

Abstract
Understanding the solvation of amino acids in biomembranes is an important step to better explain membrane protein folding. Several experimental studies have shown that polar residues are both common and important in transmembrane segments, which means they have to be solvated in the hydrophobic membrane, at least until helices have aggregated to form integral proteins. In this work, we have used computer simulations to unravel these interactions on the atomic level, and classify intramembrane solvation properties of amino acids. Simulations have been performed for systematic mutations in poly-Leu helices, including not only each amino acid type, but also every z-position in a model helix. Interestingly, many polar or charged residues do not desolvate completely, but rather retain hydration by snorkeling or pulling in water/headgroups—even to the extent where many of them exist in a microscopic polar environment, with hydration levels corresponding well to experimental hydrophobicity scales. This suggests that even for polar/charged residues a large part of solvation cost is due to entropy, not enthalpy loss. Both hydration level and hydrogen bonding exhibit clear position-dependence. Basic side chains cause much less membrane distortion than acidic, since they are able to form hydrogen bonds with carbonyl groups instead of water or headgroups. This preference is supported by sequence statistics, where basic residues have increased relative occurrence at carbonyl z-coordinates. Snorkeling effects and N-/C-terminal orientation bias are directly observed, which significantly reduces the effective thickness of the hydrophobic core. Aromatic side chains intercalate efficiently with lipid chains (improving Trp/Tyr anchoring to the interface) and Ser/Thr residues are stabilized by hydroxyl groups sharing hydrogen bonds to backbone oxygens.
INTRODUCTION
Membrane proteins play key roles in a wide range of processes in the cell, including signal transduction and molecular transport across the plasma membrane. It has been estimated that α-helical membrane proteins account for ∼25% of all proteins in a typical genome (1), and possibly as much as 50% of drug targets (2). The fundamental structural unit of this class of proteins is one or more transmembrane helices with a high fraction of hydrophobic residues. According to the two-stage model of Popot and Engelman (3), helices are first inserted independently in the bilayer environment where they are at least transiently stable as isolated structures (4), and in a second stage, they aggregate to form tightly packed integral proteins. Due to the inherent experimental difficulties in purifying and crystalizing membrane proteins, there are currently only ∼110 unique structures that have been determined (5), which seriously limits our knowledge about membrane protein folding compared to globular proteins. While some small single-helix membrane proteins can insert spontaneously into membranes (6), multihelix structures normally have to be inserted into translocon protein channels after being synthesized in the ribosomes, and then transported out into the bilayer as first suggested by Blobel and Dobberstein (7). In both cases, the transmembrane helices must be hydrophobic enough to insert stably into the membrane, but polar and even charged groups do occur in transmembrane segments and are crucially important for both membrane protein function and folding, since the chemical interactions of aliphatic side chains are quite limited (8). Statistics on membrane protein-sequence data additionally shows that these residues tend to be less mutable than others, which confirms their functional importance (9,10). Common examples include, e.g., proton transport and binding in bacteriorhodopsin (11,12) and heme group binding in cytochrome c oxidase (13). It is also known that the specific lipid composition in different cellular membranes affects selection, structure, and function of membrane proteins, although the molecular basis for this is not yet fully understood (14).
The insertion and aggregation of transmembrane helices has received considerable attention in experiments as well as theoretical studies. Recent computer simulations of helices inside the SecYEβ translocon protein illustrate how the pore ring blocks ions completely, yet seems to allow passage of pulled helices (15). Interactions between lipids and proteins have been studied, e.g., in contexts of partitioning at hydrophobic interfaces (16–18), structure and binding sites around membrane proteins in different solvents (19,20), and simulations of the KvAP potassium channel (21) and isolated S4 helix (22) that have provided valuable insight in the interplay between proteins, membrane, and water. Common packing motifs for protein aggregation such as GxxxG have been identified (23), and a number of works have highlighted the significance of polar residues to drive association of helices in the membrane (24–27). Statistical data from existing crystal structures of membrane proteins reveals that side chains of polar residues located in lipid bilayers tend to be directed away from the membrane core and extend toward the headgroup region (28–30), a result which has also been observed in experiments (31,32) and simulation studies (33). Computer simulations have further suggested that charged amino acids form hydrogen bonds with the lipid headgroups and bind water molecules (22, 34), and that the hydrogen-bonding abilities of polar residues can be pivotal for membrane helix di- and trimerization (35).
Solvation properties of different amino-acid sequences in bilayers is a particularly interesting topic since it is intimately related both to discrimination of membrane versus globular proteins as well as targeting to different membranes in the cell (14). As first observed by Wimley and White (36), the free energy of solvation in bilayers/interfacial systems can be quite a bit lower compared to purely hydrophobic environments. More recently, Hessa et al. have demonstrated practically that it is quite possible to incorporate significantly hydrophilic amino-acid sequences in transmembrane helices as long as they are counterbalanced by a sufficiently large number of nonpolar residues (37), and further used this to derive an effective in vivo hydrophobicity scale (38) that in turn differs only slightly from the classical Wimley-White water/octanol hydrophobicity scale (36). This supports the idea that insertion is determined by direct lipid-protein interactions (39), although our molecular understanding of the process and interactions is still incomplete.
Here, we present results from molecular dynamics computer simulations that enable quantitative studies of atomic scale interactions in membrane-solvated transmembrane helices. Rather than using isolated amino-acid side-chain analogs, we have elected to systematically study structural effects of amino-acid substitutions using model helix sequences similar to those of Hessa et al. (37), since we believe this is important to correctly capture and classify effects such as snorkeling, helix distortion, and backbone interactions. The simulations are primarily analyzed to explain stability of transiently solvated helices, variance with residue hydrophobicity/geometry, backbone direction, and different depths in the bilayer, but also evaluated in context of how the highly adaptive membrane environment differs from simple nonpolar solvents due to polar headgroups and ordered chains, to the extent that this explains the differences between hydrophobicity scales and how it relates to current models of membrane helix aggregation.
METHODS
System preparation
Since this work was partly inspired by the in vivo hydrophobicity scale (37), we chose to use a similar reference system: a single 27-residue transmembrane segment with the sequence GGPG-(A19)-GPGG. The GGPG motifs anchor efficiently to the membrane headgroup region, while the central poly-Ala region forms an α-helix. For each of the remaining 19 amino acids, nine different test segments were designed by symmetrically substituting Ala for pairs of amino acids in positions 1–9 from the center of the helix. Since many of these helices would not insert stably in membranes due to insufficient hydrophobicity, all pair mutations except Ile, Leu, and Val were counterbalanced with between 1 and 11 surrounding Leu residues. In practice, this is likely of little effect on nanosecond scales, but there are no real drawbacks and it makes our sequences identical to those of the experimental studies (37). Mutations are labeled with the introduced side chain and offset from the membrane center. For example, the actual sequence of the “Y7” mutation is GGPG-(A2YA5L3A5YA2)-GPGG, “K5” mutation is GGPG-(A3LKL9KLA3)-GPGG, and “M3” is GGPG-(A6MA2LA2MA6)-GPGG.
A rectangular DMPC lipid membrane system was constructed from earlier DPPC simulations (40) by removing two terminal carbons from each lipid chain followed by 25 ns of equilibration, since DMPC lipids are known to adapt liquid-disordered phase at ∼300 K. Model helices were introduced vertically in this membrane and bad van der Waals contact resolved by removing overlapping lipids and water. The positions of all helix atoms were frozen and position restraints of 1000 kJ/mol applied to the z-coordinates (membrane normal direction) of water molecules to allow lipids to pack around the protein with 10,000 steps of steepest-descent energy minimization, followed by 30 ns of equilibration simulation where the constraints were gradually relaxed, first in the membrane and later also for the helix. In addition to the membrane protein, the finished configurations consisted of 112 DMPC lipids (always 56 per monolayer) fully hydrated with roughly 3600 waters, reaching a bit over 16,000 atoms in total. For charged mutations, two Na+ or Cl− counterions were added to neutralize the overall system charge.
Simulation setup
DMPC interactions were described with the Berger force-field parameters (41), using Ryckaert-Bellemans torsions (42) for the hydrocarbon chains and nonbonded interactions parameterized to reproduce experimental area and volume per lipid accurately. This force field has been show to replicate both equilibrium and dynamical experimental properties well (43,44). Transmembrane helices were modeled with the similarly derived GROMOS96 45a3 protein parameters (45), and standard combination rules applied to nonbonded interactions between lipids and helices (ε geometric, σ arithmetic). Water molecules were represented with the simple point charge model (46).
Simulations were performed with the GROMACS package (47), using 2-fs timesteps. Bond lengths were constrained with the LINCS algorithms (48) while SETTLE (49) was used for water molecules. Twin-range cutoffs of 1.4 nm for van der Waals and 1.8 nm for electrostatic interactions were used together with 1.0-nm neighbor lists updated every 10 steps. The choice of long cutoffs instead of PME (50) was technical and actually more expensive; a related project concerns free energy calculations between these states, and it is not yet possible to separate group contributions in lattice summations. While the effects are fairly small on local structure, it can have an effect on collective properties such as area per lipid, somewhat depending on the charge groups used. Wohlert et al. (51) has discussed this in more detail, where the charge groups in this work are described as Set I. We have also performed PME simulations for Arg and Lys side chains in various positions, with little or no difference on the side-chain solvation structure. All simulations were performed at constant temperature and pressure. The temperature of the system was coupled to 303 K using the Berendsen algorithm with a time constant of τT = 0.2 ps (52). All dimensions of the simulation box were coupled independently (anisotropic scaling) to reference pressures of 1 bar with Berendsen weak coupling, a τP = 1.0 ps time constant, dispersion corrections to pressure, and a system compressibility of 4.5 × 10−5 bar−1 (52).
RESULTS
Simulations and conformational stability
The reference helix sequence, as well as 171 mutation test systems, were all simulated for at least 20 ns each. Including equilibration, the aggregated simulation time reached ≫4 μs. To rule out equilibration artifacts, water molecules that had entered the membrane were moved back to the bulk region, first after 4 ns, and then once more after 8 ns. Production data was collected from 14 ns. Further, all systems with charged substitutions (K,R,D,E) were extended to 32 ns of simulation time to ensure equilibration of retained hydration water and structural reorientation of the lipid headgroups and/or helix.
Both the protein α-helix structure and surrounding membrane remained close to ideal conformation throughout nearly all simulations. The only exceptions were mutations that introduced new acidic residues buried in the hydrophobic core, which frequently resulted in systematic bending or distortion of the helix, sometimes coupled with 1–2 lipid headgroups turning inwards to screen the charged groups on the side chain. In addition, many mutations involving large and/or polar groups affect the membrane acyl-chain ordering around the helix, which is quite expected. Lipid reorientation is slow, but since they do relax on 10-ns scales (43), the simulations are likely to have reached equilibrated states.
Table 1 summarizes the average amount of helical content for all mutations and positions, and it is further resolved as a function of time with DSSP (53) plots for a selection of residues in Fig. 1. As anticipated, small hydrophobic mutations do not affect the helix stability appreciably, but more interestingly, the same also largely holds for all polar residues and bulky aromatic side chains such as Phe, Tyr, and even Trp, and mutations to proline only introduce a very slight bending of the helix. Even amino acids with basic charge such as Arg and Lys normally only result in minor distortion, with 17–18 out of 19 residues remaining clearly within the helical region of Ramachandran plots. The remaining observed perturbation is mainly due to the structural reconfiguration of lipids and water around the charged groups, which slightly affects the helix termini. It is astonishing how stable the Arg and Lys structures are over time, even in position 1 with adjacent mutations in the hydrophobic membrane core.
TABLE 1.
Helical structure content and membrane thickness as a function of each amino-acid mutation introduced symmetrically in positions position 1–9 from the helix center
| No. of helical residues
|
Thickness
|
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AA | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | Avg |
| A | 18.8 | 18.8 | 18.8 | 18.8 | 18.8 | 18.8 | 18.8 | 18.8 | 18.8 | 2.72 | 2.95 | 2.82 | 2.60 | 2.77 | 3.02 | 2.78 | 2.88 | 3.00 | 2.84 |
| C | 18.8 | 18.6 | 17.9 | 18.9 | 18.8 | 18.8 | 18.6 | 18.6 | 18.6 | 2.70 | 3.21 | 3.67 | 3.59 | 3.36 | 3.66 | 3.47 | 3.34 | 3.53 | 3.39 |
| D | 15.5 | 13.4 | 13.2 | 18.4 | 11.0 | 14.0 | 18.0 | 13.5 | 15.3 | 3.31 | 3.56 | 3.47 | 2.98 | 2.91 | 3.45 | 2.04 | 2.79 | 3.24 | 3.08 |
| E | 18.9 | 10.5 | 13.0 | 18.9 | 16.8 | 16.3 | 18.7 | 17.7 | 14.0 | 3.30 | 3.45 | 3.32 | 3.28 | 3.36 | 3.20 | 3.40 | 3.56 | 3.36 | 3.36 |
| F | 18.9 | 18.8 | 18.9 | 18.3 | 19.0 | 18.6 | 18.9 | 19.0 | 19.0 | 2.80 | 3.37 | 3.49 | 3.49 | 3.17 | 3.42 | 3.67 | 3.35 | 3.62 | 3.37 |
| G | 18.3 | 18.9 | 18.8 | 18.9 | 18.9 | 18.9 | 18.5 | 19.0 | 18.0 | 2.94 | 3.52 | 2.93 | 2.44 | 2.96 | 3.02 | 2.35 | 2.10 | 3.03 | 2.81 |
| H | 18.9 | 18.5 | 18.9 | 18.7 | 18.9 | 18.9 | 18.8 | 18.8 | 18.8 | 3.62 | 3.49 | 3.40 | 3.40 | 3.54 | 3.66 | 3.20 | 3.64 | 3.45 | 3.49 |
| I | 18.8 | 18.8 | 18.9 | 18.9 | 18.8 | 17.9 | 18.1 | 18.1 | 18.5 | 3.51 | 3.48 | 3.64 | 3.62 | 3.53 | 3.07 | 3.15 | 2.42 | 3.07 | 3.27 |
| K | 17.7 | 11.2 | 17.1 | 18.0 | 16.2 | 18.2 | 18.9 | 17.7 | 17.1 | 2.88 | 3.27 | 3.41 | 3.39 | 3.37 | 3.49 | 3.11 | 3.29 | 3.70 | 3.33 |
| L | 18.5 | 18.9 | 18.6 | 18.9 | 18.7 | 19.0 | 18.5 | 18.8 | 18.9 | 3.60 | 3.57 | 3.91 | 3.40 | 3.75 | 3.49 | 2.68 | 2.82 | 3.28 | 3.39 |
| M | 18.6 | 18.9 | 18.9 | 18.9 | 19.0 | 18.6 | 18.5 | 17.8 | 18.9 | 3.71 | 3.25 | 3.19 | 3.10 | 3.61 | 3.86 | 3.45 | 3.60 | 3.50 | 3.47 |
| N | 18.6 | 17.9 | 18.9 | 17.9 | 18.6 | 18.8 | 19.0 | 18.1 | 18.3 | 3.48 | 3.76 | 3.09 | 2.89 | 3.26 | 3.62 | 3.40 | 3.25 | 3.70 | 3.38 |
| P | 18.9 | 17.5 | 18.9 | 18.4 | 16.4 | 17.2 | 18.7 | 17.6 | 19.0 | 3.34 | 3.36 | 3.69 | 3.46 | 3.74 | 3.52 | 3.67 | 3.00 | 3.31 | 3.45 |
| Q | 18.5 | 18.9 | 18.9 | 18.8 | 18.7 | 18.6 | 18.5 | 18.8 | 18.0 | 2.41 | 3.60 | 3.63 | 3.58 | 4.01 | 3.36 | 3.58 | 3.46 | 3.25 | 3.24 |
| R | 18.4 | 18.9 | 18.2 | 18.3 | 18.7 | 18.8 | 19.0 | 18.7 | 19.0 | 3.28 | 3.11 | 3.36 | 3.16 | 2.87 | 3.20 | 3.39 | 3.35 | 3.11 | 3.20 |
| S | 18.6 | 17.4 | 18.2 | 18.6 | 18.6 | 18.2 | 18.9 | 18.6 | 17.6 | 3.01 | 2.85 | 3.32 | 3.65 | 3.45 | 3.73 | 3.48 | 3.51 | 3.66 | 3.41 |
| T | 18.9 | 18.5 | 19.0 | 18.0 | 18.9 | 18.6 | 18.4 | 19.0 | 19.0 | 3.41 | 2.74 | 3.50 | 2.65 | 3.64 | 2.93 | 3.09 | 3.72 | 3.28 | 3.07 |
| V | 18.6 | 18.5 | 18.8 | 18.1 | 18.6 | 18.7 | 18.8 | 18.9 | 18.8 | 3.30 | 3.49 | 3.41 | 3.38 | 3.49 | 3.83 | 3.74 | 3.49 | 3.33 | 3.48 |
| W | 18.9 | 18.7 | 18.1 | 18.7 | 18.6 | 18.8 | 18.9 | 18.7 | 19.0 | 3.28 | 3.52 | 3.33 | 3.34 | 2.39 | 3.38 | 3.29 | 3.43 | 3.65 | 3.12 |
| Y | 18.9 | 18.0 | 18.8 | 18.9 | 18.0 | 19.0 | 17.7 | 17.9 | 18.8 | 3.31 | 3.56 | 3.01 | 3.51 | 3.37 | 3.75 | 3.38 | 3.76 | 3.17 | 3.42 |
The left part of the table (No. of helical residues) lists the average number of helical residues for each helix according to DSSP criteria, after the initial 14 ns of equilibration. The maximum possible number of helical residues in each sequence is 19. The right part (Thickness) indicates the local distortion/thickness of the membrane, measured as the average distance between the four lipid molecules closest to the helix on each side.
FIGURE 1.

Secondary structure of representative transmembrane helices as a function of simulation time, after equilibration. The mutation test residue is indicated to the left of each group together with helix sequence indices. The three graphs in a group correspond to symmetric substitutions into positions 1 (top), 3, and 5 (bottom) from the center of the helix. Of these, only aspartic acid substitutions result in any serious distortion, while the lysine-containing helices are remarkably stable even with two adjacent charged side chains in the hydrophobic core. Proline-mutated helices are slightly bent, but still within the α-helix region in the Ramachandran space.
The outcome of introducing the acidic residues Asp and Glu is, however, markedly different: these two side chains bend the helix backbone significantly, and in some cases stretch it to an extent where the helix secondary structure partially unwind to expose peptide bonds to the lipid environment, leaving only 10–13 residues in intact form. The table indicates that for these residues the helix is occasionally more intact when the mutations are introduced in the bilayer core, but this is purely a secondary effect from the major membrane deformation that enables water and/or lipid headgroups to permeate the hydrophobic core to screen the two proximate charges instead of stretching the helix to opposite sides.
An interesting question in this context is to what degree the membrane adapts its thickness around these polar or charged residues, in particular when their (semi-)terminal introduction results in shorter fully hydrophobic segments. We can define a local thickness from the distance between carbon atoms connecting the acyl chains in lipids on opposite sides of the membrane, and average this over the n lipids closest to the protein in each layer. The thickness results in Table 1 were calculated from n = 4, but virtually identical outcome is obtained in the range n = 5–8. Notably, the lack of trend or large variations indicates that while individual lipid headgroups close to a charged or polar side chain sometimes do penetrate the bilayer to solvate it, there is little or no systematic difference of local membrane thickness due to these mutations. This is not entirely unexpected due to the other side of the helix remaining clearly hydrophobic. The DMPC lipids in the present system were chosen to match the length of the helices; if lipids with shorter chains had been used, it is likely that the helix would naturally have adopted a tilted orientation, and if surrounded by lipids with longer chains it could be much harder for charged side chains to snorkel efficiently. It is an interesting question for future research whether this would result in more water entering the hydrophobic core, or a distortion/stretching of the helix secondary structure.
Fig. 2 displays simulation snapshots at 20 ns to highlight some of these effects: the length of the side chain as well as the basic hydrogen-bond donor group is pivotal for Lys, and to a somewhat lesser extent for Arg. It enables these residues to reach out and escape the hydrophobic core (so-called “snorkeling”) and form hydrogen-bonds with the deeply buried carbonyl oxygens even when located close to the center of the helix; note the virtually complete lack of lipid chain deformation. For the mutations where two strongly snorkeling groups such as Lys appear on opposite sides of the helix, the resulting torque can even tilt the entire helix 10–15°. In contrast, the acidic residues are both shorter and require hydrogen-bond donor partners rather than acceptors, i.e., water or choline groups. This explains the major stretching and deformation, which enables water or even lipid headgroups to enter the membrane to solvate the negative charges. Finally, the bulky aromatic rings appear to adapt to the lipid chain environment by ordering their plane along the membrane normal.
FIGURE 2.

Snapshots of simulated model systems with symmetric amino-acid substitutions in offset 5 from the helix center. From left to right: (i) Lysine residues exhibit significant snorkeling due to their length and flexibility, and form hydrogen bonds with carbonyl groups and water. (ii) Aspartic acid residues on the same side of the helix introduce major bending and results in notable distortion including headgroups/water inside the bilayer. (iii) Tyrosine (as other aromatic) side chains orient along the membrane-normal to intercalate efficiently with the lipid hydrocarbon chains.
Solvation structure, hydration, and hydrogen-bonding
The free energy cost of introducing polar groups into the hydrophobic bilayer is a combination of enthalpy loss from desolvation, and opposing entropic terms due to the perturbation of the hydrophobic core when introducing hydrophilic atoms. One important observation is that all charged, but also many polar, groups appear to attract nonnegligible amounts of water to partially preserve their hydration in the membrane, and hence more or less exist in a local solvated state with significant remaining enthalpy contributions rather than the classical view of a hydrophobic environment. Several cycles of water removal in the membrane were performed in the simulations to ensure these phenomena were not artifacts, but the same equilibrium amount of water around the amino acids reestablishes itself within a couple of nanoseconds, suggesting that intramembrane solvation water is indeed naturally occurring. There is a wide range of correlation times; water loosely associated with polar groups is exchanged with the bulk liquid on scales of 0.1–1 ns, while molecules forming hydrogen bonds directly to the side chains have residence times of 5–15 ns. The actual exchange rate can be even slower, since, after initial association, some of the latter waters are not exchanged at all with the bulk phase for the duration of the simulations. This was particularly true for buried charged residues.
The effective solvation environment for different classes of side chains is illustrated in Fig. 3 with radial distribution functions for a couple of different residues and positions. Such distribution functions are usually normalized to the average system density at long-range (bulk), but due to the anisotropic and inhomogenous membrane only relative magnitudes at shorter distances are meaningful here. By comparing Lys with Asp in the hydrophobic core (position 3, K3/D3 panels in Fig. 3), it is quite evident how the small positively charged group on Lys is interacting favorably with the deep lipid carbonyl groups, and is surrounded by well-ordered lipids (resolved peaks in the chain radial distribution functions). The acidic Asp has to rely almost exclusively on water to satisfy its solvation/hydration, which also distorts the membrane. Asp/Glu are occasionally interacting with positively charged choline groups in the lipid head, but solvating them entirely with penetrating headgroups in the hydrophobic core would not only be too costly entropically, but quite possibly rupture the bilayer. Closer to the membrane surface region, the Lys side chains can additionally form hydrogen bonds with oxygens in the phosphate group as acceptors, which explains the statistical preference for basic residues in multispanning membrane proteins to be exposed to the membrane in the headgroup region (54). In these positions, it is also easier for Asp/Glu to mix with the zwitterionic headgroups (not shown). Hydrophobic residues such as Met tend to interact only with the lipid chains irrespective of the position for the mutation (i.e., also when introduced at the interface), but has very limited effect on the membrane.
FIGURE 3.

Radial distribution of membrane and solvent groups around representative amino-acid side chains in different positions. The different groups shown are lipid heads (solid), carbonyls (dashed), the hydrocarbon chains (dot-dashed), and water (dot-dot-dashed). Densities are relative since there is no real bulk phase. Lysine in offset 3 from the helix center (top left) snorkel out to hydrogen-bond to the carbonyls, and to a lesser extent water. The same side chain in position 7 (top right) is equally surrounded by the large headgroups and carbonyls, and less dependent on water for a polar environment. Aspartic acid (lower left) cannot hydrogen-bond to carbonyls, and needs water/headgroups to interact with. Methionine is comfortable in the hydrophobic environment and does not distort the membrane.
The amount of hydration water retained around each residue was quantified by calculating the number of water molecules within a sphere of radius 0.5 nm centered on the mass center of each side chain and averaging over the trajectory, detailed in Table 2 along with the number of hydrogen bonds the side chains form with both water and lipids. The hydrogen-bond criteria used was an acceptor-donor distance of <0.35 nm, and bond angle below 30°. For most polar residues, it is sufficient with one or two waters to satisfy their hydrogen-bonding ability, with slowly increasing hydration levels as the mutation approaches the bilayer surface. In the innermost 1–2 positions (hydrophobic core), they lose both hydration water and lipid hydrogen bonds completely—which indicates the entropic cost of water penetration there is outweighing the desolvation enthalpy loss. There are, however, examples (predominantly charged side chains) where deeply buried residues pull in large amounts of water rather than being solvated by polar groups on lipids, usually accompanied with deformation of both lipids and helix. It is worth noting that the Berger force field employed does not use any partial charges for the acyl chains. The effect of them would be quite limited, since dipole-dipole interactions are weak, but if anything the present setup might slightly underestimate the amount of polar atoms in the hydrophobic core. Again, retained solvent was found to be particularly common for acidic residues, since their interactions with the positive lipid choline groups are quite weak due to the spread-out charge of the latter; the strength of choline interactions are similar to those of Cα hydrogen interactions in proteins.
TABLE 2.
Hydration levels around residues and number of hydrogen bonds to water/lipids
| No. of hydration waters
|
No. of hydrogen bonds to water/DMPC
|
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AA | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| A | – | – | – | – | – | – | 4.2 | 4.5 | 5.4 | −/− | −/− | −/− | −/− | −/− | −/− | 1/− | 1/− | */1 |
| C | – | – | – | – | – | – | 4.9 | 6.2 | 10.0 | −/− | −/− | −/− | −/− | −/− | −/− | 1/− | 1/− | 1/1 |
| D | – | 7.9 | 7.8 | 6.7 | 6.2 | 17.4 | 7.3 | 13.9 | 21.8 | −/− | 3/− | 3/− | 4/− | 3/* | 7/− | 4/1 | 6/* | 8/1 |
| E | 11.5 | – | 4.9 | 3.0 | 1.8 | 4.2 | 6.6 | 15.4 | 13.9 | 4/− | −/− | 3/− | 4/− | 2/− | 4/− | 5/1 | 7/− | 5/2 |
| F | – | – | – | – | – | 1.4 | 3.6 | 4.7 | 3.2 | −/− | −/− | −/− | −/− | −/− | 1/− | 1/− | 1/− | 1/1 |
| G | – | – | – | – | – | – | 3.0 | 6.1 | 10.3 | −/− | −/− | −/− | −/− | −/− | −/− | 1/− | −/− | 1/1 |
| H | – | – | – | – | 3.1 | 1.7 | 9.9 | 11.0 | 17.1 | −/− | −/− | */− | −/* | */− | 1/1 | 2/1 | 3/1 | 2/1 |
| I | – | – | – | – | – | – | 0.3 | 1.1 | 5.1 | −/− | −/− | −/− | −/− | −/− | −/− | 1/− | */− | 1/− |
| K | 0.5 | 0.1 | 1.5 | 1.5 | 5.7 | 4.0 | 2.8 | 13.7 | 16.1 | −/3 | */4 | 1/5 | 2/4 | 1/3 | 1/5 | 1/6 | 1/5 | 1/7 |
| L | – | – | – | – | – | – | 0.1 | 3.7 | 6.2 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | 1/− | 1/− |
| M | – | – | – | – | – | – | 4.3 | 7.0 | 7.3 | −/− | −/− | −/− | −/− | −/− | −/− | 1/− | 1/− | 2/* |
| N | – | 1.4 | 1.4 | * | 0.2 | – | 3.0 | 12.7 | 5.9 | −/− | −/− | 1/1 | −/− | −/3 | −/1 | 2/1 | 3/2 | 1/3 |
| P | – | – | – | – | 1.1 | – | * | – | 5.1 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | −/− | −/− |
| Q | – | – | – | – | 1.3 | 1.4 | – | 5.5 | 15.1 | −/− | −/− | −/− | −/− | 2/1 | 3/2 | −/2 | 2/2 | 3/3 |
| R | 3.9 | 16.9 | 10.2 | 8.9 | 3.7 | 1.2 | 16.1 | 8.5 | 13.6 | 5/2 | 2/7 | 5/3 | 2/4 | 2/3 | 2/3 | 4/6 | 2/7 | 3/7 |
| S | – | – | – | 5.1 | – | 1.7 | 0.8 | 10.3 | 15.8 | 1/1 | −/− | −/− | 1/1 | −/− | */1 | */1 | 2/− | 3/2 |
| T | – | – | – | 4.2 | – | 5.4 | 7.5 | 10.5 | 9.6 | −/− | −/− | −/− | 1/1 | −/− | 2/1 | 2/* | 2/1 | 3/* |
| V | – | – | – | 0.1 | * | – | – | 2.6 | 11.5 | −/− | −/− | −/− | −/− | */* | −/− | −/− | 1/− | 2/− |
| W | – | – | – | 0.2 | * | 2.2 | 5.7 | 2.0 | 4.7 | −/− | −/− | */1 | −/− | */1 | −/− | 2/* | 1/− | 1/1 |
| Y | – | – | 1.5 | – | 1.9 | 2.4 | 14.6 | 6.8 | 5.9 | 1/1 | −/− | 1/1 | -/1 | 1/1 | 1/2 | 2/2 | 2/2 | 1/2 |
The hydration level was quantified by the number of water molecules within a sphere of radius 0.5 nm from the side-chain mass center, reported as the sum for both mutated residues. For the hydrogen bonds, interactions with both carbonyl, phosphate, and choline groups are included, and the value rounded up. Asterisks denote nonzero values <0.1. The amount of water and the total number of hydrogen bonds are highly correlated and partly illustrate the same phenomenon, that charged or polar side chains strive hard to maintain their polar interactions and solvation enthalpy even inside the membrane.
It is quite instructive to compare the hydration level of the individual polar/charged residues with the amount of water actually entering the different regions of the membrane. We have accomplished this by calculating the z-dependent density curves for water, but to resolve the small variations from generic membrane undulation effects (40), the density calculation was restricted to a cylinder of radius 1.0-nm centered on the helix center of mass. Fig. 4 illustrates the densities for all mutations in positions 1, 3, and 5, and the small inset plots additionally indicate the integrated total amount of water penetrating into central 2.5 nm of the membrane for the remaining positions 2, 4, 6, and 8. The N-terminal side of the helix is oriented toward negative z-coordinates. In general, polar side chains are found to pull more water into the membrane the further in they are located, and the density is slightly higher on the N-terminal side for most residues. Still, the total amount of water in the actual hydrophobic core is negligible in nearly all cases. Lysine provides a remarkable example, with quite high hydration levels around the charged group as seen in Table 2, but the extremely efficient snorkeling makes it unnecessary to introduce much of this water in the membrane. For the two adjacent Glu mutations in position 1, the very high water content is due to several residues of the helix breaking up and exposing peptide groups to the environment, but this is much less pronounced in other positions for this residue. Also note how small polar side chains such as Ser and Thr do not retain any hydration at all when located in the hydrophobic core where the entropic cost would be too large, but gradually increase their hydration further out; the effect is particularly apparent for serine.
FIGURE 4.

Water density within a cylinder of a radius of 1.0 nm around the helix for different amino acids (single-letter code). Large plots display local density as a function of z-coordinate for substitutions in positions 1 (solid), 3 (dashed), and 5 (dot-dashed) from the helix center. Insets show the integrated average number of water molecules in the centermost 2.5 nm for the remaining systems, with substitution position on the horizontal and number of waters on the vertical axis. A regression is included to illustrate trends (dotted). Note how aspartic and glutamic acid retain large amounts of solvent, while arginine and most polar groups can hydrogen-bond to the deeper carbonyls. Lysine, in contrast, is extremely efficient at snorkeling and pulls in water only in position 1.
The N- versus C-terminal bias in side-chain hydration is clearly visible by separating the amount of hydration water for the two sides, and instead averaging over positions 1–8, as presented in Fig. 5. Virtually all amino acids in our simulations retain more water when located N-terminally, which agrees with snorkeling orientations determined from crystal structures (28) where most residues favor the N-terminal side. In fact, judging from the side-chain orientations discussed in the next section, it is likely the snorkeling bias that enable N-terminal residues to maintain higher hydration.
FIGURE 5.

Amount of hydration water within a 0.5-nm sphere from the side-chain mass center, separated for the N- and C-terminal mutations in the helix. The averaged values for the first eight positions on both sides are plotted for each amino acid. It is evidently easier to pull in water at the N-terminal rather than the C-terminal end of the helix, which agrees well with observed snorkeling abilities for most amino acids in the N-terminal direction.
From the hydration amounts in Table 2 it might be tempting to principally ascribe the cost of introducing polar/charged side chains in the membrane to desolvation and enthalpy loss, but that would be seriously misguided. As a counter-example, Fig. 6 shows the amount of hydrogen-bonds that Arg side chains are making to waters, headgroups, carbonyls, and even the rest of the helix backbone for different mutation positions. While there are large variations in the individual terms, the total number of hydrogen bonds is constant within a standard deviation over all positions, with an average four out of the five Arg side-chain donor hydrogens being paired. This normally also holds for polar residues such as Ser/Thr/Asn/Gln, since these side chains can share hydrogen bonds to preceding residues in the helix when located in the hydrophobic core. Thus, our results indicate that the solvation enthalpy might be reduced but certainly not lost, and entropic effects are very likely important, if not dominant, for introducing many hydrophilic amino acids in membrane helices.
FIGURE 6.

Hydrogen-bond distribution for arginine side chains. While the number of hydrogen-bonds to carbonyl groups (diamond), headgroups (cross), water (asterisk), and the rest of the helix (circle) vary appreciably, the total hydration level measured in number of hydrogen bonds (solid line) is essentially constant over the different depths in the membrane.
Classification of side-chain properties
As previously indicated, there are obvious systematic variations both in residue orientation and flexibility with the position, which we have quantified and summarized in Table 3. The orientation of each side chain was defined by the vector from the Cβ atom to the outermost heavy atom, which for obvious reasons eliminates alanine and glycine from the statistics. The average orientation and standard deviation of these vectors with respect to the membrane plane was calculated separately for the N- and C-terminal mutations. Fluctuations are clearly smaller for polar and charged residues, meaning they are not only oriented but also less mobile than their hydrophobic relatives in the interior of the bilayer. The charged residues snorkel to their respective side of the bilayer, with Lys showing extreme average angles up to 50° from the membrane plane. The large distortion and bending of the helix for the acidic residues Asp and Glu means their absolute orientation appears more random in the hydrophobic core, partly due to the unwinding of the backbone. Both Ser and Thr snorkel toward the N-terminal regardless of where the mutation is introduced, since this makes it possible to share hydrogen bonds with the residue four positions earlier in the helix. Hydrophobic residues like Leu and Met point back into the hydrophobic region when located at the surface (antisnorkeling), and in the hydrophobic core, their side chains have no prominent orientation preferences. Note how the N-/C-terminal bias is reproduced here too for, e.g., Lys and Arg; the snorkeling angles are somewhat larger on the N-terminal side, which brings them closer to the interface region as shown in Fig. 7.
TABLE 3.
Side-chain orientation and fluctuations relative to membrane plane
| Orientation N-terminal/C-terminal (mean ± SD) for each position
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| AA | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| C | 21/34(4) | 23/26(6) | 21/25(7) | 7/24(7) | 0/30(7) | −6/24(5) | −4/28(6) | 0/11(6) | 6/−1(4) |
| D | 10/31(3) | 30/30(4) | 55/−6(7) | 20/−12(8) | 20/−41(9) | 9/−33(9) | 25/−35(9) | 23/−39(9) | 24/−44(10) |
| E | 38/41(5) | 55/49(7) | 72/34(6) | 58/27(5) | 59/−9(6) | 56/−23(6) | 68/−42(5) | 49/−35(6) | 36/−27(8) |
| F | 10/−29(9) | 1/−14(6) | −21/−16(4) | −36/10(4) | −20/14(6) | 3/32(8) | 5/12(8) | −10/−5(6) | −20/−21(1) |
| H | −13/3(4) | 10/−5(6) | 19/6(7) | 10/−13(7) | −11/−3(8) | 7/−18(8) | 2/−11(6) | 4/−15(4) | −18/−13(2) |
| I | −17/−18(1) | −13/−9(4) | −13/−10(4) | −11/−8(4) | −1/−18(2) | 4/−18(2) | −6/−21(2) | −15/−21(2) | −7/−21(4) |
| K | 46/−48(2) | 47/−43(2) | 46/−45(2) | 21/−43(3) | 24/−44(3) | 20/−28(3) | 33/−23(3) | 32/−28(3) | 38/−43(2) |
| L | 33/30(7) | 20/28(8) | 21/27(8) | 28/24(9) | 35/25(6) | 10/28(6) | −2/18(8) | −17/20(9) | −14/17(7) |
| M | −7/17(8) | 5/16(6) | 25/18(6) | 21/30(8) | 12/34(8) | 1/9(8) | 3/18(6) | −6/12(6) | −23/32(7) |
| N | −18/−6(10) | −21/−19(7) | −19/−30(4) | 4/−29(5) | 11/−29(6) | 25/−23(5) | 17/−10(3) | 15/−9(3) | 3/−16(2) |
| P | 31/43(3) | 36/41(3) | 44/39(3) | 47/37(4) | 41/39(3) | 44/38(4) | 43/39(4) | 49/38(4) | 44/41(3) |
| Q | −25/37(4) | 3/31(5) | 30/18(6) | 39/20(6) | 21/6(6) | 1/−5(6) | −10/−16(6) | −1/−8(4) | 10/3(3) |
| R | 22/−16(3) | 28/−32(4) | 36/−32(5) | 43/−24(4) | 36/−11(5) | 41/−3(4) | 41/−15(5) | 45/−23(5) | 36/−43(4) |
| S | 28/38(3) | 35/32(3) | 39/28(4) | 34/26(4) | 32/28(5) | 40/28(6) | 42/30(6) | 40/29(6) | 30/29(5) |
| T | 30/34(7) | 34/28(4) | 47/32(7) | 35/36(6) | 21/35(4) | 37/23(7) | 45/30(8) | 37/27(7) | 36/30(7) |
| V | 0/3(3) | −6/−9(5) | −13/−22(5) | −9/19(6) | −15−18(4) | −18/−5(4) | −18/2(4) | −15/3(4) | −14/−9(2) |
| W | 33/−12(3) | 12/−8(3) | 4/0(3) | −13/26(2) | −13/10(3) | −3/12(4) | −2/−8(5) | 7/3(6) | −7/−9(6) |
| Y | 0/18(5) | 11/20(5) | 18/27(4) | 31/26(5) | 19/10(3) | 8/−11(4) | 23/−9(5) | 10/−9(4) | −1/−11(2) |
The orientation of each side chain is defined as the vector from Cβ to the outermost heavy atom; positive values denote side chains extending toward the N-terminal side, and negative toward the C-terminus. In each group, the first two numbers denote angles to the membrane plane for the mutation on the N-/C-terminal side, respectively, and the value in parentheses the average standard deviation.
FIGURE 7.

Snorkeling angles for N-/C-terminal mutations of methionine and arginine as a function of position. Positive values indicate side chains extending toward the N-terminal side. The hydrophobic side chains show little preference for any orientation, and are rather more flexible. Arginine, in contrast, shows obvious snorkeling, is more oriented, and exhibits N-/C-terminal bias; on average, the N-terminal snorkeling is 37°, while C-terminal only reaches 22°.
Basic side chains: lysine and arginine
The primary characteristic of all charged amino acids is that they snorkel appreciably, and pull in water and/or lipid headgroups to pair their hydrogen bonds. In the limited scale covered by the simulations, we did not observe any counterions systematically interacting with the side chains, but their small number makes it hard to draw any statistically certain conclusions. It is interesting to study the difference between Arg and Lys, where Lys side-chain mutations result in an almost intact bilayer structure while the Arg system is more affected. Due to the extended side chain and a small concentrated as well as oriented charged group, Lys can fully bury its polar atoms in the interface region by snorkeling even when the side chain is positioned in the center of the membrane. For the centermost positions, the side chain snorkels as much as 5.7 Å toward the interface regions, which corresponds to more than a full turn of the helix. Despite a maximum snorkeling distance of 5.3 Å for Arg, it snorkels less efficiently due to its two polar NH2 groups, which are considerably harder to simultaneously direct away from the nonpolar membrane and pack efficiently with the lipids than the single NH3 group in Lys. Normally, four out of five polar hydrogens in Arg form hydrogen bonds (Hε being unpaired), which leads to increased distortion when Arg retains more water. Due to the positive charge, Arg and Lys form hydrogen bonds both to carbonyl and phosphate groups of lipids, and since primarily the carbonyl group is located much further into the membrane than headgroups or water, binding to them significantly reduces the distortion of the bilayer and helix.
Acidic side chains: aspartate and glutamate
Comparable effects are observed for the acidic amino acids, but with considerably larger deformation of the system. The acidic side chains are too short to reach out to the interface region from the innermost positions (maximum observed snorkeling for Asp is 2.9 Å and for Glu, 4.3 Å). Apart from water, the negatively charged groups can only form hydrogen bonds with choline donors from the lipid headgroup. These are positioned much further out compared to the carbonyls, and the hydrogen bonds are also weaker due to the N(CH3)3+ group being a less potent donor, with strengths similar to Cα hydrogen bonds. This tends to favor water hydrogen bonds (frequently as salt bridges to the headgroups) for acidic residues, and accordingly larger distortion of the system.
Snorkeling for all charged residues is generally amplified in the N-terminal direction due to backbone geometry where the Cβ atom is directed toward the N-terminal. This bias is evident in the water density plots, with a pronounced increase on the N-terminal side. The varying potential of lipid headgroups and carbonyls as hydrogen-bond donors/acceptors depending on residue charge is intriguing, since it might provide a mechanism to control the type of proteins targeted to a particular membrane through its lipid composition, as recently reviewed by Lee (14).
Hydroxyl groups: serine and threonine
Serine and threonine are interesting exceptions to the rule that most polar residues snorkel toward the interface. Both these side chains have polar hydroxyl groups, which in our simulations orient to share the peptide oxygen four residues earlier in the helix as a hydrogen-bond acceptor, as illustrated in Fig. 8. This effectively pairs the side-chain's hydrogen, and is considerably more advantageous than paying the entropic cost of introducing lipids or water in the membrane core. The prevalent rotamer for all positions but the out-ermost for these two amino acids in our simulations is χ1 = – 60°, which is in accordance with results obtained by Chamberlain et al. (28). This means all Ser/Thr side chains are directed toward the N-terminal (supported by Table 3), and hence the amount of water and the degree of membrane distortion should be larger in the N-terminal direction, as confirmed by the water density plots in Fig. 4. Clusters of Cα–H···O hydrogen bonds have been found around these two amino acids and Gly in interfaces between transmembrane helices (55), implying their importance for helix dimerization. We believe it could be biologically significant that the cost of inserting these residues in the membrane is low enough for the insertion to occur without retained hydration water. They are transiently quite stable due to the backbone interaction, yet polar enough to prefer separate hydrogen bonds between residues on aggregated helices when given the opportunity instead of interacting with the backbone.
FIGURE 8.

Hydrogen-bonding networks of serine (left) and threonine (right) inside the bilayer. The hydroxyl group side chains, which are of great importance in helix-helix interactions, are relatively easy to solvate in the bilayer since they can form (shared) hydrogen bonds to the backbone oxygen of residue i–4. Hydrogen bonds are illustrated and distances shown in angstroms. This provides the hydroxyl group with a paired hydrogen bond, which is more advantageous than retaining polar groups like water.
Other polar: histidine, glutamine, and asparagine
The remaining polar amino acids tend to snorkel even in the innermost positions, up to 2.4 Å for Asn and 3.3 Å for Gln, which are values comparable to those of acidic residues. As the desolvation enthalpy loss is smaller than the entropic cost of retaining polar groups, the bilayer remains remarkably intact, which again is interesting considering the observed importance of these residues for driving helix aggregation (25). It is not until position 5 that the balance swings and they start retaining water, as can be seen in the densities plots, e.g., for Gln or His. Unlike the aromatic amino acids, His does not intercalate with the lipids but all three residues occasionally form hydrogen bonds to the helix backbone, just as Ser/Thr.
Hydrophobic or small side chains: Cys, Leu, Ile, Met, Val, Ala, and Gly
Structurally, these residues are mostly featureless in the sense that the system remains unaffected by the modified amino acids. The distribution of angles varies greatly with both position and time, since the side chains are comfortable in the lipid membrane and hence very flexible. There are some examples of antisnorkeling behavior (primarily Leu, Met) for the distal positions where the nonpolar side chains are oriented toward the hydrophobic part of the membrane. Still, they have another important function as unperturbed reference systems, and by comparing to the other residues it is, e.g., possible to conclude that the membrane thickness is virtually independent of the mutations.
Aromatic side chains: phenylalanine, tyrosine, and tryptophan
For all aromatic ring side chains we observe significant intercalation, i.e., they have clear propensity to align the ring plane parallel to the lipids chains, allowing for very efficient packing. To measure the degree of intercalation, the order parameters for the normal to the aromatic ring plane was used,
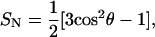 |
(1) |
where θ is the angle between the ring and membrane normal vectors. A value of 1.0 would mean the aromatic ring is horizontal, while −0.5 corresponds to vertical orientation. Both Phe and Trp exhibit very ordered rings for all positions, with average order parameters between −0.4 and −0.5, i.e., the rings are effectively fixed in vertical orientation between lipid chains. The innermost positions for Tyr show similar order parameters, but increasing slowly as the residue is placed further out in the helix. This trend is likely explained from the snorkeling of the polar Tyr when it directs the hydroxyl group toward the interface, which allows it to form hydrogen bonds with water/headgroups, and hence be positioned in the less ordered interface region where it is not necessary to intercalate. In contrast, the nonpolar Phe and Trp tend to antisnorkel for the outermost positions to solvate the aromatic rings in the lipid phase for all positions. The intercalation phenomenon seems to be an amazingly simple way for groups as bulky as Trp to be solvated in the membrane without any need for lipid distortion or significantly unfavorable entropy. There is further a double effect for Trp (and to some extent Tyr) to be locked in the interface region, since it simultaneously wants to direct its aromatic ring to intercalate in the hydrophobic core and nitrogen group toward the polar region, as illustrated in Fig. 9. This would explain why Tyr/Trp residues are so prevalent and useful as membrane helix anchors, as found in experimental studies (56).
FIGURE 9.

Efficient anchoring of tryptophan in the interface region. The tryptophan side chain strives to direct its polar nitrogen toward the interface region to pair its hydrogen bond; here, the direction and length of the hydrogen bond between this nitrogen and the carbonyl group of a lipid are shown. At the same time, the side chain wants to bury its aromatic ring in the membrane core and pack it efficiently between lipid chains by intercalation, which can also be seen from this plot. Similar effects are seen for Tyr.
Proline
Proline does not pull in any polar groups into the membrane. It is, however, well known as a helix breaker, and here it induces bending of the helix but no breakage of the backbone hydrogen bonds, which would be highly unfavorable since polar CO and NH groups would be exposed to the unpolar lipid environment. Both in theory and simulations this makes Pro quite hydrophobic and stable in membrane proteins, but since the helix bending is caused by local residue properties rather than the solvent environment it could affect the steric ability of helices to insert through the translocon.
DISCUSSION
A conspicuous observation for all amino-acid side chains studied here is how adaptive the membrane environment is as a solvent, in contrast to the two-dimensional hydrophobic solvent picture. There are essentially three different zones in the system, ranging from the fully hydrated headgroup region over the polar carbonyl groups to the mostly hydrophobic interior. As recently observed in simulations by Johnston et al. (20), membranes appear to be particularly efficient at stabilizing helices, in part due to the cost of exposing peptide bonds to the membrane (36). However, as soon as polar or charged groups are introduced in this environment, the lipid molecules reorient to satisfy hydrogen-bond pairing or ordering around bulky groups such as tryptophan. It is illustrative to think of an “effective” hydrophobic thickness: for residues such as Glu or Asp that only have limited ability to form hydrogen bonds with the lipids, the experienced membrane thickness will be close to the distance between the headgroup regions, or 3–4 nm. In the other extreme we find Lys, which not only can hydrogen-bond to the deeply located carbonyl groups, but the length of the side chain and the small hydrophilic group makes it remarkably efficient at snorkeling; it is really only in the middle 1–1.5 nm that this side chain is solvated in a hydrophobic surrounding.
Though not common inside bilayers, basic residues are critically important for some structures like KvAP ion channels, where they have been shown to bind hydration water and form salt bridges to lipids (22). The difference to acidic residues observed here is striking, in particular the significant helix distortion; it is well known that charged residues are enriched toward the surface region, but by comparing the relative occurrence of basic/acidic ones in membrane protein structures ((30); E. Granseth, 2006, personal communication) there appears to have been evolutionary pressure to select for positively charged side chains that interact favorably with the carbonyl groups in addition to the headgroups, as illustrated in Fig. 10. The different side chain-lipid interactions also suggests a possible mechanism for proteins to target different membrane compositions based on their sequence.
FIGURE 10.

Relative occurrence of acidic (solid) and basic (dashed) amino acids in membranes as a function of the z-coordinate, using statistics from 104 membrane proteins. The value z = 0 represents the center of the membrane, and the negative values are for the cytosolic side. The difference in basic versus acidic distributions after subtracting a linear positive inside trend (dot-dashed) shows two localized peaks for basic amino-acid positions. This agrees well with the carbonyl group density from our simulated systems (dotted), which indicates that the distribution might be heavily influenced by the ability to form hydrogen bonds with lipid carbonyls located deeper than the headgroups.
The atomic solvation properties provide valuable insight into why polar residues are so efficient at driving aggregation of helices (25). Superficially, charged residues should interact even better, but since these are mostly hydrated with hydrogen bonds formed even in bilayers, there is likely little relative difference in free energy. In contrast, the polar residues Asn/Gln/Ser/Thr are desolvated in the hydrophobic core, which means that any hydrogen bonding stabilizes aggregated conformations; experiments indicate values in the 2-kcal/mol range (24). This is smaller than a normal hydrogen bond, which is expected since the residues interact with the backbone in the nonaggregated state. This model is further substantiated by the position-dependence we observe: polar residues in the interface region do form hydrogen bonds with carbonyls and/or water, and corresponding experimental mutations are neither stabilizing nor destabilizing dimerization (57).
The intercalation of aromatic rings with lipid chains is a simple yet beautiful way of accommodating bulky groups in the membrane, as well as highly efficient headgroup anchoring when combined with polar groups. Interestingly, the effect is not at all prominent in studies of available membrane protein structures (28), which might be explained by these being solvated in less ordered detergents before crystallization. There are, however, a number of studies that have reported similar packing patterns for cholesterol (58), fluorescent probes (59), and disaccharides (60). Further, Aliste reports decreased mobility in simulations of Trp-containing decapeptides in lipid interfaces (18), which agrees well with our observations of more ordered states.
Comparing the level of hydration in simulations to the Hessa (37) and Wimley-White (36) hydrophobicity scales in Fig. 11 shows evident correlations. While this is mostly a qualitative observation, it strongly supports the idea that many side chains maintain significant hydration, and the free energy cost of introducing them in membrane helices could rather be due to entropic effects. The simulations also agree very well with the position-dependence in the biological hydropathy scale (37,38), with quite narrow, fully hydrophobic regions in the central bilayer, followed by a continuous trend as residues are positioned closer to the surface. It is intriguing that the simulations seem to agree somewhat better with the nonbiological scale (i.e., not involving translocons). Proline, for instance, which is important in many ion channels (61), appears quite expensive to insert in vivo, yet hydrophobic both in octanol and simulations. One possible explanation for this could be that, although hydrophobic enough, it is difficult to transport kinked helices through the narrow translocon channel. This hypothesis should be possible to test, either through simulations or with helices that spontaneously partition into membranes. The only other residues with significant differences are Asn and Gln, but in this case, we find no obvious reason why they should be harder to insert in vivo than the similar Ser/Thr.
FIGURE 11.

Comparison of the Hessa biological hydrophobicity scale and Wimley-White octanol scale with effective side-chain hydration (water within 0.5 nm) in the bilayer. While the latter is not a strict or linear hydrophobicity scale, it suggests that the free energy of insertion is largely due to entropic cost of maintaining the hydration for polar/charged residues, rather than enthalpic loss of it. Interestingly, Pro, Asn, and Gln appear more hydrophilic in vivo compared to both the Wimley-White scale and our hydration levels, which could indicate helix-helix or translocon interactions during insertion for helices containing these residues.
In closing, the observed hydrogen-bonding networks, snorkeling, intercalation, and helix interactions shows that membrane solvation is both specific and quite complex, and probably difficult to model accurately with implicit or simplified representation. Another side effect is that it is computationally very costly to perform all-atom free energy calculations, since intramembrane solvation water, helix, and membrane conformations need to be fully equilibrated in all intermediate states. Simulations of 30 ns are sufficient to give us an idea of the behavior of the system, but accurate free energies for charged side chains in helices could require an order of more data time for each state. Another factor influencing such quantitative measures is the uncertainty of protonation states, which is not always obvious (62). Nonetheless, these issues are not insurmountable, and resolving them will be important to decipher membrane protein folding.
Acknowledgments
We thank Tara Hessa and Gunnar von Heijne for stimulating discussions as well as model sequence data.
This work was supported by the Swedish Research Council, a Bio-X grant from the Swedish Foundation for Strategic Research, and computer resources provided by the Swedish National Allocations Committee.
References
- 1.Wallin, E., and G. von Heijne. 1998. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 7:1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Terstappen, G., and A. Reggiani. 2001. In silico research in drug discovery. Trends Pharmacol. Sci. 22:23–26. [DOI] [PubMed] [Google Scholar]
- 3.Popot, J. L., and D. M. Engelman. 1990. Membrane protein folding and oligomerization: the two-stage model. Biochemistry. 29:4031–4037. [DOI] [PubMed] [Google Scholar]
- 4.White, S. H., and G. von Heijne. 2005. Transmembrane helices before, during, and after insertion. Curr. Opin. Struct. Biol. 15:378–386. [DOI] [PubMed] [Google Scholar]
- 5.White, S. 2006. Membrane proteins of known structure. Http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html.
- 6.Dempsey, C. 1990. The actions of melittin on membranes. Biochim. Biophys. Acta. 1031:143–161. [DOI] [PubMed] [Google Scholar]
- 7.Blobel, G., and B. Dobberstein. 1975. Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J. Cell Biol. 67:835–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelman, D., Y. Chen, C.-N. Chi, A. Curran, A. Dixon, A. Dupuy, A. Lee, U. Lehnert, E. Matthews, Y. Reshetnyak, A. Senes, and J.-L. Popot. 2003. Membrane protein folding: beyond the two-stage model. FEBS Lett. 555:122–125. [DOI] [PubMed] [Google Scholar]
- 9.Tourasse, N. J., and W. H. Li. 2000. Selective constraints, amino acid composition, and the rate of protein evolution. Mol. Biol. Evol. 17:656–664. [DOI] [PubMed] [Google Scholar]
- 10.Arkin, I. T., and A. T. Brunger. 1998. Statistical analysis of predicted transmembrane α-helices. Biochim. Biophys. Acta. 1429:113–128. [DOI] [PubMed] [Google Scholar]
- 11.Henderson, R., J. M. Baldwin, T. A. Ceska, F. Zemlin, E. Beckmann, and K. H. Downing. 1990. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 213:899–929. [DOI] [PubMed] [Google Scholar]
- 12.Luecke, H., B. Schobert, H. T. Richter, J. P. Cartailler, and J. K. Lanyi. 1999. Structural changes in bacteriorhodopsin during ion transport at 2 Ångström resolution. Science. 286:255–261. [DOI] [PubMed] [Google Scholar]
- 13.Iwata, S., C. Ostermeier, B. Ludwig, and H. Michel. 1995. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 376:660–669. [DOI] [PubMed] [Google Scholar]
- 14.Lee, A. G. 2004. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 1666:62–87. [DOI] [PubMed] [Google Scholar]
- 15.Gumbart, J., and K. Schulten. 2006. Molecular dynamics studies of the archaeal translocon. Biophys. J. 90:2356–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White, S. H., and W. C. Wimley. 1999. Membrane protein folding and stability: Physical principles. Annu. Rev. Biophys. Biomol. Struct. 28:319–365. [DOI] [PubMed] [Google Scholar]
- 17.Hristova, K., C. E. Dempsey, and S. H. White. 2001. Structure, location, and lipid perturbations of melittin at the membrane interface. Biophys. J. 80:801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aliste, M., and D. P. Tieleman. 2005. Computer simulation of partitioning of 10 pentapeptides ACE-WLXLL at the cyclohexane/water and phospholipid/water interfaces. BMC Biochem. 6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bond, P. J., and M. S. P. Sansom. 2006. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 128:2697–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnston, J. M., G. A. Cook, J. M. Tomich, and M. S. P. Sansom. 2006. Conformation and environment of channel-forming peptides: a simulation study. Biophys. J. 90:1855–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monticelli, L., K. M. Robertson, J. L. MacCallum, and D. P. Tieleman. 2004. Computer simulation of the KVaP voltage-gated potassium channel: steered molecular dynamics of the voltage sensor. FEBS Lett. 564:325–332. [DOI] [PubMed] [Google Scholar]
- 22.Freites, J. A., D. J. Tobias, G. von Heijne, and S. H. White. 2005. Interface connections of a transmembrane voltage sensor. Proc. Natl. Acad. Sci. USA. 102:15059–15064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fleming, K. G., and D. M. Engelman. 2001. Specificity in transmembrane helix-helix interactions can define a hierarchy of stability for sequence variants. Proc. Natl. Acad. Sci. USA. 98:14340–14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gratkowski, L., J. D. Lear, and W. F. DeGrado. 2001. Polar side chains drive the association of model transmembrane peptides. Proc. Natl. Acad. Sci. USA. 98:880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou, R., B. J. Berne, and R. Germain. 2001. The free energy landscape for β hairpin folding in explicit water. Proc. Natl. Acad. Sci. USA. 98:14931–14936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hénin, J., A. Pohorille, and C. Chipot. 2005. Insights into the recognition and association of transmembrane α-helices. The free energy of α-helix dimerization in glycophorin A. J. Am. Chem. Soc. 127:8478–8484. [DOI] [PubMed] [Google Scholar]
- 27.Mottamal, M., J. Zhang, and T. Lazaridis. 2006. Energetics of the native and non-native states of the glycophorin transmembrane helix dimer. Proteins Struct. Funct. Gen. 62:996–1009. [DOI] [PubMed] [Google Scholar]
- 28.Chamberlain, A. K., Y. Lee, S. Kim, and J. U. Bowie. 2004. Snorkeling preferences foster an amino acid composition bias in transmembrane helices. J. Mol. Biol. 339:471–479. [DOI] [PubMed] [Google Scholar]
- 29.Chamberlain, A. K., and J. U. Bowie. 2004. Analysis of side-chain rotamers in transmembrane proteins. Biophys. J. 87:3460–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Granseth, E., G. von Heijne, and A. Elofsson. 2005. A study of the membrane-water interface region of membrane proteins. J. Mol. Biol. 346:377–385. [DOI] [PubMed] [Google Scholar]
- 31.Strandberg, E., S. Morein, D. T. S. Rijkers, R. M. J. Liskamp, P. r. C. A. van der Wel, and J. A. Killian. 2002. Lipid dependence of membrane anchoring properties and snorkeling behavior of aromatic and charged residues in transmembrane peptides. Biochemistry. 41:7190–8. [DOI] [PubMed] [Google Scholar]
- 32.Strandberg, E., and J. A. Killian. 2003. Snorkeling of lysine side chains in transmembrane helices: how easy can it get? FEBS Lett. 544:69–73. [DOI] [PubMed] [Google Scholar]
- 33.Deol, S. S., P. J. Bond, C. Domene, and M. S. P. Sansom. 2004. Lipid-protein interactions of integral membrane proteins: a comparative simulation study. Biophys. J. 87:3737–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kandasamy, S. K., and R. G. Larson. 2005. Molecular dynamics study of the lung surfactant peptide SP-B1–25 with DPPC monolayers: insights into interactions and peptide position and orientation. Biophys. J. 88:1577–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stockner, T., W. L. Ash, J. L. MacCallum, and D. P. Tieleman. 2004. Direct simulation of transmembrane helix association: role of asparagines. Biophys. J. 87:1650–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wimley, W. C., and S. H. White. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3:842–848. [DOI] [PubMed] [Google Scholar]
- 37.Hessa, T., S. H. White, and G. von Heijne. 2005. Membrane insertion of a potassium-channel voltage sensor. Science. 307:1427. [DOI] [PubMed] [Google Scholar]
- 38.Hessa, T., H. Kim, K. Bihlmaier, C. Lundin, J. Boekel, H. Andersson, I. Nilsson, S. H. White, and G. von Heijne. 2005. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 433:377–381. [DOI] [PubMed] [Google Scholar]
- 39.Heinrich, S., W. Mothes, J. Brunner, and T. Rapoport. 2000. The SEC61p complex mediates the integration of a membrane protein by allowing lipid partitioning of the transmembrane domain. Cell. 102:233–244. [DOI] [PubMed] [Google Scholar]
- 40.Lindahl, E., and O. Edholm. 2000. Mesoscopic undulations and thickness fluctuations in lipid bilayers from molecules dynamics simulations. Biophys. J. 79:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berger, O., O. Edholm, and F. Jähnig. 1997. Molecular dynamics simulation of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure and constant temperature. Biophys. J. 72:2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryckaert, J., and A. Bellemans. 1975. Molecular dynamics of liquid n-butane near its boiling point. Chem. Phys. Lett. 30:123–125. [Google Scholar]
- 43.Lindahl, E., and O. Edholm. 2001. Molecular dynamics simulation of NMR relaxation rates and slow dynamics in lipid bilayers. J. Chem. Phys. 115:4938–4950. [Google Scholar]
- 44.Benz, R., F. Castro-Roman, D. Tobias, and S. White. 2005. Experimental validation of molecular dynamics simulations of lipid bilayers: a new approach. Biophys. J. 88:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuler, L. D., X. Daura, and W. F. van Gunsteren. 2001. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J. Comput. Chem. 22:1205–1218. [Google Scholar]
- 46.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, and J. Hermans. 1981. Interaction models for water in relation to protein hydration. In Intermolecular Forces. B. Pullman, editor. D. Reidel Publishing Company, Dordrecht, The Netherlands. 331–342.
- 47.Lindahl, E., B. A. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Mod. 7:306–317. [Google Scholar]
- 48.Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- 49.Miyamoto, S., and P. A. Kollman. 1992. SETTLE: An analytical version of the SHAKE and RATTLE algorithms for rigid water models. J. Comput. Chem. 13:952–962. [Google Scholar]
- 50.Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8592. [Google Scholar]
- 51.Wohlert, J., and O. Edholm. 2004. The range and shielding of dipole-dipole interactions in phospholipid bilayers. Biophys. J. 87:2433–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berendsen, H. J. C., J. P. M. Postma, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 53.Kabsch, W., and C. Sander. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 54.Adamian, L., V. Nanda, W. F. DeGrado, and J. Liang. 2005. Empirical lipid propensities of amino acid residues in multispan alpha-helical membrane proteins. Proteins Struct. Funct. Gen. 59:496–509. [DOI] [PubMed] [Google Scholar]
- 55.Senes, A., I. Ubarretxena-Belandia, and D. M. Engelman. 2001. The Cα-H…O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc. Natl. Acad. Sci. USA. 98:9056–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Planque, M. R., B. B. Bonev, J. A. Demmers, D. V. Greathouse, R. E. Koeppe, F. Separovic, A. Watts, and J. A. Killian. 2003. Interfacial anchor properties of tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry. 42:5341–5348. [DOI] [PubMed] [Google Scholar]
- 57.Lear, J. D., H. Gratkowski, L. Adamian, J. Liang, and W. F. DeGrado. 2003. Position-dependence of stabilizing polar interactions of asparagine in transmembrane helical bundles. Biochemistry. 42:6400–6407. [DOI] [PubMed] [Google Scholar]
- 58.Hofsäss, C., E. Lindahl, and O. Edholm. 2003. Molecular dynamics simulations of phospholipid bilayers with cholesterol. Biophys. J. 84:2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Repáková, J., J. M. Holopainen, M. Karttunen, and I. Vattulainen. 2006. Influence of pyrene-labeling on fluid lipid membranes. J. Phys. Chem. B. 110:15403–15410. [DOI] [PubMed] [Google Scholar]
- 60.Sum, A. K., R. Faller, and J. J. de Pablo. 2003. Molecular simulation study of phospholipid bilayers and insights of the interactions with disaccharides. Biophys. J. 85:2830–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woolfson, D. N., R. J. Mortishire-Smith, and D. H. Williams. 1991. Conserved positioning of proline residues in membrane-spanning helices of ion-channel proteins. Biochim. Biophys. Acta. 175:733–737. [DOI] [PubMed] [Google Scholar]
- 62.Varma, S., S.-W. Chiu, and E. Jakobsson. 2006. The influence of amino acid protonation states on molecular dynamics simulations of the bacterial porin OmpF. Biophys. J. 90:112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]