

Abstract
Chromatin assembly is condensed by histone tail-tail interactions and other nuclear proteins into a highly compact structure. Using an optical trap modulation force spectroscopy, we probe the effect of tail interactions on local chromatin fluidity. Chromatin fibers, purified from mammalian cells, are tethered between a microscope coverslip and a glass micropipette. Mechanical unzipping of tail interactions, using the micropipette, lead to the enhancement of local fluidity. This is measured using an intensity-modulated optically trapped bead positioned as a force sensor on the chromatin fiber. Enzymatic digestion of the histone tail interactions of tethered chromatin fiber also leads to a similar increase in fluidity. Our experiments show that an initial increase in the local fluidity precedes chromatin decompaction, suggesting possible mechanisms by which chromatin-remodeling machines access regulatory sites.
INTRODUCTION
Chromatin structure in a eukaryotic cell nucleus is organized by various histone proteins to achieve a packing ratio of the order of 105 (1). The nucleosome assembly, the fundamental unit of the chromatin, is formed by the winding of ∼146 bp of DNA around the 10-nm histone octamer complex driven primarily by the electrostatic interactions (2,3). The core and linker histone tail interactions further compact the nucleosome array into higher order chromatin fibers (4–6). The N-terminal domain (NTD) of core histone H4 on one nucleosome interacts with a charged patch on the surface of core histone H2A on adjacent nucleosome and is necessary for 30-nm chromatin secondary structure formation (7). The 30-nm fiber and its higher order structures are further stabilized by the linker histones, which consist of a globular winged helix domain, a short unstructured NTD, and an unstructured C-terminal domain (CTD) of ∼100 amino acid residues that are highly basic (8–13). Digestion of the tail regions by limited trypsinization has revealed nucleosomal instability and decompaction of the chromatin fibers (14–16).
Access to DNA requires the disruption of the chromatin assembly, which is achieved in vivo by a complex set of histone-modifying and chromatin-remodeling enzymes (17). The histone-modifying enzymes change the local charge on the nucleosome by acetylation, methylation, or other modifications of specific residues on the histone tails and hence alter the nucleosome stability (18,19). The chromatin-remodeling enzymes are known to physically remodel the nucleosomes in an ATP-dependent manner (1), the specific mechanisms of which are yet to be elucidated. Both of the above groups of enzymes are responsible for altering the chromatin structure and are recruited specifically to chromatin regions required to be decondensed. In vivo, regions of condensed and decondensed chromatin states are actively maintained, and altered when required, by various proteins that tune the local fluidity and hence the accessibility of the DNA to proteins. Mechanical unfolding experiments on chromatin fibers have provided a measure of the forces that stabilize the nucleosome arrays and its higher order structure (20–23). In addition, unfolding experiments have demonstrated the role of histone tails and their modifications in the stability of the chromatin structure (24).
In this article we provide, for the first time to our knowledge, a map of the local fluidity of higher order chromatin structure. For this a micropipette-based manipulation method is used to disassemble chromatin isolated from HeLa cells, as shown in Fig. 1. A phase-sensitive optical trap modulation force spectroscopy technique is developed to probe the local chromatin fluidity as a function of its decompaction. The local fluidity displays an initial increase followed by a reduction upon unfolding the chromatin fiber by mechanical tension. At a fixed unfolded state, trypsin digestion of the chromatin fiber leads to similar enhancement in local fluidity.
FIGURE 1.

Cartoon of the experimental geometry, where the micropipette (tip size ∼0.5 μm) is used to pull out chromatin fibers from isolated chromatin adhered onto a poly-D-lysine-coated glass coverslip. The optically trapped bead is adhered onto the chromatin fiber nonspecifically. Inset shows a fluorescence image of the chromatin fiber pulled out from an isolated chromatin piece (with the exogeneous H2B-EGFP protein), using a micropipette.
MATERIALS AND METHODS
Intensity-modulated optical trap
The optical trap was built on an inverted microscope (Model IX70 Olympus, Tokyo, Japan) using a current-controlled 830-nm diode laser (GaAlAs diode, model 5430; SDL, San Jose, CA; and controller model LDC-3724B; ILX Lightwave, Bozeman, MT) as described in Roopa et al. (25). A 5-mW diode laser, 635 nm (model 31-0128; Coherent, Auburn, CA) was used to track the bead position in the trap by imaging the back-scattered light onto a photodetector. The photodetector current was amplified by two low-noise current preamplifiers (SR570, Stanford Research Systems, Sunnyvale, CA), the difference in the voltages giving the position of the bead from the trap center. A function generator (DS345, Stanford Research Systems) was used to modulate the controller current, hence periodically varying the trap potential. The voltage outputs of the two preamplifiers were used as input to the lock-in amplifier (Model SRS530, Stanford Research Systems), locked to the modulating signal by a TTL synchronous pulse from the function generator. The lock-in amplifier convolutes the signals with the reference signal to give the phase-sensitive response of the bead at the frequency of modulation. The time series of the amplitude and phase of the bead fluctuations at the modulation frequency were measured (200 points at acquisition rate of 3 Hz; the lock-in low pass filter time constant being 300 ms). Data acquisition and analysis were done using a data acquisition board (PCI-MIO-16XE-10, National Instruments, Austin, TX) and LabView (Version 5.1, National Instruments).
Chromatin extraction from He-La cells
He-La cells were stably transfected with histone H2B-EGFP fusion protein, with Lipofectamine-2000 (Invitrogen, Carlsbad, CA) and Blasticidin as the selection drug (1 μg/ml of culture medium—Dulbecco's Modified Eagle's Medium (Gibco, Grand Island, NY) with 5% fetal bovine serum (Gibco)) in an incubator maintained at 37°C temperature and with 5% CO2. For chromatin extraction, the freshly harvested cells were washed with M1 buffer (50 mM of Tris-Cl pH 7.5, 100 mM of MgCl2, 100 mM of NH4Cl, and 4% w/v of PEG3350) and centrifuged (1500 × g, 20 min). The cells were mechanically sheared using an insulin syringe (30-gauge needle) to lyse the cell membrane, and the nuclei were centrifuged (13,400 × g, 5 min). The nuclei were resuspended in M1 buffer and sonicated (5 min) to obtain the chromatin pieces. The chromatin pieces were then sorted from the remaining debris in a Fluorescence Assisted Cell Sorter (BD FACSVantage SE System, BD, Rockville, MD), using the (488-nm excitation, 520-nm emission) fluorescence of the exogenous H2B-EGFP fusion protein. The chromatin samples were stored at 4° in phosphate buffer saline buffer (1×, pH 7.4) and used over a week.
The experiments were performed in 50 mM NaCl solution; the chromatin is known to exist mostly as bundles of 30-nm fibers at this salt concentration. The chromatin sample was adhered onto a poly-D-Lysine-coated coverglass and mounted on the microscope. The fluorescence of the H2B-EGFP, shown in Fig. 1, was used to identify a chromatin blob free of debris, and the sample was rinsed with 50 mM NaCl before the experiment. A 0.5-μm tip sized micropipette coated with poly-D-Lysine and mounted on a motorized stage (ESP300 Newport, Irvine, CA) was used to pull out the fibers from the chromatin. Fig. 1 shows the experimental arrangement to probe the coupling between internucleosomal interactions and the local fluidity.
Estimation of chromatin fiber stiffness
In Fig. 2 we compare the position distributions of the trapped bead in solution with that of a bead adhered onto the chromatin fiber. The standard deviation of the position histograms was used to calculate the optical trap stiffness and the effective stiffness of the chromatin fiber. The values of the standard deviation are ∼39 nm for a free bead in solution and ∼8 nm for the bead adhered onto the fiber under an extension of ∼8 μm. The effective stiffness can be calculated by assuming the chromatin-trap system as springs in parallel and using 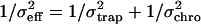 , where σeff = 8 nm is the effective position standard deviation of the bead in the chromatin-trap system, σtrap = 39 nm is the position standard deviation of the bead in the trap, and σchro is the position standard deviation of the bead due to chromatin fiber. Using this we estimated the ktrap ∼ 2.6 × 10−6 N/m, keff ∼ 0.63 × 10−4 N/m, and the kchro to be ∼0.59 × 10−4 N/m. In the inset to Fig. 2 we plot the position distributions for a trapped bead adhered onto the chromatin fiber at two different extensions of the fiber to show that the conventional optical trap is less sensitive to decipher subtle changes in local fluidity of chromatin.
, where σeff = 8 nm is the effective position standard deviation of the bead in the chromatin-trap system, σtrap = 39 nm is the position standard deviation of the bead in the trap, and σchro is the position standard deviation of the bead due to chromatin fiber. Using this we estimated the ktrap ∼ 2.6 × 10−6 N/m, keff ∼ 0.63 × 10−4 N/m, and the kchro to be ∼0.59 × 10−4 N/m. In the inset to Fig. 2 we plot the position distributions for a trapped bead adhered onto the chromatin fiber at two different extensions of the fiber to show that the conventional optical trap is less sensitive to decipher subtle changes in local fluidity of chromatin.
FIGURE 2.

Histograms for the trapped bead position fluctuations in solution (open stars) and a trapped bead adhered onto the chromatin fiber pulled out with the micropipette (open circles). The position time series is acquired at a sampling rate of 5000 Hz. The Gaussian fits give the standard deviation values of 39 nm for trapped bead in solution (open stars) and 8 nm for trapped bead adhered onto the chromatin fiber (open circles). Inset shows position distribution of the trapped bead adhered to the chromatin fiber at extensions of ∼10 μm (open circles) and ∼20 μm (open stars). The distributions are fit to Gaussian functions and show no variation in the standard deviation values for the above extensions.
RESULTS
Optical trap modulation force spectroscopy as a probe of local viscosity
We have developed a sensitive method to map differential changes in viscosity using an intensity-modulated optical trap. This enabled us to decipher the small variations in chromatin fluidity arising due to structural changes. In this method the trap stiffness was modulated by sinusoidal modulation of the trapping laser intensity (see Materials and Methods). The lock-in amplifier gives the imaginary (VI) and real (VR) components of the response at the modulating signal frequency from which the amplitude 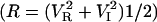 and the phase (P = tan−1 (VI/VR)) of the bead oscillation were calculated (25,26). Response and phase time series were measured for a trapped bead in solutions of varying viscosities achieved by mixing an appropriate glycerol concentration in water. We find that the standard deviation of the amplitude and phase time series is a measure of the local viscosity and sensitivity when compared to the position fluctuation distributions of the trapped bead. The amplitude output, R, of the lock-in amplifier gives the strength of the bead fluctuations at the modulating frequency. The mean amplitude decreases with increasing solution viscosities, as expected due to greater damping (data not shown) (25,26). The mean phase and the standard deviation of the phase histograms for varying solution viscosities are plotted in Fig. 3. The phase standard deviation (PSD) is ∼30° for a 2-μm diameter polystyrene bead in water (γ = 0.3 × 10−7 Ns/m) in a potential well of stiffness ktrap = 2.6 × 10−6 N/m, the amplitude of modulation k0 being ±0.8 × 10−6 N/m (at 100-Hz frequency). With increase in viscosity, γ = 1.7 × 10−7 Ns/m (40% glycerol in water) the PSD increases to ∼70° (Fig. 3).
and the phase (P = tan−1 (VI/VR)) of the bead oscillation were calculated (25,26). Response and phase time series were measured for a trapped bead in solutions of varying viscosities achieved by mixing an appropriate glycerol concentration in water. We find that the standard deviation of the amplitude and phase time series is a measure of the local viscosity and sensitivity when compared to the position fluctuation distributions of the trapped bead. The amplitude output, R, of the lock-in amplifier gives the strength of the bead fluctuations at the modulating frequency. The mean amplitude decreases with increasing solution viscosities, as expected due to greater damping (data not shown) (25,26). The mean phase and the standard deviation of the phase histograms for varying solution viscosities are plotted in Fig. 3. The phase standard deviation (PSD) is ∼30° for a 2-μm diameter polystyrene bead in water (γ = 0.3 × 10−7 Ns/m) in a potential well of stiffness ktrap = 2.6 × 10−6 N/m, the amplitude of modulation k0 being ±0.8 × 10−6 N/m (at 100-Hz frequency). With increase in viscosity, γ = 1.7 × 10−7 Ns/m (40% glycerol in water) the PSD increases to ∼70° (Fig. 3).
FIGURE 3.

Mean phase and the standard deviation of phase time series (PSD) of trapped bead plotted for varying solution viscosities. The optical trap stiffness (ktrap = 2.6 × 10−6 N/m) is sinusoidaly modulated (k0 = 0.8 × 10−6 N/m), and the bead fluctuations are measured by using a back-scattered red laser (635 nm). The back-scattered laser is imaged onto a photodiode partitioned into two halves to give two voltages proportional to the intensity of the laser on each of the partitions. The above voltages, amplified using low-noise preamplifiers, were input to the lock-in amplifier, which is frequency locked at the trap modulation frequency (100 Hz). The lock-in amplifier gives the amplitude and phase of the bead position at the trap modulation frequency (the time constant of the low pass filter in the lock-in amplifier = 300 ms). Data from the lock-in amplifier are acquired using a PCI board at a sampling rate of 3 Hz.
Mapping local chromatin fluidity using trap modulation
The internucleosomal interactions, mediated by the core and linker histone protein tails, determine the local stability of the chromatin assembly. Mechanical tension applied on the chromatin leads to unfolding of the chromatin fiber due to the breaking of the internucleosomal interactions and hence could modulate the local viscoelasticity. To decipher these structural changes in the chromatin, we used the optical trap modulation technique as a local probe of fluidity combined with micromanipulation to mechanically unfold the chromatin assembly (Fig. 1). The trapped bead in the modulated potential was adhered onto the chromatin fiber, and the response and phase was measured as the chromatin was unfolded by mechanical tension. The representative phase distributions with increase in applied tension are shown in Fig. 4 a. In Fig. 4 b we plot the PSD as a function of chromatin fiber extension. With tension, the chromatin structure is unfolded due to disruption of the internucleosomal interactions in the chromatin, resulting in increase in the relative viscosity and hence the PSD. We observed a nonmonotonous enhancement in the PSD as a function of tether extension. The PSD increases from ∼2° at an extension of ∼8 μm to ∼4° at ∼25 μm. Further increase in tether extension (∼25–50 μm) leads to a decrease in the PSD. The PSD remains constant for larger extensions >50 μm. Extension of the tether length beyond 80 μm results in rupture of the chromatin fiber. The error bars in the curves have been obtained over six experiments. The inset to Fig. 4 b shows a similar experiment on chromatin isolated from apoptotic cells (see Discussion).
FIGURE 4.

(a) Histogram of phase time series of trapped bead adhered onto the chromatin fiber pulled out with the micropipette. The unnormalized histograms with five-point adjacent averaging are plotted for increasing extensions of the chromatin fiber: ∼10 μm (open circles) to ∼20 μm (open stars). The trap stiffness was 2.6 × 10−6 N/m with a change in stiffness of ±0.8 × 10−6 N/m due to modulation at 100 Hz. The lines joining the symbols represent Gaussian fits to the probability distributions giving standard deviations of 16° for 10 μm and 25° for 20 μm extensions of the chromatin fiber. (b) Standard deviation of the phase time series of trapped bead adhered onto the chromatin fiber pulled out with the micropipette as a function of increasing extension. The chromatin fiber was extended up to ∼80 μm beyond which the fiber ruptured. An initial increase was observed in the PSD calculated from the phase time series of the lock-in output (sampling rate was 3 Hz with the lock-in time constant set at 300 ms). The line joining the symbols represents spline fit to the data. Inset shows PSD as a function of extension of chromatin isolated from apoptotic cells. The maximum length the fiber can be extended before rupture is ∼20 μm. The line joining the symbols represents spline fit to the data.
Trypsin digestion of chromatin leads to enhanced chromatin fluidity
In the previous section, internucleosome interactions and hence the chromatin structure was disrupted by mechanical tension. Here we use enzymatic unfolding of tethered chromatin fiber held at fixed tension to test if the internucleosomal interactions determine the local chromatin fluidity. Trypsin digestion of histone tails are known to decondense chromatin due to disruption of its higher order organization (14,15). This is analogous to tension-induced decondensation of the chromatin higher order structure. In this experiment trypsin was added in the sample well and the response and phase time series were measured with the fiber length held constant at ∼10 μm, hence fixing the tension. The phase histograms were plotted for different time points after addition of trypsin as shown in Fig. 5. Decondensation leads to loosening of the fiber and an increase in the PSD of the trapped bead adhered onto the fiber. The PSD increases by ∼20°, over many experiments, after incubating with trypsin for ∼10 min. Representative distributions at t = 0 min and t = 10 min are shown in Fig. 5. This increase in local fluidity is analogous to the elastic-viscous transition observed with mechanical tension applied on the chromatin. The inset to Fig. 5 shows the typical step-like jumps (marked by arrows) in the mean position of the bead in the trap, which reflects strand breakages due to trypsin digestion. We observe that beyond 10 min of digestion the fiber length increases and is finally ruptured. These results directly suggest the contribution of internucleosomal tail interactions to chromatin rigidity and its fluidity.
FIGURE 5.

Histograms of the phase time series of a bead adhered onto the chromatin fiber being digested with trypsin. A bundle of chromatin fiber is pulled out to a length of ∼10 μm using the micropipette, and an optically trapped bead is adhered onto it, trap stiffness being 2.6 × 10−6 N/m with a change in stiffness of ±0.8 × 10−6 N/m due to modulation at 100 Hz. The figure compares the histograms before trypsin digestion and at ∼10 min after trypsin addition in the sample well. At later time points the chromatin fiber relaxes, indicating an increase in length, and is eventually ruptured. The lines joining the symbols represent Gaussian fits to the distributions, the standard deviations of the distribution being 16.5° and 7.5° for the two time points. Inset shows a typical position time series of the bead adhered to the chromatin, held at a constant length of ∼10 μm and trypsin digested. There are sudden jumps in the bead position, indicating strand breakages.
DISCUSSION
The 30-nm higher order chromatin structure is maintained by the interactions mediated by the core histone and linker histone terminal regions (6–12). Remodeling of the 30-nm fiber requires decompaction of the chromatin assembly and this is achieved in vivo by ATP-dependent remodeling enzymes and the modification enzymes (17–19). Mechanical disruption of in vitro reconstituted nucleosomal arrays using optical tweezers revealed a reversible multistage release of DNA (20–23). Further, micromanipulation of chromatin devoid of the terminal tails of the histones revealed lower rupture forces (24). Acetylation of specific residues on the terminal regions of the histones also resulted in lower rupture force, indicating that the tail interactions determine the higher order chromatin structure (24). In this work the decompaction of higher order chromatin structure was achieved by both mechanical tension and enzymatic digestion of the internucleosomal interactions. Using a trapped bead adhered onto the fiber as a sensor, the chromatin structural change as a function of tension and trypsin digestion was measured.
The standard deviation values of position distributions of a bead in a conventional optical trap decrease with increasing solution viscosities. These distributions are, however, not sensitive to small changes in local viscosity when the bead is adhered onto the fiber, which provides a large stiffness as compared to the trap. To measure the small fluidity changes resulting from chromatin structural decompaction, we developed an optical trap modulation method, where the intensity and hence the trap stiffness was modulated. Although spatially modulated optical traps have been used to measure the viscoelastic properties (27–29), the measurements are homogeneous, assuming no nonspecific adhesion of the trapped bead to the viscoelastic media. Hence spatially modulated optical trap is not appropriate when the trapped bead is firmly adhered to the sample, as is the case in our experiments.
The phase time series was measured for solutions of varying viscosity to characterize the method. The PSD changes from ∼30° to ∼70° for a sixfold change in solution viscosity, providing a large dynamic range. The PSDs were sensitive to the local chromatin fluidity changes, as compared to that of the amplitude histograms. The measured changes in PSD for chromatin unfolded with tension is <8° in all our experiments. Our results reveal a regime where the nucleosomal arrays are more mobile when the histone tail interactions are disrupted by tension, as shown in the model in Fig. 6. We observed similar behavior with tension-induced decompaction of chromatin isolated from apoptotic cells, though in this case the length to which the fiber could be extruded before fiber rupture was at least fourfold smaller than the length achieved in normal samples (inset to Fig. 4 b). To verify this regime of increased fluidity achieved by decompaction, we fixed the tension on the chromatin fiber and induced decompaction by trypsin digestion of the histone tails. When the chromatin fiber is digested with trypsin, while the fiber length and hence the stiffness is held constant, the fluidity increases with time. Eventually the fiber stiffness decreases as the excess DNA gets released as a consequence of decompaction, leading to larger changes in PSD unlike the case of tension-induced decondensation.
FIGURE 6.

Schematic showing the tension-induced rupture of the histone tail interactions leading to decompaction of the chromatin assembly. For a fixed tension, trypsinization of the histone tails results in a similar increase in the fluidity of the chromatin.
Our experiment takes advantage of the fluctuations in the measured phase lag signal to probe the viscoelastic coupling of the trapped bead with the surrounding fluid and the chromatin fiber. The trapped bead response is dominated by the chromatin fiber as is clear when we compare the standard deviation values of the position histograms of the trapped bead in solution (standard deviation of 39 nm) and trapped bead adhered onto the chromatin fiber (standard deviation of 8 nm). As a function of chromatin extension or the enzymatic disruption, the change in the distribution in phase lag (PSD) measures the changes in the viscoelastic coupling of the bead with the chromatin fiber. The fact that we observe an initial increase and a decrease in PSD as a function of chromatin extension suggests that both the viscous and elastic part of the trapped bead coupling to chromatin is altered. This could be either due to changes in unharmonic elasticity of the chromatin fiber or the viscoelastic coupling between the bead and the chromatin fiber surrounded by the fluid.
In summary the initial loosening of chromatin fiber upon disruption of tail-tail interactions suggests a mechanistic basis for chromatin remodeling in vivo by remodeling enzymes. The current understanding of chromatin remodeling involves charge modifications, for example acetylation, of specific residues on the histone tails by chromatin-modifying enzymes (17). The modifications result in weakening of the interactions between adjacent nucleosomal tails and loosening of nucleosomal arrays (30,31). The remodeling enzymes, like the SWI-SNF complexes, could use the loosened chromatin fiber as substrates for physical remodeling of DNA around the histone octamer (19). Our work presents a methodology to measure such small changes in chromatin fluidity and could be applied to study chromatin structural changes achieved by remodeling enzymes in vitro.
Acknowledgments
We thank the NCBS flow-cytometry facility for chromatin sample preparation.
We thank the Nanomaterials Science & Technology Initiative of the Department of Science & Technology, Government of India for financial support.
References
- 1.Khorasanizadeh, S. 2004. The nucleosome: from genomic review organization to genomic regulation. Cell. 116:259–272. [DOI] [PubMed] [Google Scholar]
- 2.Dorigo, B., T. Schalch, A. Kulangara, S. Duda, R. R. Schroeder, and T. J. Richmond. 2004. Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science. 306:1571–1573. [DOI] [PubMed] [Google Scholar]
- 3.Schalch, T., S. Duda, D. F. Sargent, and T. J. Richmond. 2005. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 436:138–141. [DOI] [PubMed] [Google Scholar]
- 4.Luger, K., and J. C. Hansen. 2005. Nucleosome and chromatin fiber dynamics. Curr. Opin. Struct. Biol. 15:188–196. [DOI] [PubMed] [Google Scholar]
- 5.Zlatanova, J., and S. H. Leuba, editors. 2004. Chromatin Structure and Dynamics: State-of-the-Art. Elsevier, Amsterdam, The Netherlands.
- 6.Luger, K., and T. J. Richmond. 1998. The histone tails of the nucleosome. Curr. Opin. Genet. Dev. 8:140–146. [DOI] [PubMed] [Google Scholar]
- 7.Dorigo, B., T. Schalch, K. Bystricky, and T. J. Richmond. 2003. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 327:85–96. [DOI] [PubMed] [Google Scholar]
- 8.Hendzel, M. J., M. A. Lever, E. Crawford, and J. P. Th'ng. 2004. The C-terminal domain is the primary determinant of histone H1 binding to chromatin in vivo. J. Biol. Chem. 279:20028–20034. [DOI] [PubMed] [Google Scholar]
- 9.Carruthers, L., and J. C. Hansen. 2000. The core histone N termini function independently of linker histones during chromatin condensation. J. Biol. Chem. 275:37285–37290. [DOI] [PubMed] [Google Scholar]
- 10.Fletcher, T. M., and J. C. Hansen. 1995. Core histone tail domains mediate oligonucleosome folding and nucleosomal DNA organization through distinct molecular mechanisms. J. Biol. Chem. 270:25359–25362. [DOI] [PubMed] [Google Scholar]
- 11.Schwarz, P. M., A. Felthauser, T. M. Fletcher, and J. C. Hansen. 1996. Reversible oligonucleosome self-association: dependence on divalent cations and core histone tail domains. Biochemistry. 35:4009–4015. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Ramirez, M., F. Dong, and J. Ausio. 1992. Role of the histone “tails” in the folding of oligonucleosomes depleted of histone H1. J. Biol. Chem. 267:19587–19595. [PubMed] [Google Scholar]
- 13.Angelov, D., J. M. Vitolo, V. Mutskov, S. Dmitrov, and J. J. Hayes. 2001. Preferential interaction of the core histone tail domains with linker DNA. Proc. Natl. Acad. Sci. USA. 98:6599–6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leuba, S. H., C. Bustamante, J. Zlatanova, and K. van Holde. 1998. Contributions of linker histones and histone H3 to chromatin structure: scanning force microscopy studies on trypsinized fibers. Biophys. J. 74:2823–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ausio, J., F. Dong, and K. E. van Holde. 1989. Use of selectively trypsinized nucleosome core particles to analyze the role of the histone ‘tails’ in the stabilization of the nucleosome. J. Mol. Biol. 206:451–463. [DOI] [PubMed] [Google Scholar]
- 16.Tse, C., and J. C. Hansen. 1997. Hybrid trypsinized nucleosomal arrays: identification of multiple functional roles of the H2A/H2B and H3/H4 N-termini in chromatin fiber compaction. Biochemistry. 36:11381–11388. [DOI] [PubMed] [Google Scholar]
- 17.Cosgrove, M. S., J. D. Boeke, and C. Wolberger. 2004. Regulated nucleosomal mobility and the histone code. Nat. Struct. Mol. Biol. 11:1037–1043. [DOI] [PubMed] [Google Scholar]
- 18.Hamiche, A., J. G. Kang, C. Dennis, H. Xiao, and C. Wu. 2001. Histone tails modulate nucleosomal mobility and regulate ATP-dependent nucleosome sliding by NURF. Proc. Natl. Acad. Sci. USA. 98:14316–14321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hassan, A. H., K. E. Neely, and J. L. Workman. 2001. Histone acetyltransferase complexes stabilize SWI/SNF binding to promoter nucleosomes. Cell. 104:817–827. [DOI] [PubMed] [Google Scholar]
- 20.Bennink, M. L., S. H. Leuba, G. H. Leno, J. Zlatanova, B. G. Grooth, and J. Greve. 2001. Unfolding individual nucleosomes by stretching single chromatin fibers with optical tweezers. Nat. Strut. Mol. Biol. 8:606–610. [DOI] [PubMed] [Google Scholar]
- 21.Brower-Toland, B. D., C. L. Smith, R. C. Yeh, J. C. Lis, C. L. Peterson, and M. D. Wang. 2002. Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proc. Natl. Acad. Sci. USA. 99:1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui, Y., and C. Bustamante. 2000. Pulling a single chromatin fiber reveals the forces that maintain its higher-order structure. Proc. Natl. Acad. Sci. USA. 97:127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pope, L. H., M. L. Bennink, K. A. van Leijenhorst-Groener, D. Nikova, J. Greve, and J. F. Marko. 2005. Single chromatin fiber stretching reveals physically distinct populations of disassembly events. Biophys. J. 88:3572–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brower-Toland, B., D. A. Wacker, R. M. Fulbright, J. T. Lis, W. Lee Kraus, and M. D. Wang. 2005. Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J. Mol. Biol. 346:135–146. [DOI] [PubMed] [Google Scholar]
- 25.Roopa, T., N. Kumar, S. Bhattacharya, and G. V. Shivashankar. 2004. Dynamics of membrane nanotubulation and DNA self-assembly. Biophys. J. 84:974–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soni, G. V., G. Ananthakrishna, and G. V. Shivashankar. 2004. Probing collective dynamics of active particles using modulation force spectroscopy. Appl. Phys. Lett. 85:2414–2416. [Google Scholar]
- 27.Ou-Yang, H. D. 1999. Design and applications of oscillating optical tweezers for direct measurements of colloidal forces. In Colloid-Polymer Interactions: From Fundamentals to Practice. R. S. Farinato and P. L. Dubin, editors. John Wiley & Sons, New York. 385–406.
- 28.Valentine, M. T., L. E. Dewalt, and H. D. Ou-Yang. 1996. Forces on a colloidal particle in a polymer solution: a study using optical tweezers. J. Phys. Condens. Matter. 8:9477–9482. [Google Scholar]
- 29.Nemet, B. A., and M. Cronin-Golomb. 2003. Measuring microscopic viscosity with optical tweezers as a confocal probe. Appl. Opt. 42:1820–1832. [DOI] [PubMed] [Google Scholar]
- 30.Polach, K. J., P. T. Lowary, and J. Widom. 2000. Effects of core histone tail domains on the equilibrium constants for dynamic DNA site accessibility in nucleosomes. J. Mol. Biol. 298:211–223. [DOI] [PubMed] [Google Scholar]
- 31.Tse, C., T. Sera, A. P. Wolffe, and J. C. Hansen. 1998. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol. Cell. Biol. 18:4629–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]