

Abstract
High passive stiffness is one of the characteristic properties of the asynchronous indirect flight muscle (IFM) found in many insects like Drosophila. To evaluate the effects of two thick filament protein domains on passive sarcomeric stiffness, and to investigate their correlation with IFM function, we used microfabricated cantilevers and a high resolution imaging system to study the passive IFM myofibril stiffness of two groups of transgenic Drosophila lines. One group (hinge-switch mutants) had a portion of the endogenous S2 hinge region replaced by an embryonic version; the other group (paramyosin mutants) had one or more putative phosphorylation sites near the N-terminus of paramyosin disabled. Both transgenic groups showed severely compromised flight ability. In this study, we found no difference (compared to the control) in passive elastic modulus in the hinge-switch group, but a 15% reduction in the paramyosin mutants. All results were corroborated by muscle fiber mechanics experiments performed on the same lines. The fact that myofibril elasticity is unaffected by hinge switching implies alternative S2 hinges do not critically affect passive sarcomere stiffness. In contrast, the mechanical defects observed upon disrupting paramyosin phosphorylation sites in Drosophila suggests that paramyosin phosphorylation is important for maintaining high passive stiffness in IFM myofibrils, probably by affecting paramyosin's interaction with other sarcomeric proteins.
INTRODUCTION
Drosophila indirect flight muscle (IFM) is an asynchronous muscle (1) characterized by low isometric tension, high passive stiffness, and pronounced stretch activation (2). These properties probably evolved out of the distinctive requirement for the IFM's high frequency oscillatory work. Mutations in Drosophila muscle proteins that lead to reduced stretch activation and/or reduced passive stiffness have been reported to impair the insect's flight ability (3–6), suggesting the importance of high passive stiffness and stretch activation.
During flight, the Drosophila thorax vibrates at its resonant frequency (∼240 Hz), driving the wings to beat at the same frequency over a span of ∼170° with high efficiency (7). Two sets of perpendicularly placed IFM work in tandem; when one set actively contracts, the other is stretched. Kinetic energy (stiffness times the square of the length change) is stored in elongated molecular “springs” consisting of connecting filaments and other elastic elements in the sarcomere, including thick filaments (8). The stored energy is released to facilitate the wing beat stroke when the opposing set of IFM deactivates itself (5). For fast vibrations, sarcomere-length changes cannot be large; therefore, small length changes (∼3.5% in Drosophila melanogaster: (9)) require high passive stiffness to store significant energy. The high tension generated from stretching connecting filaments is also thought to be a prerequisite for stretch activation (10).
The high passive stiffness of IFM can be largely explained by short connecting filaments (C-filaments) that anchor the thick filaments to the Z-disk in the sarcomere (11). In Drosophila IFM, C-filaments consist of the proteins projectin (12) and kettin (13,14). Other proteins are thought to form cross-links between thick and thin filaments to further strengthen the sarcomere in Drosophila IFM, including troponin H (specifically, the C-terminal extension: (15)), the myosin regulatory light chain (the N-terminal extension: (5,6)), and, possibly, flightin (3). Weak actomyosin cross bridges have also been implicated (16).
In this study, we examined the myofibril passive stiffness of two previously constructed lines of transgenic Drosophila that showed compromised flight ability compared to their positive controls. One group (hereafter referred to as hinge-switch lines) had the central portion of its endogenous S2 hinge (15a) in IFM replaced by the embryonic version (15b) (17). As 15b is expressed in slower and presumably more compliant muscles than IFM, it is of great interest to investigate whether alternative hinge regions modulate sarcomere stiffness. In the other group (paramyosin mutants), one or more serines (putative phosphorylation sites) near the N-terminus of paramyosin's nonhelical region were replaced by alanines (18). Previous muscle fiber mechanics studies on the paramyosin lines found a significant reduction in the passive, active, and rigor elastic modulus (18). We designed this study to test whether similar differences in passive stiffness occur at the level of the myofibril, the smallest subdivision of muscle that retains the organized myofilament lattice.
A comparison of hinge-switch and paramyosin mutants at the myofibril and muscle fiber levels showed marked differences in passive stiffness. Although alternative hinge regions have different propensities for forming a coiled coil, the hinge mutants exhibit the same passive stiffness as the control. This result shows that swapping the S2 hinges does not affect passive sarcomeric stiffness. The paramyosin phosphorylation-site mutants, in contrast, have a significantly lower passive stiffness compared to control. The reduced stiffness suggests paramyosin interacts via phosphorylation with other sarcomeric proteins (probably projectin and/or kettin), which help maintain high passive stiffness in IFM myofibrils.
MATERIALS AND METHODS
Fly stocks and transgenic construction
Wild-type or mutant versions of the Drosophila myosin heavy chain or paramyosin gene were expressed in mutant lines that fail to express their endogenous myosin heavy chain (20) or paramyosin (18) genes. Construction of the transgenes and preparation of transgenic lines by P element-mediated transformation have been described previously (17,18). Briefly, the 15b-47 and 15b-108 lines express the same embryonic version of the endogenous S2 hinge, except with different transgene insertion points. The pmS18A line has a single serine to alanine substitution (serine 18) in the N-terminus of paramyosin's non-helical region, whereas pmS-A4 has four substitutions (serine 9, 10, 13, and 18). The appropriate positive controls for the hinge and paramyosin mutant lines are pwMhc2 and pm, respectively.
Single myofibril mechanics
A single myofibril, immersed in a physiological relaxing solution, was attached between the tips of a glass needle and a microfabricated cantilever working as a force transducer (stiffness, 12 pN/nm). The myofibril was then incrementally stretched by a total of ∼2–4%. Force and sarcomere length data were collected 1–2 min after each stretch increment. The slope of the linear fit to the force versus sarcomere length plot was taken as the sarcomere stiffness, which means the amount of tension (in nN) a single sarcomere develops per unit length (in nm) of stretch. Myofibril diameter was estimated from the width of the myofibril captured in CCD video images. Sarcomere length (SL) was the slope of the linear fit of A-band peak positions versus their index numbers. The experiments were performed at room temperature. The relaxing solution (pCa 8) was 20 mM BES, 15 mM creatine phosphate, 240 units/ml creatine phosphokinase, 1 mM DTT, 5 mM EGTA, 1 mM free Mg2+, 5 mM MgATP, and 8 mM Pi at pH 7.0 and an ionic strength of 200 mEq adjusted with sodium methane sulfate. Details of the method for preparing single myofibrils and measuring passive (resting) stiffness are given elsewhere (19).
Elastic modulus (nN/μm2) of each tested myofibril was calculated from (stiffness/CSA) × SL0, in which SL0 means initial sarcomere length when tension is zero, and stiffness/CSA is sarcomere stiffness divided by cross-sectional area (CSA). The phase contrast imaging technique for measuring myofibril diameter underestimates the true values by roughly 24%, thereby producing an underestimate of true myofibril cross-sectional area by roughly 58% (19). To account for this area underestimation, the uncorrected value of the elastic modulus was multiplied by 0.58 to obtain the corrected value.
Muscle fiber mechanics
A chemically skinned muscle fiber was secured at both ends with aluminum T-clips and mounted between a strain gauge force transducer and a piezo-motor. After measuring the initial length (L0) when the specimen was just taut, and the cross-sectional area (CSA), the fiber was prestretched incrementally to 1.05L0 in relaxing solution at 15°C. Sinusoidal perturbation of amplitude 0.125%L0 was applied at 47 frequencies (0.5–1000 Hz) and the tension (T) signal was recorded. The complex ratio (with both amplitude and phase) of stress (T/CSA) to strain (0.125%) was taken as the dynamic modulus of the passive muscle fiber, which was decomposed into elastic (in-phase) and viscous (out-of-phase) components. The relaxing solution (pCa 8) was the same as used for the myofibril mechanics. A detailed description of the preparation, experimental equipment, and method of sinusoidal analysis are given elsewhere (21). Elastic modulus values obtained at the lowest oscillation frequency (0.5 Hz) are directly compared to the myofibril data since this slow oscillation best simulates the static methods used to determine the single myofibril stiffness. The frequency dependence of both elastic and viscous moduli was measured in the skinned fiber since phenotypical differences may only appear under dynamic conditions. Dynamic measurements with myofibrils were not feasible because of the technical difficulty in characterizing the high frequency viscoelastic properties of the attachments to the motor and strain gauge.
Transmission electron microscopy
After completion of muscle fiber mechanics, wild-type fibers were fixed for 2 h in Karnovsky's fixative (2.5% glutaraldehyde and 1.0% paraformaldehyde in 0.1 M Millonig's phosphate buffer, pH 7.2). After removal of their T-clips, fixed fibers were embedded in 2.5% SeaPrep Agarose, chilled for 15 min at 4°C, immersed in Karnovsky's fixative for 15 min at 4°C, and rinsed 3 times for 10 min each in Millonig's buffer. Samples were postfixed in 1% OsO4 for 45 min at 4°C, then washed 3 times for 5 min and subsequently stored for 24 h in 0.1 M Millonig's buffer at 4°C. Details of the dehydration, infiltration, embedding, sectioning, and imaging are given elsewhere (22). Image analysis was performed using ImageJ software version 1.36b (National Institutes of Health, Bethesda, MD). Myofibril area per total fiber cross-sectional area, an important factor for making comparisons between myofibril and fiber studies, was calculated by darkening the myofibrils, thresholding the entire image, and calculating the percentage of total area covered by myofibrils in 18 × 18 μm fields.
COILS test
COILS is a program that predicts the probability of a sequence to form a coiled coil based on the similarity of the sequence in question with a database of known parallel two-stranded coiled-coils (23). Amino acid sequences were fed to COILS version 2.2 program on line (http://www.ch.embnet.org/software/COILS_form.html). Default parameters were chosen whenever possible, i.e., matrix: MTIDK; no weighing on positions a and d, and window width: 21.
Statistical analysis
Statistical analysis was carried out with SPSS v.11 (SPSS, Chicago, IL). Test results were considered significant at the p < 0.05 level. For the myofibril data, one-way analysis of variance (ANOVA) tests were performed to determine the effects of different strains. If differences were found to be significant, the least significant difference (LSD) post hoc test was performed and used to determine which means differed. For the fiber data, since the elastic and viscous modulus were examined across various oscillation frequencies, a repeated-measures ANOVA with frequency as the repeated measure was performed first to determine the effects of the different transgenic and control strains. If a significant strain effect was found between subjects, then one-way ANOVAs were performed at each frequency to determine significant differences.
RESULTS
Transmission electron microscopy
Myofibril area per total skinned fiber cross-sectional area was 36.6 ± 1.4% (n = 12), significantly less than the 55% reported previously for intact fibers (18). The reduced myofibril area in skinned fibers results from skinned mitochondria occupying more area than intact mitochondria due to membrane rupture and mitochondrial swelling. To directly compare stiffness moduli from the skinned fibers and myofibrils, the moduli obtained from the fiber measurements were divided by 0.37, i.e., the factor that converts skinned fiber cross-sectional area to total myofibrillar cross-sectional area.
Validation of positive controls
The positive controls, pwMhc2 for hinge-switch mutants and pm for paramyosin mutants, underwent the same transformation and genetic manipulations as their corresponding mutants, except that the wild-type versions of the protein supplied by the transgenes were crossed into the null mutant backgrounds. We compared myofibrils from the wild-type strain yw (19) to the two positive controls (Table 1). The elastic moduli of the two positive controls and wild-type lines were similar, indicating the genetic transformations themselves did not alter the passive mechanical properties of the IFM. Interestingly, the myofibril diameter increased in both positive controls and the sarcomere stiffness was statistically higher in pwMhc2. These differences disappear, however, once the data are normalized for cross-sectional area, as shown by the stiffness/CSA and elastic modulus values.
TABLE 1.
Myofibril statistics of yw wild-type and two positive controls: pwMhc2 for hinge mutants and pm for paramyosin mutants
| Sarcomere stiffness (nN/nm)* | Diameter (μm)* | Stiffness/CSA‡ (nN/nm/μm2)* | Initial SL (μm)* | Elastic modulus§ (nN/μm2)* | Corrected elastic modulus¶ (nN/μm2)* | n | |
|---|---|---|---|---|---|---|---|
| yw‖ | 0.658 ± 0.035 | 1.68 ± 0.03 | 0.307 ± 0.016 | 3.65 ± 0.07 | 1094 ± 66 | 635 ± 38 | 13 |
| pwMhc2 | 0.849 ± 0.023† | 1.88 ± 0.04† | 0.308 ± 0.012 | 3.61 ± 0.10 | 1114 ± 52 | 646 ± 30 | 9 |
| pm | 0.735 ± 0.042 | 1.77 ± 0.03† | 0.296 ± 0.013 | 3.68 ± 0.06 | 1095 ± 59 | 635 ± 34 | 15 |
| p-value | 0.009 | 0.001 | 0.802 | 0.843 | 0.932 | 0.932 |
Values are mean ± SE.
Indicates value significantly different (p < 0.05) from that of yw.
Stiffness/CSA = sarcomere stiffness / (π × (diameter/2)2).
Elastic modulus = (stiffness / CSA) × initial SL.
Corrected elastic modulus = 0.58 × elastic modulus.
Wild-type strain yw values reproduced from Hao et al. (19). Myofibril data from all lines (yw, pwMhc2, pm, 15b-47, 15b-108, pmS18A, and pmS-A4) were collected during the same time period.
Hinge-switch mutants
Myofibril stiffness measurements were conducted on two hinge-switch transgenic lines, namely 15b-47 and 15b-108. The elastic moduli of the two hinge-switch lines were similar to the positive control (Table 2), despite both lines having severely impaired flight ability (17). Both hinge-switch transgenic lines have reduced sarcomere stiffness compared to the positive control, which can be accounted for by their smaller myofibril diameters, as demonstrated by the good agreement between the stiffness/CSA values. The modest increase in initial sarcomere length of the 15b-47 line does not appear to affect the passive mechanical properties of the myofibril since the stiffness/CSA and elastic modulus values agree among all three lines.
TABLE 2.
Myofibril statistics of pwMhc2 positive control and two 15b trangenics
| Sarcomere stiffness (nN/nm)* | Diameter (μm)* | Stiffness/CSA‡ (nN/nm/μm2)* | Initial SL (μm)* | Elastic modulus§ (nN/μm2)* | Corrected elastic modulus¶ (nN/μm2)* | n | |
|---|---|---|---|---|---|---|---|
| pwMhc2 | 0.849 ± 0.023 | 1.88 ± 0.04 | 0.308 ± 0.012 | 3.61 ± 0.10 | 1114 ± 52 | 646 ± 30 | 9 |
| 15b-47 | 0.692 ± 0.018† | 1.68 ± 0.03† | 0.314 ± 0.011 | 3.95 ± 0.10† | 1245 ± 68 | 722 ± 39 | 9 |
| 15b-108 | 0.729 ± 0.019† | 1.78 ± 0.03† | 0.293 ± 0.015 | 3.81 ± 0.04 | 1115 ± 58 | 647 ± 34 | 8 |
| p-value | 0.004 | 0.000 | 0.502 | 0.031 | 0.226 | 0.226 |
Values are mean ± SE.
Indicates values are significantly different (p < 0.05) from the hinge-switch positive control, pwMhc2.
Stiffness/CSA = sarcomere stiffness / (π × (diameter/2)2).
Elastic modulus = (stiffness / CSA) × initial SL.
Corrected elastic modulus = 0.58 × elastic modulus.
Muscle fibers had no significant differences in either elastic or viscous modulus across the frequency range measured (Fig. 1). In comparison with the myofibril data, the elastic modulus at 0.5 Hz showed no significant differences between the hinge-switch mutants and the positive control (622 ± 64 nN/μm2 for pwMhc2, n = 12; 710 ± 66 nN/μm2 for 15b-47, n = 12; and 734 ± 94 nN/μm2 for 15b-108, n = 15). Therefore, the trends and magnitude of the elastic moduli found in both the myofibril and muscle fiber results are consistent among these three lines.
FIGURE 1.

Elastic (A) and viscous modulus (B) values for passive (pCa 8) IFM fibers across muscle oscillation frequencies for hinge-switch mutants (15b-47 and 15b-108) and their positive control (pwMhc2). Values are mean ± SE. No significant differences were found between the three lines. Temperature = 15°C.
Paramyosin mutants
Myofibril stiffness tests were performed on two paramyosin transgenic lines, namely pmS18A and pmS-A4, which have one and four N-terminal serines replaced by alanines, respectively. Both lines had shown severely impaired flight ability whereas their ultrastructure was found to be normal (18). The myofibril elastic moduli of both paramyosin mutants were decreased by 14–16% compared to the positive control (Table 3). Note neither the sarcomere stiffness values nor the diameters were significantly different among the lines. However, when normalized by cross-sectional area (stiffness/CSA and elastic modulus), the differences in passive mechanical properties become evident. The result of this adjustment underlines the importance of normalizing the stiffness of each individual myofibril to its CSA as well as the sensitivity of our method.
TABLE 3.
Myofibril statistics of pm positive control and paramyosin transgenics
| Sarcomere stiffness (nN/nm)* | Diameter (μm)* | Stiffness/CSA‡ (nN/nm/μm2)* | Initial SL (μm)* | Elastic modulus§ (nN/μm2)* | Corrected elastic modulus¶ (nN/μm2)* | n | |
|---|---|---|---|---|---|---|---|
| pm | 0.735 ± 0.042 | 1.77 ± 0.04 | 0.296 ± 0.013 | 3.68 ± 0.06 | 1095 ± 59 | 635 ± 34 | 15 |
| pmS18A | 0.692 ± 0.037 | 1.83 ± 0.05 | 0.258 ± 0.007† | 3.55 ± 0.11 | 917 ± 46† | 532 ± 27† | 10 |
| pmS-A4 | 0.638 ± 0.031 | 1.78 ± 0.03 | 0.256 ± 0.008† | 3.67 ± 0.07 | 938 ± 33† | 544 ± 19† | 11 |
| p-value | 0.212 | 0.516 | 0.018 | 0.469 | 0.030 | 0.030 |
Values are mean ± SE.
Indicate values are significantly different (p < 0.05) from the paramyosin positive control, pm.
Stiffness/CSA = sarcomere stiffness / (π × (diameter/2)2).
Elastic modulus = (stiffness / CSA) × initial SL.
Corrected elastic modulus = 0.58 × elastic modulus.
Muscle fibers from the two paramyosin transgenic lines had a significant reduction in elastic modulus between 0.5 and 180 Hz when compared to the pm control fibers, but no change in viscous modulus (Fig. 2), similar to previous results (18). In comparison with the myofibril data, the elastic modulus at 0.5 Hz of the two paramyosin transgenic lines was significantly (p < 0.05) reduced by 29–36% when compared to the paramyosin positive control (669 ± 59 nN/μm2 for pm, n = 10; 430 ± 49 nN/μm2 for pmS18A, n = 12; and 474 ± 65 nN/μm2 for pmS-A4, n = 12). As with the hinge-switch lines, the trends in the paramyosin lines were similar between the myofibril and fiber results and the magnitude of the elastic modulus was similar between the control lines. However, the magnitude of the elastic modulus decrease in the paramyosin transgenic lines compared to controls was 14–16% in myofibrils versus 29–36% in fibers.
FIGURE 2.

Elastic (A) and viscous modulus (B) values for passive (pCa 8) IFM fibers across muscle oscillation frequencies for paramyosin mutants (pmS18A and pmSA4) and their positive control (pm). Values are mean ± SE. * indicates a span of frequencies over which there is a significant difference (p < 0.05) between the pm and the pmS18A and pmSA4 lines. Temperature = 15°C.
Alternative S2 hinge sequences and COILS test
A portion of the Drosophila myosin heavy chain S2 hinge region is encoded by mutually exclusive alternative exons 15a (adult) and 15b (embryonic), that are 26 amino acids long and differ by 72% (20). The amino acid sequences for 15a and 15b are AEHDRQTCHNELNQTRTACDQLGRDK and AEKEKNEYYGQLNDLRAGVDHITNEK, respectively. The COILS program determined that, on average, the propensity of 15a to form a coiled coil is 59% whereas that of 15b is 91%.
DISCUSSION
Using advanced methods for measuring passive myofibril mechanical properties, we evaluated the effects of two thick filament protein domains in Drosophila melanogaster. The positive controls created for the two thick filament protein domains (S2 hinge and paramyosin) were mechanically similar to wild-type, indicating that the genetic transformations did not affect passive muscle properties. Two different hinge-switch mutants, which have a portion of the endogenous S2 hinge region replaced by an embryonic version, were similar to their positive control, suggesting this domain has no effect on passive mechanical properties. However, the two paramyosin mutants, which have one or four putative phosphorylatable serine sites near the N-terminus switched to alanines, have a significant decrease in stiffness compared to their positive control. In the discussion that follows, we start with an evaluation of our techniques to detect passive IFM properties, as a basis for our subsequent interpretation of data.
Probing passive sarcomere stiffness
When a relaxed myofibril is stretched, most of its extension comes from the elongation of C- filaments that connect thick filaments to the Z-disk (11). The C-filaments have recently been shown to consist of the long extensible proteins projectin (12) and kettin (13). Although thick and thin filaments are also extensible (24–27), they are much stiffer than the C-filaments.
In the “stretch and hold” protocol from which the myofibril mechanics data were derived, each sarcomere was incrementally stretched by ∼100 nm (SL 3.6 μm × ∼3%). Because any possible cross-links (weakly attached myosin heads (16), myosin regulatory light chain N-terminal extension (5,6), the myosin associated protein flightin (3), and troponin-H isoform 34 (15)) likely detach and reattach during the long-range stretch (instead of being elongated by 100 nm without breaking), it is unlikely that the cross-links contribute significantly to the stiffness of the myofibril. Thus, the passive compliance (1/stiffness) of the half sarcomere is equal to the sum of the thick and C-filament compliance, i.e.,
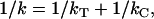 |
(1) |
where k is the stiffness of a half sarcomere, and kT and kC are the stiffnesses of the thick and C-filaments, respectively.
A recent x-ray study of Drosophila flight muscle in vivo (8) showed the thick filament backbone undergoes an ∼0.2% strain during each work-producing wing beat, as indexed by a strong 7.2-nm periodic reflection off the thick filament. Since the sarcomere length of Drosophila IFM changes by ∼3.5% during each wing beat (9), the ratio of the two length changes suggests that the thick filament is ∼17× (= 3.5%/0.2%) stiffer than that of the C-filament. Therefore, we conclude that under passive conditions Drosophila IFM thick filaments are relatively inextensible compared to C-filaments.
Hinge-switch study
Drosophila has a single gene encoding the muscle myosin heavy chain; isoforms of the protein result from alternative splicing of the primary RNA transcript (28). Alternative exons 15a and 15b encode the central 26 amino acids of the S2 hinge, which is the region located between the N-terminus of light meromyosin and the C-terminus of short S2 (29) and may be part of the thick filament backbone. 15a and 15b hinges have different properties of charge, hydrophobicity, and propensities toward forming a coiled-coil (15a has a 59% probability; 15b, 91%).
In spite of the structural differences, we found no difference in elastic modulus between the two mutants and the positive (wild-type) control. Because passive sarcomere stiffness is determined primarily by C-filament stiffness, as noted above, the lack of a difference in resting stiffness between the hinge mutants and the control suggests that the alternative hinges do not interact (or do not vary significantly in their interaction) with the C-filaments (or other structures that may link thick and thin filaments). We conclude, therefore, that alternative hinges do not modulate passive sarcomere stiffness.
Although passive stiffness appears to be unaffected by the hinge substitutions, it is possible that hinge switches do affect sarcomere stiffness in active fibers. The thick filament is measurably extensible in working muscles (8); thus, it is possible that differences in extensibility due to hinge differences may underlie the severely impaired flight ability seen previously in transgenic lines expressing an IFM myosin isoform with the “slow” hinge 15b compared to that with the native “fast” hinge 15a (17). Clearly, a comparison of sarcomere stiffness in active, working IFM from the hinge mutants and controls is necessary to fully resolve the question whether hinge differences play a significant role in flight muscle stiffness.
Paramyosin phosphorylation study
Paramyosin, a major structural protein of invertebrate thick filaments, is a rod-like molecule with a central α-helical region and two nonhelical terminal domains (30,31). In vivo phosphorylation of paramyosin has been reported in Drosophila (32) as well as in other species (33,34). In Drosophila IFM, paramyosin, despite its low concentration, is uniformly distributed along the core of the thick filament (35,36).
To test whether paramyosin phosphorylation in Drosophila plays an important role in muscle function, several transgenic lines were constructed in which one or more phosphorylatable serine residues near the N-terminus of paramyosin were replaced by alanines. Two of the resulting lines (pmS18A and pmS-A4) showed compromised flight ability, whereas the ultrastructure of their IFM was normal (18).
In this study we examined the passive stiffness of Drosophila IFM myofibrils from the two mutant lines and their positive (wild-type) control. Myofibrils from the two transgenic lines with impaired flight ability had a 15% reduction in passive stiffness compared to control. The finding of reduced passive stiffness in the paramyosin mutants was surprising since, from Eq. 1, thick filament stiffness would have to diminish by 76% to accommodate a reduction in sarcomere stiffness of 15%, assuming an initial ratio of thick-filament to C-filament stiffness of ∼17. Thus, it is possible that the paramyosin molecule contributes directly and massively to thick filament stiffness, and that disruption of the phosphorylation sites directly affects thick filament stiffness. However, in light of the exceptionally large changes in thick filament stiffness that would have to occur, it is more likely that paramyosin plays a role in anchoring kettin and/or projectin to the thick filament, and that disruption of the phosphorylation sites disrupts the anchoring. It is worth noting that any anchoring model would have to accommodate the low molar ratio of paramyosin to myosin in Drosophila IFM (molar ratio, ∼1:34: (32)), and its putative location within the core of the thick filaments (37).
Our myofibril measurements agree well with fiber measurements from a previous study (18), which reported significant reductions in passive, active, and rigor elastic modulus of muscle fibers from the same paramyosin mutants. The authors of the previous study suggested that paramyosin phosphorylation most likely contributes to thick filament stiffness by interacting with myosin rods and/or stabilizing the thick filament's connection to the M-line. Because thick filament compliance cannot be neglected in the calculation of sarcomere stiffness under active or rigor conditions (25,27), Liu and colleagues propose, in essence, that thick filament stiffness is reduced in the phosphorylation site mutants, thereby accounting for the reduced elastic moduli observed in active and rigor fibers. Although this may be the case for active and rigor fibers, our analysis indicates that the reduction in passive stiffness of the sarcomere in the phosphorylation site mutants is most likely due to altered C-filament anchoring.
The fractional reduction in elasticity in active (and rigor) muscles is greater than that in passive muscle from the paramyosin phosphorylation site mutants (18). Thus, it is likely that any weakened paramyosin interactions with C-filament proteins contributes to reduced active stiffness as well, consistent with notions advanced by previous research (10,11,16). We propose that both mechanisms, altered anchoring and reduced thick filament stiffness, give rise to the reduced flight ability of the paramyosin phosphorylation site mutants.
Myofibril versus fiber mechanics
Our results show that the mutation-related trends of both lines were similar between myofibrils and fibers. The magnitudes of the myofibril and fiber elastic modulus were similar between controls and hinge-switch lines, but differences were observed between myofibrils and fibers in the magnitude of the changes observed in the paramyosin transgenic lines. The elastic modulus in the paramyosin transgenic lines compared to controls was reduced 14–16% in myofibrils (using a 2–4% stretch) versus 29–36% in fibers (using a 0.125% sinusoidal length perturbation at 0.5 Hz). Although this suggests a possible methodological difference (stretching versus sinusoidal perturbation), a previous fiber study showed a 25–33% decrease in isometric tension for the paramyosin phosphorylation-site mutants (18). Since similar magnitude decreases in performance are observed at the fiber level, independent of measurement technique, differences between myofibril and fiber data are most likely not due to methodological differences, but rather to the distinct structural architectures of the two systems.
CONCLUSION
We report here measurements of passive IFM stiffness in two groups of transgenic Drosophila strains. In one, the myosin S2 hinge was replaced by a version expressed in slower muscles, whereas in the other the putative paramyosin phosphorylation sites were disrupted. Although alternative hinge regions have marked differences in amino acid sequence and tissue-specificity, the IFM passive stiffness of the hinge mutants was not significantly different than that of the control, implying that the S2 hinge does not modulate passive sarcomeric stiffness. In contrast, the IFM passive stiffness of the paramyosin mutants is significantly reduced compared to that of control, leading to the suggestion that paramyosin contributes to passive sarcomere stiffness in Drosophila IFM by interacting with other sarcomeric proteins (most likely C-filaments).
Acknowledgments
The authors gratefully acknowledge Dr. Jim Vigoreaux and Jennifer Suggs for helpful discussions. We thank Jeff Magula for technical assistance and Jennifer Suggs for logistical support.
Funding was provided by National Institutes of Health grants AR43396 to S.I.B. and R01049425 to D.W.M.
Douglas M. Swank's present address is Dept. of Biology, Rensselaer Polytechnic Institute, 110 8th St., Troy, NY 12180.
Hongjun Liu's present address is Cardiovascular Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bldg. 10-CRC, Rm. 5-3288, 10 Center Dr., Bethesda, MD 20892.
References
- 1.Josephson, R. K., J. G. Malamud, and D. R. Stokes. 2000. Asynchronous muscle: a primer. J. Exp. Biol. 203:2713–2722. [DOI] [PubMed] [Google Scholar]
- 2.Peckham, M., J. E. Molloy, J. C. Sparrow, and D. C. White. 1990. Physiological properties of the dorsal longitudinal flight muscle and the tergal depressor of the trochanter muscle of Drosophila melanogaster. J. Muscle Res. Cell Motil. 11:203–215. [DOI] [PubMed] [Google Scholar]
- 3.Henkin, J. A., D. W. Maughan, and J. O. Vigoreaux. 2004. Mutations that affect flightin expression in Drosophila alter the viscoelastic properties of flight muscle fibers. Am. J. Physiol. Cell Physiol. 286:C65–C72. [DOI] [PubMed] [Google Scholar]
- 4.Moore, J. R., J. O. Vigoreaux, and D. W. Maughan. 1999. The Drosophila projectin mutant, bentD, has reduced stretch activation and altered indirect flight muscle kinetics. J. Muscle Res. Cell Motil. 20:797–806. [DOI] [PubMed] [Google Scholar]
- 5.Moore, J. R., M. H. Dickinson, J. O. Vigoreaux, and D. W. Maughan. 2000. The effect of removing the N-terminal extension of the Drosophila myosin regulatory light chain upon flight ability and the contractile dynamics of indirect flight muscle. Biophys. J. 78:1431–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tohtong, R., H. Yamashita, M. Graham, J. Haeberle, A. Simcox, and D. Maughan. 1995. Impairment of muscle function caused by mutations of phosphorylation sites in myosin regulatory light chain. Nature. 374:650–653. [DOI] [PubMed] [Google Scholar]
- 7.Maughan, D. W., and J. O. Vigoreaux. 1999. An integrated view of insect flight muscle: genes, motor molecules, and motion. News Physiol. Sci. 14:87–92. [DOI] [PubMed] [Google Scholar]
- 8.Dickinson, M., G. Farman, M. Frye, T. Bekyarova, D. Gore, D. Maughan, and T. Irving. 2005. Molecular dynamics of cyclically contracting insect flight muscle in vivo. Nature. 433:330–334. [DOI] [PubMed] [Google Scholar]
- 9.Chan, W. P., and M. H. Dickinson. 1996. In vivo length oscillations of indirect flight muscles in the fruit fly Drosophila virilis. J. Exp. Biol. 199:2767–2774. [DOI] [PubMed] [Google Scholar]
- 10.Granzier, H. L., and K. Wang. 1993. Passive tension and stiffness of vertebrate skeletal and insect flight muscles: the contribution of weak cross-bridges and elastic filaments. Biophys. J. 65:2141–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White, D. C. 1983. The elasticity of relaxed insect fibrillar flight muscle. J. Physiol. 343:31–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saide, J. D., S. Chin-Bow, J. Hogan-Sheldon, L. Busquets-Turner, J. O. Vigoreaux, K. Valgeirsdottir, and M. L. Pardue. 1989. Characterization of components of Z-bands in the fibrillar flight muscle of Drosophila melanogaster. J. Cell Biol. 109:2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kulke, M., C. Neagoe, B. Kolmerer, A. Minajeva, H. Hinssen, B. Bullard, and W. A. Linke. 2001. Kettin, a major source of myofibrillar stiffness in Drosophila indirect flight muscle. J. Cell Biol. 154:1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lakey, A., C. Ferguson, S. Labeit, M. Reedy, A. Larkins, G. Butcher, K. Leonard, and B. Bullard. 1990. Identification and localization of high molecular weight proteins in insect flight and leg muscle. EMBO J. 9:3459–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clayton, J. D., R. M. Cripps, J. C. Sparrow, and B. Bullard. 1998. Interaction of troponin-H and glutathione S-transferase-2 in the indirect flight muscles of Drosophila melanogaster. J. Muscle Res. Cell Motil. 19:117–127. [DOI] [PubMed] [Google Scholar]
- 16.Granzier, H. L., and K. Wang. 1993. Interplay between passive tension and strong and weak binding cross-bridges in insect indirect flight muscle. A functional dissection by gelsolin-mediated thin filament removal. J. Gen. Physiol. 101:235–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suggs, J. A. 2003. The function of the S2 hinge of myosin heavy chain in Drosophila melanogaster. Masters thesis. San Diego State University, San Diego, CA.
- 18.Liu, H., M. S. Miller, D. M. Swank, W. A. Kronert, D. W. Maughan, and S. I. Bernstein. 2005. Paramyosin phosphorylation site disruption affects indirect flight muscle stiffness and power generation in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA. 102:10522–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hao, Y., S. I. Bernstein, and G. H. Pollack. 2004. Passive stiffness of Drosophila IFM myofibrils: a novel, high accuracy measurement method. J. Muscle Res. Cell Motil. 25:359–366. [DOI] [PubMed] [Google Scholar]
- 20.Collier, V. L., W. A. Kronert, P. T. O'Donnell, K. A. Edwards, and S. I. Bernstein. 1990. Alternative myosin hinge regions are utilized in a tissue-specific fashion that correlates with muscle contraction speed. Genes Dev. 4:885–895. [DOI] [PubMed] [Google Scholar]
- 21.Dickinson, M. H., C. J. Hyatt, F. O. Lehmann, J. R. Moore, M. C. Reedy, A. Simcox, R. Tohtong, J. O. Vigoreaux, H. Yamashita, and D. W. Maughan. 1997. Phosphorylation-dependent power output of transgenic flies: an integrated study. Biophys. J. 73:3122–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barton, B., G. Ayer, N. Heymann, D. W. Maughan, F. O. Lehmann, and J. O. Vigoreaux. 2005. Flight muscle properties and aerodynamic performance of Drosophila expressing a flightin transgene. J. Exp. Biol. 208:549–560. [DOI] [PubMed] [Google Scholar]
- 23.Lupas, A., M. Van Dyke, and J. Stock. 1991. Predicting coiled coils from protein sequences. Science. 252:1162–1164. [DOI] [PubMed] [Google Scholar]
- 24.Dunaway, D., M. Fauver, and G. Pollack. 2002. Direct measurement of single synthetic vertebrate thick filament elasticity using nanofabricated cantilevers. Biophys. J. 82:3128–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huxley, H. E., A. Stewart, H. Sosa, and T. Irving. 1994. X-ray diffraction measurements of the extensibility of actin and myosin filaments in contracting muscle. Biophys. J. 67:2411–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu, X., and G. H. Pollack. 2002. Mechanics of F-actin characterized with microfabricated cantilevers. Biophys. J. 83:2705–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wakabayashi, K., Y. Sugimoto, H. Tanaka, Y. Ueno, Y. Takezawa, and Y. Amemiya. 1994. X-ray diffraction evidence for the extensibility of actin and myosin filaments during muscle contraction. Biophys. J. 67:2422–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.George, E. L., M. B. Ober, and C. P. Emerson, Jr. 1989. Functional domains of the Drosophila melanogaster muscle myosin heavy-chain gene are encoded by alternatively spliced exons. Mol. Cell. Biol. 9:2957–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu, R. C. 1980. Identification of a region susceptible to proteolysis in myosin subfragment-2. Proc. Natl. Acad. Sci. USA. 77:2010–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Becker, K. D., P. T. O'Donnell, J. M. Heitz, M. Vito, and S. I. Bernstein. 1992. Analysis of Drosophila paramyosin: identification of a novel isoform which is restricted to a subset of adult muscles. J. Cell Biol. 116:669–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kagawa, H., K. Gengyo, A. D. McLachlan, S. Brenner, and J. Karn. 1989. Paramyosin gene (unc-15) of Caenorhabditis elegans. Molecular cloning, nucleotide sequence and models for thick filament structure. J. Mol. Biol. 207:311–333. [DOI] [PubMed] [Google Scholar]
- 32.Vinos, J., A. Domingo, R. Marco, and M. Cervera. 1991. Identification and characterization of Drosophila melanogaster paramyosin. J. Mol. Biol. 220:687–700. [DOI] [PubMed] [Google Scholar]
- 33.Schriefer, L. A., and R. H. Waterson. 1989. Phosphorylation of the N-terminal region of Caenorhabditis elegans paramyosin. J. Mol. Biol. 207:451–454. [DOI] [PubMed] [Google Scholar]
- 34.Watabe, S., T. Tsuchiya, and D. J. Hartshorne. 1989. Phosphorylation of paramyosin. Comp. Biochem. Physiol. B. 94:813–821. [DOI] [PubMed] [Google Scholar]
- 35.Maroto, M., J. Arredondo, D. Goulding, R. Marco, B. Bullard, and M. Cervera. 1996. Drosophila paramyosin/miniparamyosin gene products show a large diversity in quantity, localization, and isoform pattern: a possible role in muscle maturation and function. J. Cell Biol. 134:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Royuela, M., R. Garcia-Anchuelo, M. I. Arenas, M. Cervera, B. Fraile, and R. Paniagua. 1996. Immunocytochemical electron microscopic study and western blot analysis of paramyosin in different invertebrate muscle cell types of the fruit fly Drosophila melanogaster, the earthworm Eisenia foetida, and the snail Helix aspersa. Histochem. J. 28:247–255. [DOI] [PubMed] [Google Scholar]
- 37.Epstein, H. F., G. Y. Lu, P. R. Deitiker, I. Oritz, and M. F. Schmid. 1995. Preliminary three-dimensional model for nematode thick filament core. J. Struct. Biol. 115:163–174. [DOI] [PubMed] [Google Scholar]