrPSc infection. Only Mab 6D11, which had the strongest therapeutic effect in N2a/22L cells, was also effective in preventing infection. These experiments illustrate additional therapeutic mechanisms of anti-PrP Mabs which appear to be relevant to in vivo treatment as demonstrated in an experiment where mice were treated with a single dose of 7D9 shortly after exposure. These treated mice showed a significantly prolonged symptom-free incubation period.
The therapeutic mechanisms of anti-PrP Mabs which include: (i) antigen-specific interference of PrPC–PrPSc interaction; (ii) PrPSc neutralization; and (iii) PrPC sequestration on the cell surface, are not mutually exclusive but may synergistically potentiate the therapeutic effects of Mabs. All three mechanisms should be considered in the development of therapeutic strategies, mindful that the disease process in a living organism is far more complex than that in infected cell lines. In animal or human diseases, the formation of PrPSc in already-infected cells occurs concurrently with a constant process of infecting other cells (Weissmann et al., 2002). Therefore, the ability of Mabs to reduce prion infectivity is desirable in addition to their primary function, which is antigen-specific blocking of PrPSc formation. 6D11 is an example of an antibody with all three properties. Our studies demonstrate that anti-PrP Mabs constitute a safe and feasible therapeutic approach for the treatment of prion infections. Information regarding antigen-specific inhibition of PrPSc formation is important for the rational design of humoral therapy for prion infection in animals and eventually in humans.
Acknowledgments
Supported by AG24847 (M.S.), NS47433 (T.W.), AG15408 (T.W.), NS45981 (M.S.S.), US Department of Army contract (DAMD17-03-1-0286 (M.S.S.) and NIH contract N01-NS-02327 (R.J.K.).
References
- Adler V, Zeller B, Kryukov V, Kascsak R, Rubenstein R, Grossman A. Small, highly structured RNAs participate in the conversion of human recombinant Prp (Sen) to Prp (Res) in vitro . J Mol Biol. 2003;332:47–57. doi: 10.1016/s0022-2836(03)00919-7. [DOI] [PubMed] [Google Scholar]
- Balter M. Spongiform disease. Experts downplay new vCJD fears. Science. 2002;289:1866–1867. doi: 10.1126/science.289.5485.1663b. [DOI] [PubMed] [Google Scholar]
- Bosque PJ, Prusiner SB. Cultured cell sublines highly susceptible to prion infection. J Virol. 2000;74:4377–4386. doi: 10.1128/jvi.74.9.4377-4386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carp RI, Meeker HC, Rubenstein R, Sigurdarson S, Papini M, Kascsak RJ, Kozlowski PB, Wisniewski HM. Characteristics of scrapie isolates derived from hay mites. J Neurovirol. 2000;6:137–144. doi: 10.3109/13550280009013157. [DOI] [PubMed] [Google Scholar]
- DeArmond SJ, Kretzschmar H, Prusiner SB. Prion diseases. In: Graham DI, Lantos P, editors. Greenfield’s Neuropathology. Arnold; London: 2002. pp. 273–324. [Google Scholar]
- Eklund CM, Kennedy RC, Hadlow WJ. Pathogenesis of scrapie virus infected mice. J Infect Dis. 1967;117:15–22. doi: 10.1093/infdis/117.1.15. [DOI] [PubMed] [Google Scholar]
- Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci USA. 2001a;98:9295–9299. doi: 10.1073/pnas.151242598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci USA. 2001b;98:9295–9299. doi: 10.1073/pnas.151242598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feraudet C, Morel N, Simon S, Volland H, Frobert Y, Creminon C, Vilette D, Lehmann S, Grassi J. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem. 2005;280:11247–11258. doi: 10.1074/jbc.M407006200. [DOI] [PubMed] [Google Scholar]
- Goni F, Knudsen E, Schreiber F, Scholtzova H, Pankiewicz J, Carp R, Meeker HC, Rubenstein R, Brown DR, Sy MS, Chabalgoity JA, Sigurdsson EM, Wisniewski T. Mucosal vaccination delays or prevents prion infection via an oral route. Neuroscience. 2005;133:413–421. doi: 10.1016/j.neuroscience.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Musahl C, Arrighi I, Klein MA, Rulicke T, Oesch B, Zinkernagel RM, Kalinke U, Aguzzi A. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science. 2001;294:178–182. doi: 10.1126/science.1063093. [DOI] [PubMed] [Google Scholar]
- Herzog C, Sales N, Etchegaray N, Charbonnier A, Freire S, Dormont D, Deslys JP, Lasmezas CI. Tissue distribution of bovine spongiform encephalopathy agent in primates after intravenous or oral infection. Lancet. 2004;363:422–428. doi: 10.1016/S0140-6736(04)15487-1. [DOI] [PubMed] [Google Scholar]
- Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Penney M, Ritchie D, Ironside JW. Accumulation of prion protein in tonsil and appendix: review of tissue samples. Br Med J. 2002;325:633–634. doi: 10.1136/bmj.325.7365.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, Penney M, Hegazy D, Ironside JW. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol. 2004a;203:733–739. doi: 10.1002/path.1580. [DOI] [PubMed] [Google Scholar]
- Hilton DA, Sutak J, Smith MEF, Penney M, Conyers L, Edwards P, McCardle L, Ritchie D, Head MW, Wiley CA, Ironside JW. Specificity of lymphoreticular accumulation of prion protein for variant Creutzfeldt–Jakob disease. J Clin Pathol. 2004b;57:300–302. doi: 10.1136/jcp.2003.012278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Caughey B. Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J. 1999;18:3193–3203. doi: 10.1093/emboj/18.12.3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston F, Foster JD, Chong A, Hunter N, Bostock CJ. Transmission of BSE by blood transfusion in sheep. Lancet. 2000;356:999–1000. doi: 10.1016/s0140-6736(00)02719-7. [DOI] [PubMed] [Google Scholar]
- Hundt C, Peyrin JM, Haik S, Gauczynski S, Leucht C, Rieger R, Riley ML, Deslys JP, Dormont D, Lasmezas CI, Weiss S. Identification of interaction domains of the prion protein with its 37-kDa/67-kDa laminin receptor. EMBO J. 2001;20:5876–5886. doi: 10.1093/emboj/20.21.5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Huete A, Lievens PMJ, Vidal R, Piccardo P, Ghetti B, Tagliavini F, Frangione B, Prelli F. Endogenous proteolytic cleavage of normal and disease-associated isoforms of the human prion protein in neural and non-neural tissues. Am J Pathol. 1998;153:1561–1572. doi: 10.1016/S0002-9440(10)65744-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, Cohen FE, Prusiner SB. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci USA. 1997;94:10069–10074. doi: 10.1073/pnas.94.19.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kascsak RJ, Rubenstein R, Merz PA, Carp RI, Robakis NK, Wisniewski HM, Diringer H. Immunological comparison of scrapie-associated fibrils isolated from animals infected with four different scrapie strains. J Virol. 1986;59:676–683. doi: 10.1128/jvi.59.3.676-683.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987;61:3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CL, Karino A, Ishiguro N, Shinagawa M, Sato M, Horiuchi M. Cell-surface retention of PrPC by anti-PrP antibody prevents protease-resistant PrP formation. J Gen Virol. 2004;85:3473–3482. doi: 10.1099/vir.0.80113-0. [DOI] [PubMed] [Google Scholar]
- Kocisko DA, Priola SA, Raymond GJ, Chesebro B, Lansbury PT, Jr, Caughey B. Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc Natl Acad Sci USA. 1995;92:3923–3927. doi: 10.1073/pnas.92.9.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA. 2001;98:9836–9841. doi: 10.1073/pnas.161274798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leucht C, Simoneau S, Rey C, Vana K, Rieger R, Lasmezas CI, Weiss S. The 37 kDa/67 kDa laminin receptor is required for PrPSc propagation in scrapie-infected neuronal cells. EMBO Reports. 2003;4:290–295. doi: 10.1038/sj.embor.embor768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Zwingman T, Li R, Pan T, Wong BS, Petersen RB, Gambetti P, Herrup K, Sy MS. Differential expression of cellular prion protein in mouse brain as detected with multiple anti-PrP monoclonal antibodies. Brain Res. 2001;896:118–129. doi: 10.1016/s0006-8993(01)02050-9. [DOI] [PubMed] [Google Scholar]
- Ma JY, Lindquist S. Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science. 2002;298:1785–1788. doi: 10.1126/science.1073619. [DOI] [PubMed] [Google Scholar]
- Ma JY, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science. 2002;298:1781–1785. doi: 10.1126/science.1073725. [DOI] [PubMed] [Google Scholar]
- Madore N, Smith KL, Graham CH, Jen A, Brady K, Hall S, Morris R. Functionally different GPI proteins are organized in different domains on the neuronal surface. EMBO J. 1999;18:6917–6926. doi: 10.1093/emboj/18.24.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JAR, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Nishida N, Harris DA, Vilette D, Laude H, Frobert Y, Grassi J, Casanova D, Milhavet O, Lehmann S. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J Virol. 2000;74:320–325. doi: 10.1128/jvi.74.1.320-325.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after A beta 42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Pan T, Li RL, Kang SC, Pastore M, Wong BS, Ironside J, Gambetti P, Sy MS. Biochemical fingerprints of prion diseases: scrapie prion protein in human prion diseases that share prion genotype and type. J Neurochem. 2005;92:132–142. doi: 10.1111/j.1471-4159.2004.02859.x. [DOI] [PubMed] [Google Scholar]
- Pan T, Li RL, Kang SC, Wong BS, Wisniewski T, Sy MS. Epitope scanning reveals gain and loss of strain specific antibody binding epitopes associated with the conversion of normal cellular prion to scrapie prion. J Neurochem. 2004;90:1205–1217. doi: 10.1111/j.1471-4159.2004.02582.x. [DOI] [PubMed] [Google Scholar]
- Pan T, Li RR, Wong BS, Liu T, Gambetti P, Sy MS. Heterogeneity of normal prion protein in two-dimensional immunoblot: presence of various glycosylated and truncated forms. J Neurochem. 2002;81:1092–1101. doi: 10.1046/j.1471-4159.2002.00909.x. [DOI] [PubMed] [Google Scholar]
- Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet. 2004;364:527–529. doi: 10.1016/S0140-6736(04)16811-6. [DOI] [PubMed] [Google Scholar]
- Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412:739–743. doi: 10.1038/35089090. [DOI] [PubMed] [Google Scholar]
- Perrier V, Solassol J, Crozet C, Frobert Y, Mourton-Gilles C, Grassi J, Lehmann S. Anti-PrP antibodies block PrPSc replication in prion-infected cell cultures by accelerating PrP degradation. J Neurochem. 2004;89:454–463. doi: 10.1111/j.1471-4159.2004.02356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priola SA, Chesebro B. A single hamster Prp amino-acid blocks conversion to protease-resistant Prp in scrapie-infected mouse neuroblastoma-cells. J Virol. 1995;69:7754–7758. doi: 10.1128/jvi.69.12.7754-7758.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein R, Merz PA, Kascsak RJ, Scalici CL, Papini MC, Carp RI, Kimberlin RH. Scrapie-infected spleens – analysis of infectivity, scrapie-associated fibrils, and protease-resistant proteins. J Infectious Dis. 1991;164:29–35. doi: 10.1093/infdis/164.1.29. [DOI] [PubMed] [Google Scholar]
- Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li YS, Schmidt SD, Mathews PM, Fryer JD, Holtzman DM, Sigurdsson EM, Wisniewski T. A synthetic peptide blocking the apolipoprotein E/beta-amyloid binding mitigates beta-amyloid toxicity and fibril formation in vitro and reduces beta-amyloid plaques in transgenic mice. Am J Pathol. 2004a;165:937–948. doi: 10.1016/s0002-9440(10)63355-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski M, Pankiewicz J, Scholtzova H, Tsai J, Li YS, Carp RI, Meeker HC, Gambetti P, Debnath M, Mathis CA, Shao L, Gan WB, Klunk WE, Wisniewski T. Targeting prion amyloid deposits in vivo. J Neuropath Exp Neurol. 2004b;63:775–784. doi: 10.1093/jnen/63.7.775. [DOI] [PubMed] [Google Scholar]
- Sadowski M, Tang CY, Aguinaldo JG, Carp R, Meeker HC, Wisniewski T. In vivo micro magnetic resonance imaging signal changes in scrapie infected mice. Neurosci Lett. 2003;345:1–4. doi: 10.1016/s0304-3940(03)00319-7. [DOI] [PubMed] [Google Scholar]
- Sadowski M, Wisniewski T. Vaccines for conformational disorders. Expert Rev Vaccines. 2004;3:89–94. doi: 10.1586/14760584.3.3.279. [DOI] [PubMed] [Google Scholar]
- Schenk D. Opinion: Amyloid-beta immunotherapy for Alzheimer’s disease: the end of the beginning. Nat Rev Neurosci. 2002;3:824–828. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- Schwarz A, Kratke O, Burwinkel M, Riemer C, Schultz J, Henklein P, Bamme T, Baier M. Immunisation with a synthetic prion protein-derived peptide prolongs survival times of mice orally exposed to the scrapie agent. Neurosci Lett. 2003;350:187–189. doi: 10.1016/s0304-3940(03)00907-8. [DOI] [PubMed] [Google Scholar]
- Scott M, Groth D, Foster D, Torchia M, Yang SL, DeArmond SJ, Prusiner SB. Propagation of prions with artificial properties in transgenic mice expressing chimeric Prp genes. Cell. 1993;73:979–988. doi: 10.1016/0092-8674(93)90275-u. [DOI] [PubMed] [Google Scholar]
- Shashidhar S, Lorente G, Nagavarapu U, Nelson A, Kuo J, Cummins J, Nikolich K, Urfer R, Foehr ED. GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene. 2005;24:1673–1682. doi: 10.1038/sj.onc.1208395. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Brown DR, Daniels M, Kascsak RJ, Kascsak R, Carp R, Meeker HC, Frangione B, Wisniewski T. Immunization delays the onset of prion disease in mice. Am J Pathol. 2002a;161:13–17. doi: 10.1016/S0002-9440(10)64151-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Brown DR, Daniels M, Kascsak RJ, Kascsak R, Carp RI, Meeker HC, Frangione B, Wisniewski T. Vaccination delays the onset of prion disease in mice. Am J Pathol. 2002b;161:13–17. doi: 10.1016/S0002-9440(10)64151-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Scholtzova H, Mehta P, Frangione B, Wisniewski T. Immunization with a nontoxic/nonfibrillar amyloid-β homologous peptide reduces Alzheimer’s disease associated pathology in transgenic mice. Am J Pathol. 2001;159:439–447. doi: 10.1016/s0002-9440(10)61715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Sy MS, Li RL, Scholtzova H, Kascsak RJ, Kascsak R, Carp R, Meeker HC, Frangione B, Wisniewski T. Anti-prion antibodies for prophylaxis following prion exposure in mice. Neurosci Lett. 2003;336:185–187. doi: 10.1016/s0304-3940(02)01192-8. [DOI] [PubMed] [Google Scholar]
- Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003;22:3591–3601. doi: 10.1093/emboj/cdg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiveron MC, Nostenbertrand M, Jani H, Garnett D, Hirst EMA, Grosveld F, Morris RJ. The mode of anchorage to the cell-surface determines both the function and the membrane location of Thy-1 glycoprotein. J Cell Sci. 1994;107:1783–1796. doi: 10.1242/jcs.107.7.1783. [DOI] [PubMed] [Google Scholar]
- UK Department of Health Monthly CJD Statistics. [Accessed on 5-May-2006];2006 [ http://www.dh.gov.uk/PublicationsAndStatistics/PressReleases/PressReleasesNotices/fs/en]
- Wadsworth JDF, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet. 2001;358:171–180. doi: 10.1016/s0140-6736(01)05403-4. [DOI] [PubMed] [Google Scholar]
- Weissmann C, Enari M, Klohn PC, Rossi D, Flechsig E. Transmission of prions. Proc Natl Acad Sci USA. 2002;99:16378–16383. doi: 10.1073/pnas.172403799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AR, Enever P, Tayebl M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422:80–83. doi: 10.1038/nature01457. [DOI] [PubMed] [Google Scholar]
- Wong BS, Li R, Sassoon J, Kang SC, Liu T, Pan T, Greenspan NS, Wisniewski T, Brown DR, Sy MS. Mapping the antigenicity of copper-treated cellular prion protein with the scrapie isoform. Cell Mol Life Sci. 2003;60:1224–1234. doi: 10.1007/s00018-003-3057-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara O, Hanai K, Ohkubo I, Sasaki M, McGeer PL, Kimura H. Expression of cystatin C in rat, monkey and human brains. Brain Res. 1993;628:85–92. doi: 10.1016/0006-8993(93)90941-f. [DOI] [PubMed] [Google Scholar]
- Zanusso G, Liu DC, Ferrari S, Hegyi I, Yin XH, Aguzzi A, Hornemann S, Liemann S, Glockshuber R, Manson JC, Brown P, Petersen RB, Gambetti P, Sy MS. Prion protein expression in different species: Analysis with a panel of new mAbs. Proc Natl Acad Sci USA. 1998;95:8812–8816. doi: 10.1073/pnas.95.15.8812. [DOI] [PMC free article] [PubMed] [Google Scholar]