

Abstract
Infrared difference spectroscopy analysis of the purified melibiose permease of Escherichia coli reconstituted into liposomes was carried out as a function of the presence of the two symporter substrates (Na+, melibiose) in either H2O or in D2O media. Essentially, the data first show that addition of Na+ induces appearance of peaks assigned to changes in the environment and/or orientation of α-helical domains of purified melibiose permease. Likewise, melibiose addition in the presence of Na+ produces peaks corresponding to additional changes of α-helix environment or tilt. In addition to these changes, a pair of peaks (1599 (+) cm−1/1576 (−) cm−1) appearing in the Na+-induced difference spectrum is assigned to the antisymmetric stretching of COO− groups, since they show practically no shift upon H/D exchange. It is proposed that these acidic groups participate in Na+ coordination. A corresponding pair of peaks, again fairly insensitive to H/D substitution (1591 (−) cm−1/1567 (+) cm−1), appear in the melibiose-induced difference spectra, and may again be assigned to COO− groups. The latter carboxyl groups may correspond to part or all of the acidic residues interacting with Lys or Arg in the resting state that become free upon melibiose binding.
INTRODUCTION
Melibiose permease (MelB) of Escherichia coli transports the disaccharide melibiose to the cell interior coupled to the downhill electrochemical ion gradient of Na+, Li+, or H+ (1). Melibiose (6-OR-galactopyranosyl-D-glucose) and coupling ions bind to the transporter in a 1:1 ratio. Furthermore, the coupling ions compete for the same binding site and enhance the cotransporter affinity for melibiose (2,3). Conversely, melibiose binding enhances MelB affinity for the coupling ion (4). Several works including immunological studies (5), proteolytic mapping (6), two-dimensional crystallization (7), and Fourier transform infrared (FTIR) studies (8) are consistent with a structure containing 12 transmembrane α-helical domains. Cation binding site would be localized in the N-terminal domain and the sugar-binding site in the C-terminal domain (9–13). It has also been suggested that helix IV connects the two substrate binding sites (14) and that loops IV–V and X–XI are important for the MelB function (6,15–17). Site-directed mutagenesis has highlighted the importance of Asp and Glu residues in cation and melibiose binding/translocation (10,16,18–20). Other residues such as Asn (21), Arg (15,22), and Tyr (20) have also been shown to be important in these processes.
Evidence for changes in MelB structure occurring upon substrate binding/translocation has been obtained by different biophysical techniques. Thus, it was described that the coupling cations induce changes in the fluorescence of Trp located in the N-terminal domain, whereas melibiose interaction preferentially influences the fluorescence of Trp located in the C-terminal domain (12,23). Also, fluorescence resonance energy transfer experiments suggest that interaction of Na+ (or Li+) induces a structural change in the sugar-binding site environment or in its immediate vicinity (13,24). An electrophysiological approach revealed fast transient currents (in the 20-ms range) associated to either Na+ or to melibiose binding (4). The melibiose-induced charge transfer is most probably consecutive to structural changes involving movement of charged amino acids and/or a reorientation of helix dipoles (25). Finally, Fourier transform infrared spectroscopy provided information on the secondary structure components of MelB and suggested changes in α-helix structures upon substrate binding (8). Existence of substrate-induced structural changes were corroborated in H/D exchange experiments, where MelB incubated with sugar and either of the cations was less accessible to solvent than transporters incubated with the corresponding cations alone. Furthermore, β-sheet structures were found to be protected against H/D exchange by sugar binding (26).
Recently, attenuated total reflection (ATR)-FTIR difference spectroscopy allowed detection of smaller Na+- or melibiose-induced structural changes than those previously observed in FTIR deconvoluted spectra (27). Peak assignment in the Amide I interval in difference spectra suggested that all types of secondary structures (α-helix, β-sheets, and turns) are involved in the substrate-induced structural changes. In addition, variations of signals at the level of peaks assigned to Tyr, Asp, and Glu side chains were reported (27).
In the present work, we pursue this ATR-FTIR study by comparing difference spectra recorded in H2O and D2O media either with or without MelB cosubstrates, with a threefold aim: 1), to improve previous peak assignments to given secondary structure components; 2), to assess their accessibility to the solvent; and 3), to further investigate signals potentially arising from carboxylic side chains or from other amino acids such as Tyr. It is indeed well accepted that comparison of IR difference spectra in D2O and in H2O is of significant value to better identify and characterize peaks arising from different secondary structure components or from given amino acids (reviewed in (28)). The different isotopic shifts of secondary structures and side chains allow removing ambiguities in the assignment of peaks, which is of fundamental importance in advancing comprehension of the transporting mechanism. For example, secondary structures such as α-helices or β-sheets undergo slight downshift (1–2 cm−1) in their wavenumber position due to isotopic exchange (29–31). Turns and β-sheets of high wavenumber can shift up to 10–15 cm−1 (31,32), whereas unordered structures can shift up to 15–20 cm−1 (33). Finally, the largest shifts are observed in peaks arising from amino acids with an exchangeable proton (34). For the sake of space, we will essentially emphasize below the signal variations arising from α-helical components and acidic amino acids. The data strongly suggest the implication of distinct α-helical components to the structural changes resulting from interaction of MelB with its cosubstrates and are consistent with the participation of acidic amino acids to these interactions.
MATERIALS AND METHODS
Sample preparation and data acquisition
MelB production and purification, and preparation of MelB proteoliposomes were carried out as described by Dave et al. (26). Experimental setup was the same as that described in León et al. (27) with some modifications. A sample of 20 μl of a proteoliposome suspension (∼150 μg of protein, at a lipid/protein ratio 2:1 by weight) was spread homogeneously on a ZnSe ATR crystal (50 × 10 × 2 mm, yielding 12 internal reflections at the sample side; obtained from Harrick, Ossining, NY) and dried under a stream of nitrogen. The film was exposed to deuterated buffer without substrate for 1000 min to achieve maximum H/D exchange (26). The substrate containing deuterated buffer and the reference deuterated buffer (with or without MelB substrates) were then alternatively perfused over the proteoliposome film at a rate of ≈1.5 ml/min. The film was exposed to the substrate-containing buffer for 2 min and washed with the reference buffer for 10 min. A Bio-Rad FTS-6000 infrared spectroscopy instrument (Hercules, CA) was used for spectra acquisition. For each cycle, 1000 scans at a resolution of 4 cm−1 were recorded and 10 spectra were taken and averaged to increase the signal/noise ratio, i.e., a total of 10,000 scans for every difference spectrum. A minimum of two separate experiments using newly prepared films were done for each condition. The same differences were made in H2O to compare band shift. A total of 25,000 scans were averaged in this case. Incidentally, it should be pointed out that the difference spectra taken in H2O using the ZnSe crystal are quite similar to those reported previously for the Ge crystal (27).
Degree of H/D exchange of the MelB film
The degree of global H/D exchange in MelB was calculated as described (26). The H/D exchange of the structures giving rise to the difference spectra was calculated from the ratio of the areas amide II/amide I (H2O difference) versus amide II/amide I (D2O difference); and amide A/amide I (dry film) versus amide A/amide I (D2O difference). The range of integration for amide I, amide II, and amide A were 1696–1615 cm−1, 1565–1525 cm−1, and 3450–3200 cm−1, respectively, with some modification that allowed the integration of all bands with some contribution in the limits of its respective amide region.
Data corrections and manipulations
Spectra corrections were essentially carried out as described previously (27). For amide A baseline correction, a baseline simulating band water fluctuation was subtracted from difference spectra. This fluctuation has a maximum at 3420 cm−1 and a minimum at 3620 cm−1. Deconvolution by the maximum entropy method was applied to the difference spectra as previously described (27,35).
Contribution of melibiose to the difference spectra
Substrates can absorb in the infrared. Therefore, changes in substrate vibrations due to substrate binding could lead to changes in its absorption bands (36) and to the appearance of peaks in the difference spectrum not corresponding to changes in protein structure. Melibiose in H2O has two peaks at 1350 and 1150 cm−1. No appreciable peaks are observed in the difference spectra due to melibiose binding in H2O in these regions. Furthermore, no melibiose peaks appear in the Amide I or Amide II region. Therefore, peaks corresponding to melibiose itself, due to its binding to MelB in the presence of Na+, can be discarded.
About differences in the level of H/D exchange induced by substrates
H/D exchange properties of MelB depend on the substrate bound to the permease (26). Before taking differences, extensive H/D exchange is carried out without the presence of substrates (i.e., in the presence of H+)—a condition that, as in the presence of Na+, leads to the highest exchange in the permease (26). Then, difference spectra are acquired changing the D-buffer carrying one substrate or the other. In general, change of substrate could modify the exchange level of peptide bonds and induce peaks in the difference spectra due to any increase of H/D exchange between the two conditions. However, because substrates incubation of MelB decreases the accessibility of the protein to the solvent, no further H/D exchange will take place. Indeed, no changes were observed between consecutive difference spectra acquired—either in the intensity of peaks absorbing in the Amide II region or in the position of peaks absorbing in the Amide I region. As a result, the difference spectra in D2O are due only to substrate binding, without any contribution of additional deuteration.
Estimation of spectrum intensity
To compare difference spectra between them, it is useful to normalize the area of the spectra. Then it is possible to observe not only peak shifts but also compare peak intensity between the two conditions. However, to normalize spectra it is necessary to find a reference. Because in our experiments there are two kinds of differences (Na+ binding and melibiose binding in the presence of Na+), the reference to be used in the normalization should be the same in both differences. For these reasons, the difference spectra were normalized by the area of the peaks absorbing in the Amide I region. Although there is a peak shift that leads to the increase or decrease of intensity due to peak overlap or vice versa, if all peaks after isotopic exchange continue absorbing in the amide I region, the area of the amide I region would be the same. However, such a normalization procedure does not take into account: 1), the difference in the absorption coefficient of the different structures that contribute to amide I in D2O and in H2O (37); 2), the difference of absorption coefficient of amino-acid side chains absorbing in the amide I region in D2O and in H2O media; and 3), that residues such as Arg and Lys absorb in the amide I region in H2O and out of the amide I region in D2O. Despite these potential sources of error, difference spectra present a relative good ratio after normalization, indicating that this is a good choice.
RESULTS AND DISCUSSION
Degree of H/D exchange
Extensive H/D exchange of the MelB film (1000 min) with the transporter in the H+-MelB state produces an important decrease of the amide II peak intensity and a concomitant increase in amide II′ (Fig. 1). The accessibility of the protein to D2O can be estimated by the comparison of the corrected amide II areas (amide II/amide I), giving an exchange of 56% of the hydrogen atoms. This value compares well with that of 54% previously measured by a different experimental approach, i.e., by flushing a dry film with N2 gas saturated with D2O (26).
FIGURE 1.

ATR-FTIR absorption spectrum of a dry film of MelB (shaded line) and ATR-FTIR absorption spectrum of a wet film in D2O after 1000 min of exchange (solid line). This spectrum was corrected for film inflation using amide I area (1696–1600 cm−1).
The percentage of H/D exchange corresponding to structures giving peaks in the difference spectra may not be the same as the global percentage, because the difference spectra show only the peaks corresponding to changes induced by substrate binding. These peaks may correspond to accessible or to nonaccessible structures or side chains. To estimate the accessibility level of the structures or side chains that show changes upon substrate binding, H/D exchange degree was calculated for both the cation- and the melibiose-induced difference spectra. The area in the amide I and amide II regions of difference spectra were measured and the percentage of hydrogen exchange was calculated as for the absorbance spectra, using the amide I area of the difference spectrum as a normalizing factor (Figs. 2 and 3). In the Na+-induced difference spectrum, the amount of H+ exchanged is ∼80%, whereas in the melibiose-induced difference spectrum, it is 55%. H/D exchange was also calculated from amide A vibration (see Fig. 4). Because no amide A peaks can be detected in the H2O differences due to noise, the exchange was calculated by using amide A and amide I bands of the dry film instead of a film covered with buffer. For Na+-induced difference spectrum we obtained an H/D exchange of 76%, whereas for melibiose-induced difference spectrum, an H/D exchange of 58% was obtained. Both values agree well with those obtained using amide II. Altogether, these data indicate that whereas Na+ binding preferentially induces change at the level of accessible structures, melibiose binding effects, to similar extent, both the accessible and nonaccessible structures (see also (31,38)).
FIGURE 2.

Difference spectrum due to Na+ binding. (A) (Solid line) ATR-FTIR difference spectrum of a film of MelB in D2O in 20 mM MES, 100 mM KCl, 10 mM NaCl, pH 6.6; minus the film in 20 mM MES and 110 mM KCl, pH 6.6. (Shaded line) The same difference in H2O. (B) (Solid line) Deconvoluted spectrum of the difference in D2O shown in panel A. (Shaded line) Deconvoluted spectrum of the difference in H2O shown in panel A. The wavenumbers that coincide in both spectra are in bold.
FIGURE 3.

Difference spectrum due to melibiose interaction. (A) (Solid line) ATR-FTIR difference spectrum of a film of MelB in D2O in 20 mM MES, 100 mM KCl, 10 mM NaCl, 10 mM melibiose, pH 6.6; minus the film in 20 mM MES, 100 mM KCl, 10 mM NaCl, pH 6.6. (Shaded line) The same difference in H2O. (B) (Solid line) Deconvoluted spectrum of the difference in D2O shown in panel A. (Shaded line) Deconvoluted spectrum of the difference in H2O shown in panel A. The wavenumbers that coincide in both spectra are in bold.
FIGURE 4.

Difference spectrum due to substrate binding in amide A region. (A) ATR-FTIR difference spectrum of a film of MelB in D2O in 20 mM MES, 100 mM KCl, 10 mM NaCl, pH 6.6; minus the film in 20 mM MES, 110 mM KCl, pH 6.6, after baseline correction. (B) ATR-FTIR difference spectrum of a film of MelB in D2O in 20 mM MES, 100 mM KCl, 10 mM NaCl, 10 mM melibiose, pH 6.6; minus the film in 20 mM MES, 100 mM KCl, and 10 mM NaCl, pH 6.6, after baseline correction.
Na+-induced spectral differences in H2O and D2O
Fig. 2 (top) compares the Na+-induced difference spectrum (Na+·MelB versus D+·MelB) in D2O and in H2O. Overall the difference spectrum in the amide I region in D2O differs significantly from that in H2O, in keeping with the high percentage of H/D exchange. To better distinguish the bands, the difference spectra were deconvoluted using a maximum entropy approach for the band-narrowing of absorbance and difference spectra, which uses the generalized negative Burg-entropy (35). As can be seen in Fig. 2 (bottom), the deconvoluted spectra show high signal/noise ratio, assuring that no false peaks may appear due to noise. In the following, we describe and discuss the most relevant peaks.
The 1800–1700 cm−1 region
The positive peak at 1742 cm−1 (H2O) has only a shift of ∼1 cm−1 to lower wavenumbers due to H/D exchange. Asp and Glu residues absorbing in this region have been shown to downshift 5–10 cm−1 upon H/D exchange (34). Hence, the peak at 1742 cm−1 corresponds likely to lipid contribution. These peaks may arise from lipids surrounding the protein that are perturbed due to conformational changes of the protein, as has been demonstrated for the Anabaena sensory rhodopsin (39). The negative peak at 1725 cm−1 (H2O) (Fig. 2 A) shifts down to a broader peak at 1716 cm−1 (D2O) that is resolved in small ones after deconvolution (Fig. 2 B). This downshift is typical of carboxylic acid residues (34). However, contribution of lipid surrounding MelB at these wavenumbers is also possible, as indicated by the peak at 1718 cm−1 in the deconvoluted spectrum, which seems not to shift upon H/D exchange.
The amide I region (1700–1620 cm−1)
As it is seen in Fig. 2 (bottom), the peak at 1700 cm−1 (H2O) shifts down to 1690 cm−1. This 1700 cm−1 peak is a candidate for either reverse turn structures accessible to solvent or carboxylic residues (Asp or Glu) (40–42). On the other hand, this shift goes over the negative peak at 1687 cm−1 (H2O), masking it and giving rise to its apparent disappearance in the D2O difference spectrum.
The positive peak at 1678 cm−1 (D2O) could arise from turn structures (31,43). This peak may absorb at ∼1682 cm−1 in H2O difference spectrum and may be masked by the negative peak at 1687 cm−1 (H2O). The peak at 1671 cm−1 (H2O) has a slight shift to lower wavenumbers (1670 cm−1). Therefore, this peak may correspond to nonaccessible helix αII (44) or to nonaccessible reverse turns (45).
The peaks at [H2O (D2O)] 1657 (1657) and 1651 (1651) cm−1 (Fig. 2 B) arise from α-helix structures not accessible to solvent because: 1), they appear in the α-helical region of amide I; and 2), they do not change their position after H/D exchange. The negative peak at 1662 cm−1 does not change its position on going from H2O to D2O, although its intensity decreases. Therefore, it should be assigned in part to nonexchangeable α-helices and in part to some exchanged structure (see below). These three peaks may correspond to changes in the environment or to changes in the tilt angle of α-helices. The negative peak at 1646 cm−1 (D2O) (Fig. 2 B) revealed by deconvolution may correspond to open loop structures that shift from part of the 1662 cm−1 signal in H2O. The peak at 1641 cm−1 (H2O) was previously assigned to β-sheet structures (27). Part of this peak may shift to 1639 cm−1 upon H/D exchange (45). Overall, the amount of peaks in the amide I that suffer shifts upon H/D exchange is clearly greater to those that do not shift, in agreement with the previous estimation of an exchange level of 76–80% in the amide I (see above).
Carboxylic antisymmetric stretching region (1600–1550 cm−1)
In Fig. 2 A, one can note the presence of peaks at 1576 (−) and 1599 cm−1 (+) in the Na+-induced difference spectrum in H2O that virtually do not exhibit shift in D2O. These peaks are typical of the antisymmetric stretching of COO− groups of Asp or Glu (34). Additionally, and as noted in the spectra, these stretching peaks suffer an increase in the absorption coefficient on going from H2O to D2O (34). Therefore, this pair of peaks in the MelB signal can be interpreted as a shift of an absorbing band at 1576 cm−1 in H2O to 1599 cm−1 upon Na+ binding, corresponding to antisymmetric COO− stretching. It has been previously reported that free COO− of Asp side chain displays a signal in the 1574–1579 cm−1 range, whereas the free COO− of Glu has a band ∼1560 cm−1 (34). Moreover, interaction of Na+ leads to change in these frequencies. It has been reported that monodentate interaction increases the COO− stretching frequency as compared to the noninteracting group, whereas the bidentate interaction decreases its frequency (46); in the pseudo-bridging mode, one oxygen is interacting with the cation and the other one is hydrogen-bonded to a water molecule, leading to a slight frequency increase (47). On this basis, the observed upshift of the peak from 1576 cm−1 to 1599 cm−1 most likely traduces Na+ interaction with negatively charged amino acids (Asp or Glu) of MelB in the unidentate or the pseudo-bridging mode. Besides, potential interactions between COO− groups and Arg or Lys side chains (22,48,49) may also lead to the antisymmetric band upshift. These two important observations will be further commented below.
The positive peak at 1403 cm−1 (H2O) corresponds to the symmetric COO− stretching and may be partially masked by amide II′ (1500–1400 cm−1) after H/D exchange. However, the carboxylic negative peak at 1384 cm−1 (H2O) is outside the possible influence of the amide II′ band, and has a small upshift as it was expected for a signal arising from carboxylic groups (34). Therefore, these peaks and the other one previously observed at 1725 cm−1 (H2O) are most likely indicative of changes in the protonation and/or environment of one or more of the Asp side chains that are suspected to participate directly or indirectly in cation binding (50).
Melibiose-induced spectral differences in H2O and D2O
Fig. 3 shows difference spectra of melibiose·Na+·MelB versus Na+·MelB in D2O and in H2O. The D2O spectrum presents variations with respect to the H2O spectrum, as in Na+-induced differences. However, there are no large dissimilarities in the amide I region and only small or no shifts are observed. Hence, melibiose binding in the presence of Na+ principally involves change of secondary structures not accessible to the solvent (in the core of the protein) and structures with small shifts upon deuteration (38).
The 1800–1700 cm−1 region
As in the Na+-induced difference spectrum, we assign the peak(s) around 1738 cm−1 mainly to lipids because there is a very low sensitivity to H/D exchange (Fig. 3 A). The negative peak at 1703 cm−1 (H2O) (Fig. 3 B) disappears completely. This peak has several possible assignments, i.e., Asp/Glu (34), Asn/Gln (34), or turns (42). However, it is not possible to discern between these possibilities because the location of this peak in D2O is not clear.
The amide I region (1700–1620 cm−1)
The positive peak near 1688 cm−1 (H2O) (Fig. 3 B) appears to suffer a shift of 2 cm−1 downward and an intensity decrease upon H/D exchange (Fig. 3). This small shift should correspond to a secondary structure that, given the location of the peak, could be due to turn or β-sheet with limited accessibility. On the other hand, the two peaks at 1680 cm−1 and 1674 cm−1 are insensitive to exchange and hence could be due to nonexchangeable β-sheet or turn structures.
As a general principle, H/D exchange induces downshift in peak position, whereas peaks not exchanged will remain at the same wavenumber. However, in melibiose·Na+·MelB versus Na+·MelB differences, three peaks present an unexpected small upshift: [H2O (D2O)] 1680 (1681), 1652 (1653), and 1643 (1645) cm−1 (Fig. 3 B). A likely explanation is that other peaks of low intensity overlapping with them suffer a downshift, resulting in an apparent small upshift of the nonexchanged peak. This is supported by the fact that the MelB difference spectra due to Na+ binding does not present any upshift of the 1651 cm−1 peak (see Fig. 2, bottom).
The most intense positive amide I peak appearing at 1668 cm−1 in H2O, shifts down to 1666 cm−1 (Fig. 3 B). It could correspond to partially exchanged α-helix or turn structure, giving rise to a peak with a frequency in between those of fully protonated and fully deuterated amide groups, or could be due to the disappearance of a small overlapped band upon H/D exchange. In this case, the peak could correspond to nonexchanged α-helix or turns (27). The peaks at 1659 and 1652/1653 cm−1 are insensitive to isotopic exchange and, as in Na+-induced difference spectrum, they are assigned to nonaccessible transmembrane α-helices. The negative peak at 1643/1645 cm−1 is also unaffected by H/D exchange and would therefore correspond to non-solvent-accessible structures such as 310-helices or β-sheets (8). The negative peak at 1630/1631 cm−1 does not shift, and hence, can be assigned to non-solvent-accessible β-sheet (43).
Carboxylic stretching vibrations region (1600–1550 cm−1)
A prominent change between the H2O and D2O spectra is the appearance of a positive peak at 1567 cm−1 in D2O (Fig. 3 A). This effect is most likely due to a shift of a negative peak at ∼1570 cm−1 in H2O to 1460–1470 cm−1 in D2O, a region that changes from slightly positive to negative. Therefore, the intense pair of negative and positive peaks at 1591 and 1567 cm−1 in D2O are already present in the H2O difference spectrum, although it is not possible to know its exact position or intensity in H2O due to the overlapping with amide II peaks (1580–1520 cm−1). As for the Na+-induced difference spectrum, this pair of negative/positive peaks can be assigned to the antisymmetric stretching of Asp/Glu side chains (34) arising from a shift of a band from 1591 cm−1 to 1567 cm−1 upon melibiose binding. It can be noted by comparing the difference spectra, that there is an apparent intensity increase of these peaks on going from H2O to D2O, for two reasons: 1), these peaks are unmasked after H/D exchange (carboxylic acids only experiment a slight up shift), as indicated above; and 2), in general, the carboxylic antisymmetric vibrations at ∼1570 cm−1 have higher absorption coefficients in D2O than in H2O (34,43). As discussed below, this melibiose-induced downshift may be attributed to the loss of previous interactions established between COO− groups and Arg/Lys side chains, or between COO− groups and Na+ (47).
These peaks plus those appearing in H2O at 1703 (+), 1404 (+), and 1390 (−) cm−1 and other putative bands hidden by lipid C=O absorption around 1740 cm−1 can be assigned to carboxylic residues interacting with the substrates and/or suffering protonations/deprotonations upon melibiose binding. This is in keeping with previous works demonstrating that both Asp and Glu residues take part directly or indirectly in melibiose binding/translocation. These carboxylic residues may be those putatively located in various domains of the protein (i.e., in helices I, II, and IV (10,19), loops IV–V (51), and loops X–XI (16)). Furthermore, Asp side chains involved in cation binding could be affected by melibiose binding. For this reason, the merging of changes of protonation with changes of the interaction with Na+ or of the environment of the acidic residues of MelB may give rise to a complex pattern of peaks. For example, the antisymmetric COO− peaks at 1591 and 1567 cm−1 seem to be composed by some bands partially resolved by deconvolution (Fig. 3 B). This is also apparent in the deconvoluted spectrum of Fig. 2 B.
Tyrosine region (1520–1513 cm−1)
Peaks at 1518 (−) and 1513 (+) cm−1 (D2O) (1520 cm−1 (−) and 1515 cm−1 (+) in H2O) are very likely to correspond to Tyr side chains, as previously suggested (27). Two facts support this assignment: 1), Upon H/D exchange, their intensity increase is due to downshift of amide II peaks and also to the increase of Tyr absorption coefficient (34). 2), A downshift of 1–2 cm−1 was expected, as observed. Tyrosine perturbations are in line with site-directed mutagenesis studies (20).
Substrate-induced peaks in the amide-A region
Infrared difference spectroscopy usually focuses on amide I and amide II vibrations. However, amide A vibrations absorbing between 3320 and 3270 cm−1 are also sensitive to changes in protein secondary structure and side chains (52). A difficulty is that water also absorbs in this region, decreasing the signal/noise ratio. This problem does not take place in D2O media. Amide A mode is mainly due to N-H (stretching) vibration; therefore upon H/D exchange the amide A bands contain exclusively nonexchanged N-H groups. Fig. 4 shows the difference spectra in the amide A region of Na+·MelB versus D+·MelB (top) and melibiose·Na+·MelB versus Na+·MelB (bottom). In both spectra, in spite of the baseline distortion, positive and negative peaks are clearly seen above the noise level. To have a clearer view, baseline correction was made in both spectra (see Materials and Methods). Confirming the conclusions obtained from amide I and II regions, the spectra exhibit a different pattern of positive and negative peaks. Furthermore, the melibiose-induced difference spectrum also shows higher intensity than Na+-induced difference spectrum. In addition, positive and negative peaks at 3462 and 3472 cm−1 can be attributed to the indole N-H stretch of Trp (52,53). These Trp-linked signals may be related to changes in the environment of some Trp occurring upon sugar binding (12,23) and/or to their potential catalytic implication. It has thus been reported that mutation of given Trp of MelB (W116, W128) impairs sugar affinity and/or cosubstrate translocation (14). Also, one or more of these Trp residues may have a role analogous to Trp151 in the lactose permease in stabilizing the galactopyranosyl ring in the sugar-binding site (54). In the Na+-induced difference spectrum, the negative peak at 3252 cm−1 can be tentatively assigned to β-sheet and those at higher wavenumbers to α-helices (55).
BIOLOGICAL IMPLICATIONS OF THE SPECTRAL DIFFERENCES
Comparison of the substrate-induced difference ATR-FTIR spectra recorded from MelB proteoliposomes in H2O and in D2O improves the identification of several discrete and sharp absorbance variations that bring valuable information on the relationships between Na+-coupled sugar symport activity and the underlying structural and molecular events. Of particular interest are the data related to 1), the participation of different populations of α-helical domains in the structural rearrangements occurring upon cosubstrate binding; and 2), the molecular mechanism(s) by which some of the acidic residues of MelB may contribute the transport process.
As illustrated in Results and Discussion, Na+ binding and subsequent melibiose binding induce different sets of changes in the Amide I interval that comprise, in particular, signals arising from distinct types (or populations) of α-helical components. Each α-helix population is clearly defined by its peak position and associated H/D exchange characteristics (Tables 1 and 2). The currently available knowledge on MelB structure derived from two-dimensional crystallization of MelB (7,56) suggests that transmembrane helical domains are the predominant secondary structure components. Recent resolution of three-dimensional structure of several other bacterial secondary membrane transporters (54,57–60) indicates that this is a general feature of this class of membrane transporters. More important for this discussion is the evidence that the membrane helices of all crystallized proteins exhibit varying degrees of tilt and of length and are either lining the aqueous solvent pathway (or the internal cavity), or are totally shielded from the solvent. Of particular interest is to stress that two Na+-linked transporters, NhaA and LeuTa (57,61), as well as the Ca+2-ATPase (62), possess some short membrane helical domains arising from interruption by unwound sequences where the pumped or coupling ion interacts with carbonyls of the peptidic backbone. On this basis, one can expect that various subtypes of α-helices with different IR signatures should contribute the difference ATR-FTIR spectra of these transporters. Moreover, the catalytic models proposed for all these transporters are based on the alternate site exposure principle, implying structural changes associated to readjustments of the organization of the membrane helical domains upon substrate interaction. Changes in helix tilting and/or solvent accessibility are generally invoked (63–66). They are most probably major factors leading to the complex pattern of substrate-induced changes in α-helix signals observed here with the Na+ (or H+) coupled symporter MelB. It is worth mentioning here that direct support for change in MelB helix tilt consecutive to interaction of MelB with its substrates is currently being sought using polarized light to assess MelB helix tilt properties (unpublished data). Interestingly, the dichroic data of the absorbance spectra clearly suggest the presence of several oriented structures in the 1665–1645 cm−1 interval as well as changes in their tilt angle upon interaction of the substrates. Taken together with indications that substrate binding also induces changes of IR signals linked to other secondary structural components (turns, β-structure), the observed changes of α-helix signals upon successive addition of the cosubstrate lend support to previous claims (23,24,56) that MelB modifies its conformation at different stages of the transport reaction (substrate binding and translocation steps).
TABLE 1.
Tentative assignment of the infrared peaks induced by Na+ binding to MelB
| H2O | D2O | Assignment |
|---|---|---|
| 1742 | 1741 | Lipid |
| 1725 | 1716 | COOH (with a possible lipid contribution) |
| 1700 | 1690 | Asp/Glu/solvent-accessible reverse turn |
| 1687 | 1687 | β-sheet |
| — | 1678 | Solvent-accessible turn-like structure |
| 1671 | 1670 | Turn/αII-helix |
| 1662 | 1662 | α-helix |
| 1657 | 1657 | α-helix |
| 1651 | 1651 | α-helix |
| — | 1646 | Open loops |
| 1641 | 1639 | β-sheet |
| 1599 | 1599 | COO− (antisymmetric stretching) |
| 1576 | 1576 | COO− (antisymmetric stretching) |
| 1551 | 1450 | Amide II vibration |
| 1540 | 1540 | Amide II vibration |
| 1403 | 1403 | COO− (symmetric stretching) |
| 1384 | 1385 | COO− (symmetric stretching) |
TABLE 2.
Tentative assignment of the infrared peaks induced by melibiose binding to MelB, in the presence of Na+
| H2O | D2O | Assignment |
|---|---|---|
| 1703 | 1693 | Asp/Glu/turn |
| 1703 | — | Asn |
| 1693 | 1693 | β-sheet |
| 1693 | 1683 | Solvent-accessible turn |
| 1688 | 1686 | Turn/β-sheet |
| 1680 | 1681 | β-sheet |
| 1674 | 1674 | β-sheet |
| 1668 | 1666 | α-helix/turn |
| 1659 | 1659 | α-helix |
| 1652 | 1653 | α-helix |
| 1643 | 1645 | β-sheet/310-helix/open loops |
| 1631 | 1630 | β-sheet |
| 1620 | 1616 | Secondary structure/aromatic side chain |
| 1611 | 1609 | Secondary structure/aromatic side chain |
| 1595 | 1591 | COO− (antisymmetric stretching) |
| — | 1567 | COO− (antisymmetric stretching) |
| 1518/1513 | 1520/1515 | Tyr |
| 1404 | 1402 | COO− (symmetric stretching) |
| 1390 | 1389 | COO− (symmetric stretching) |
An evident involvement of COO− groups in the substrate binding mechanism has been established in this work. In the first step of the transport reaction, Na+ binding causes a shift of antisymmetric stretching bands of COO− groups from ∼1576 cm−1 (assigned to free or ionic groups interacting with one or more H3O+) to higher wavenumbers (1599 cm−1). As indicated above, this shift is in keeping with cation coordination to these groups in the unidentate or pseudobridging mode ((47); see Fig. 5). Most probably, these COO− groups could belong to one or more of Asp19, Asp55, Asp59, and Asp124, which have been considered as potential ligands for cation in its binding site (10,19,50). In this line, acidic residues have also been suspected to participate in cation recognition in other Na+-dependent bacterial secondary transporters, including the Na+/H+ antiporter NhaA (57), the proline transporter PutP (67), or in biogenic amine transporters (see (61)). New salt-bridge interactions established between COO− groups and Arg or Lys side chains upon Na+ binding could also contribute to these peaks (68). Existence of such types of interaction in MelB and of their functional importance has been previously suggested based on second site revertant studies or mutation analyses (22,48,49). Therefore, the existence of these interactions either in the MelB resting state (i.e., the H+-MelB complex) or their formation upon Na+ binding, seems possible. In addition, peaks at ∼1725 cm−1 (−), and at 1384 (−) and 1403 cm−1 (+) indicate that protonation(s)-deprotonation(s) or changes in the environment of additional COO− groups may take place.
FIGURE 5.

Proposed changes occurring in acidic side chains upon cosubstrate interaction. The wavenumbers inside the boxes indicate the location of absorbing bands, whereas wavenumbers on the right side, outside the boxes, indicate the corresponding peaks in the difference spectra. COO− indicates one or more acidic side chains. 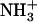 indicates protonated Arg or Lys side chains. The species present are indicated on the right side, outside the boxes.
indicates protonated Arg or Lys side chains. The species present are indicated on the right side, outside the boxes.
In the second step of the transport, melibiose binding to the Na+·MelB binary complex gives rise to prominent peaks that are fairly insensitive to H/D exchange and occur at wavenumbers characteristic of the antisymmetric stretch of COO− groups (1591 cm−1 (−)/1567 cm−1 (+)). The positive signal at 1567 cm−1 may result from a shift to lower wavenumbers of bands initially located at ∼1591 cm−1. It is remarkable that these sugar-induced peaks and those triggered by Na+ binding in the same wavenumber interval have opposite signs, albeit with different position.
A likely attractive speculation is to assume that these peaks originate from salt bridges of Asp or Glu with Arg or Lys that are already present in the resting state of the permease. As indicated in Fig. 5, these salt bridges could be broken due to melibiose binding, giving rise to the observed peaks. According to previous works, Asp59 and Asp124 are supposed to form salt bridges with Lys377 connecting helix II and helix IV with helix IX, and Asp19 and Asp55 may form another salt bridge with Arg52 (22,48,49). In this line, it is interesting to note that the loops IV–V and X–XI have been implicated in the translocation step, as possible reentrant loops (15,17). Therefore, acidic side chains present in these loops could form new hydrogen bonds with melibiose, losing salt bridges they formed initially with Arg or Lys. In any instance, it is noteworthy that our results raise the possibility of the formation of interactions between some COO− groups and Na+ or with Arg/Lys side chains upon Na+ binding, and the breaking of interactions of COO− groups upon melibiose binding in the presence of Na+. An important role of salt bridges in transporter function has already been proposed in the lactose permease (69). Identification of these side chains will provide a major insight into the transport mechanism of the melibiose permease. More generally, it is expected that analyzing how the substrate-dependent FTIR spectral variations are modified in thoroughly characterized MelB mutants will provide valuable information for the understanding of the transport mechanism of MelB and of Na+-coupled symporters.
Acknowledgments
We thank Drs. Natàlia Dave and Víctor Lórenz-Fonfría for helpful discussions and suggestions.
This work was supported by grant No. Bio4-CT97-2119 from the European Commission (to G.L. and E.P.) and grant No. BMC2003-04941 from the Dirección General de Investigación (MCYT).
References
- 1.Cohn, D. E., and H. R. Kaback. 1980. Mechanism of the melibiose porter in membrane vesicles of Escherichia coli. Biochemistry. 19:4237–4243. [DOI] [PubMed] [Google Scholar]
- 2.Damiano-Forano, E., M. Bassilana, and G. Leblanc. 1986. Sugar binding properties of the melibiose permease in Escherichia coli membrane vesicles. Effects of Na+ and H+ concentrations. J. Biol. Chem. 261:6893–6899. [PubMed] [Google Scholar]
- 3.Bassilana, M., T. Pourcher, and G. Leblanc. 1988. Melibiose permease of Escherichia coli. Characteristics of co-substrates release during facilitated diffusion reactions. J. Biol. Chem. 263:9663–9667. [PubMed] [Google Scholar]
- 4.Ganea, C., T. Pourcher, G. Leblanc, and K. Fendler. 2001. Evidence for intraprotein charge transfer during the transport activity of the melibiose permease from Escherichia coli. Biochemistry. 40:13744–13752. [DOI] [PubMed] [Google Scholar]
- 5.Botfield, M. C., and T. H. Wilson. 1989. Peptide-specific antibody for the melibiose carrier of Escherichia coli localizes the carboxyl terminus to the cytoplasmic face of the membrane. J. Biol. Chem. 264:11649–11652. [PubMed] [Google Scholar]
- 6.Gwizdek, C., G. Leblanc, and M. Bassilana. 1997. Proteolytic mapping and substrate protection of the Escherichia coli melibiose permease. Biochemistry. 36:8522–8529. [DOI] [PubMed] [Google Scholar]
- 7.Hacksell, I., J. L. Rigaud, P. Purhonen, T. Pourcher, H. Hebert, and G. Leblanc. 2002. Projection structure at 8 Å resolution of the melibiose permease, an Na-sugar co-transporter from Escherichia coli. EMBO J. 21:3569–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dave, N., A. Troullier, I. Mus-Veteau, M. Duñach, G. Leblanc, and E. Padrós. 2000. Secondary structure components and properties of the melibiose permease from Escherichia coli: a Fourier transform infrared spectroscopy analysis. Biophys. J. 79:747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hama, H., and T. H. Wilson. 1993. Cation-coupling in chimeric melibiose carriers derived from Escherichia coli and Klebsiella pneumoniae. The amino-terminal portion is crucial for Na+ recognition in melibiose transport. J. Biol. Chem. 268:10060–10065. [PubMed] [Google Scholar]
- 10.Pourcher, T., M. L. Zani, and G. Leblanc. 1993. Mutagenesis of acidic residues in putative membrane-spanning segments of the melibiose permease of Escherichia coli. I. Effect on Na+-dependent transport and binding properties. J. Biol. Chem. 268:3209–3215. [PubMed] [Google Scholar]
- 11.Wilson, D. M., and T. H. Wilson. 1992. Asp-51 and Asp-120 are important for the transport function of the Escherichia coli melibiose carrier. J. Bacteriol. 174:3083–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mus-Veteau, I., and G. Leblanc. 1996. Melibiose permease of Escherichia coli: structural organization of cosubstrate binding sites as deduced from tryptophan fluorescence analyses. Biochemistry. 35:12053–12060. [DOI] [PubMed] [Google Scholar]
- 13.Cordat, E., I. Mus-Veteau, and G. Leblanc. 1998. Structural studies of the melibiose permease of Escherichia coli by fluorescence resonance energy transfer. II. Identification of the tryptophan residues acting as energy donors. J. Biol. Chem. 273:33198–33202. [DOI] [PubMed] [Google Scholar]
- 14.Cordat, E., G. Leblanc, and I. Mus-Veteau. 2000. Evidence for a role of helix IV in connecting cation- and sugar-binding sites of Escherichia coli melibiose permease. Biochemistry. 39:4493–4499. [DOI] [PubMed] [Google Scholar]
- 15.Abdel-Dayem, M., C. Basquin, T. Pourcher, E. Cordat, and G. Leblanc. 2003. Cytoplasmic loop connecting helices IV and V of the melibiose permease from Escherichia coli is involved in the process of Na+-coupled sugar translocation. J. Biol. Chem. 278:1518–1524. [DOI] [PubMed] [Google Scholar]
- 16.Ding, P. Z. 2003. An investigation of cysteine mutants on the cytoplasmic loop X/XI in the melibiose transporter of Escherichia coli by using thiol reagents: implication of structural conservation of charged residues. Biochem. Biophys. Res. Commun. 307:864–869. [DOI] [PubMed] [Google Scholar]
- 17.Ding, P. Z. 2004. Loop X/XI, the largest cytoplasmic loop in the membrane-bound melibiose carrier of Escherichia coli, is a functional re-entrant loop. Biochim. Biophys. Acta. 1660:106–117. [DOI] [PubMed] [Google Scholar]
- 18.Pourcher, T., M. Deckert, M. Bassilana, and G. Leblanc. 1991. Melibiose permease of Escherichia coli: mutation of aspartic acid 55 in putative helix II abolishes activation of sugar binding by Na+ ions. Biochem. Biophys. Res. Commun. 178:1176–1181. [DOI] [PubMed] [Google Scholar]
- 19.Zani, M. L., T. Pourcher, and G. Leblanc. 1993. Mutagenesis of acidic residues in putative membrane-spanning segments of the melibiose permease of Escherichia coli. II. Effect on cationic selectivity and coupling properties. J. Biol. Chem. 268:3216–3221. [PubMed] [Google Scholar]
- 20.Zani, M. L., T. Pourcher, and G. Leblanc. 1994. Mutation of polar and charged residues in the hydrophobic NH2-terminal domains of the melibiose permease of Escherichia coli. J. Biol. Chem. 269:24883–24889. [PubMed] [Google Scholar]
- 21.Franco, P. J., and T. H. Wilson. 1996. Alteration of Na+-coupled transport in site-directed mutants of the melibiose carrier of Escherichia coli. Biochim. Biophys. Acta. 1282:240–248. [DOI] [PubMed] [Google Scholar]
- 22.Franco, P. J., and T. H. Wilson. 1999. Arg-52 in the melibiose carrier of Escherichia coli is important for cation-coupled sugar transport and participates in an intrahelical salt bridge. J. Bacteriol. 181:6377–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mus-Veteau, I., T. Pourcher, and G. Leblanc. 1995. Melibiose permease of Escherichia coli: substrate-induced conformational changes monitored by tryptophan fluorescence spectroscopy. Biochemistry. 34:6775–6783. [DOI] [PubMed] [Google Scholar]
- 24.Maehrel, C., E. Cordat, I. Mus-Veteau, and G. Leblanc. 1998. Structural studies of the melibiose permease of Escherichia coli by fluorescence resonance energy transfer. I. Evidence for ion-induced conformational change. J. Biol. Chem. 273:33192–33197. [DOI] [PubMed] [Google Scholar]
- 25.Meyer-Lipp, K., C. Ganea, T. Pourcher, G. Leblanc, and K. Fendler. 2004. Sugar binding induced charge translocation in the melibiose permease from Escherichia coli. Biochemistry. 43:12606–12613. [DOI] [PubMed] [Google Scholar]
- 26.Dave, N., V. A. Lórenz-Fonfría, J. Villaverde, R. Lemonnier, G. Leblanc, and E. Padrós. 2002. Study of amide-proton exchange of Escherichia coli melibiose permease by attenuated total reflection-Fourier transform infrared spectroscopy: evidence of structure modulation by substrate binding. J. Biol. Chem. 277:3380–3387. [DOI] [PubMed] [Google Scholar]
- 27.León, X., V. A. Lórenz-Fonfría, R. Lemonnier, G. Leblanc, and E. Padrós. 2005. Substrate-induced conformational changes of melibiose permease from Escherichia coli studied by infrared difference spectroscopy. Biochemistry. 44:3506–3514. [DOI] [PubMed] [Google Scholar]
- 28.Barth, A., and C. Zscherp. 2002. What vibrations tell us about proteins. Q. Rev. Biophys. 35:369–430. [DOI] [PubMed] [Google Scholar]
- 29.Susi, H., S. N. Timasheff, and L. Stevens. 1967. Infrared spectra and protein conformations in aqueous solutions. I. The amide I band in H2O and D2O solutions. J. Biol. Chem. 242:5460–5466. [PubMed] [Google Scholar]
- 30.Goormaghtigh, E., V. Cabiaux, and J. M. Ruysschaert. 1994. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. III. Secondary structures. Subcell. Biochem. 23:405–450. [DOI] [PubMed] [Google Scholar]
- 31.Baenziger, J. E., and J. P. Chew. 1997. Desensitization of the nicotinic acetylcholine receptor mainly involves a structural change in solvent-accessible regions of the polypeptide backbone. Biochemistry. 36:3617–3624. [DOI] [PubMed] [Google Scholar]
- 32.Arrondo, J. L., and F. M. Goñi. 1999. Structure and dynamics of membrane proteins as studied by infrared spectroscopy. Prog. Biophys. Mol. Biol. 72:367–405. [DOI] [PubMed] [Google Scholar]
- 33.Arrondo, J. L., A. Muga, J. Castresana, and F. M. Goñi. 1993. Quantitative studies of the structure of proteins in solution by Fourier-transform infrared spectroscopy. Prog. Biophys. Mol. Biol. 59:23–56. [DOI] [PubMed] [Google Scholar]
- 34.Barth, A. 2000. The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Biol. 74:141–173. [DOI] [PubMed] [Google Scholar]
- 35.Lórenz-Fonfría, V. A., and E. Padrós. 2005. Maximum entropy deconvolution of infrared spectra: use of a novel entropy expression without sign restriction. Appl. Spectrosc. 59:474–486. [DOI] [PubMed] [Google Scholar]
- 36.Baenziger, J. E., K. W. Miller, and K. J. Rothschild. 1993. Fourier transform infrared difference spectroscopy of the nicotinic acetylcholine receptor: evidence for specific protein structural changes upon desensitization. Biochemistry. 32:5448–5454. [DOI] [PubMed] [Google Scholar]
- 37.Barth, A., and W. Mäntele. 1998. ATP-Induced phosphorylation of the sarcoplasmic reticulum Ca2+ ATPase: molecular interpretation of infrared difference spectra. Biophys. J. 75:538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kluge, T., J. Olejnik, L. Smilowitz, and K. J. Rothschild. 1998. Conformational changes in the core structure of bacteriorhodopsin. Biochemistry. 37:10279–10285. [DOI] [PubMed] [Google Scholar]
- 39.Bergo, V. B., M. Ntefidou, V. D. Trivedi, J. J. Amsden, J. M. Kralj, K. J. Rothschild, and J. L. Spudich. 2006. Conformational changes in the photocycle of Anabaena sensory rhodopsin: absence of the Schiff base counterion protonation signal. J. Biol. Chem. 281:15208–15214. [DOI] [PubMed] [Google Scholar]
- 40.Brown, L. S., J. Sasaki, H. Kandori, A. Maeda, R. Needleman, and J. K. Lanyi. 1995. Glutamic acid 204 is the terminal proton release group at the extracellular surface of bacteriorhodopsin. J. Biol. Chem. 270:27122–27126. [DOI] [PubMed] [Google Scholar]
- 41.Gregoriou, V. G., V. Jayaraman, X. Hu, and T. G. Spiro. 1995. FT-IR difference spectroscopy of hemoglobins A and Kempsey: evidence that a key quaternary interaction induces protonation of Aspβ99. Biochemistry. 34:6876–6882. [DOI] [PubMed] [Google Scholar]
- 42.Krimm, S., and J. Bandekar. 1986. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 38:181–364. [DOI] [PubMed] [Google Scholar]
- 43.von Germar, F., A. Barth, and W. Mäntele. 2000. Structural changes of the sarcoplasmic reticulum Ca2+-ATPase upon nucleotide binding studied by Fourier transform infrared spectroscopy. Biophys. J. 78:1531–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Furutani, Y., Y. Sudo, N. Kamo, and H. Kandori. 2003. FTIR spectroscopy of the complex between Pharaonis phoborhodopsin and its transducer protein. Biochemistry. 42:4837–4842. [DOI] [PubMed] [Google Scholar]
- 45.Tatulian, S. A. 2003. Attenuated total reflection Fourier transform infrared spectroscopy: a method of choice for studying membrane proteins and lipids. Biochemistry. 42:11898–11907. [DOI] [PubMed] [Google Scholar]
- 46.Deacon, G. B., and R. J. Phillips. 1980. Relationships between the carbon-oxygen stretching frequencies of carboxylate complexes and the type of carboxylate coordination. Coord. Chem. Rev. 33:227–250. [Google Scholar]
- 47.Nara, M., M. Tasumi, M. Tanokura, T. Hiraoki, M. Yazawa, and A. Tsutsumi. 1994. Infrared studies of interaction between metal ions and Ca2+-binding proteins. Marker bands for identifying the types of coordination of the side-chain COO− groups to metal ions in Pike parvalbumin (pI = 4.10). FEBS Lett. 349:84–88. [DOI] [PubMed] [Google Scholar]
- 48.Franco, P. J., A. B. Jena, and T. H. Wilson. 2001. Physiological evidence for an interaction between helices II and XI in the melibiose carrier of Escherichia coli. Biochim. Biophys. Acta. 1510:231–242. [DOI] [PubMed] [Google Scholar]
- 49.Hastings Wilson, T., and D. M. Wilson. 1998. Evidence for a close association between helix IV and helix XI in the melibiose carrier of Escherichia coli. Biochim. Biophys. Acta. 1374:77–82. [DOI] [PubMed] [Google Scholar]
- 50.Poolman, B., J. Knol, C. van der Does, P. J. Henderson, W. J. Liang, G. Leblanc, T. Pourcher, and I. Mus-Veteau. 1996. Cation and sugar selectivity determinants in a novel family of transport proteins. Mol. Microbiol. 19:911–922. [DOI] [PubMed] [Google Scholar]
- 51.Séry, N. 2002. Role of cytoplasmic loops 4–5 and 10–11 in the Na+- symport mechanism catalyzed by the melibiose permease of Escherichia coli. Diploma of Advanced Studies, Nice University, Nice, France.
- 52.Kandori, H., and A. Maeda. 1995. FTIR spectroscopy reveals microscopic structural changes of the protein around the rhodopsin chromophore upon photoisomerization. Biochemistry. 34:14220–14229. [DOI] [PubMed] [Google Scholar]
- 53.Rath, P., F. Delange, W. J. Degrip, and K. J. Rothschild. 1998. Hydrogen bonding changes of internal water molecules in rhodopsin during metarhodopsin I and metarhodopsin II formation. Biochem. J. 329:713–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abramson, J., I. Smirnova, V. Kasho, G. Verner, H. R. Kaback, and S. Iwata. 2003. Structure and mechanism of the lactose permease of Escherichia coli. Science. 301:610–615. [DOI] [PubMed] [Google Scholar]
- 55.Goormaghtigh, E., V. Cabiaux, and J. M. Ruysschaert. 1994. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. I. Assignments and model compounds. Subcell. Biochem. 23:329–362. [DOI] [PubMed] [Google Scholar]
- 56.Purhonen, P., A. K. Lundback, R. Lemonnier, G. Leblanc, and H. Hebert. 2005. Three-dimensional structure of the sugar symporter melibiose permease from cryo-electron microscopy. J. Struct. Biol. 152:76–83. [DOI] [PubMed] [Google Scholar]
- 57.Hunte, C., E. Screpanti, M. Venturi, A. Rimon, E. Padan, and H. Michel. 2005. Structure of a Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature. 435:1197–1202. [DOI] [PubMed] [Google Scholar]
- 58.Hirai, T., and S. Subramaniam. 2004. Structure and transport mechanism of the bacterial oxalate transporter OxlT. Biophys. J. 87:3600–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang, Y., M. J. Lemieux, J. Song, M. Auer, and D. N. Wang. 2003. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 301:616–620. [DOI] [PubMed] [Google Scholar]
- 60.Yamashita, A., S. K. Singh, T. Kawate, Y. Jin, and E. Gouaux. 2005. Crystal structure of a bacterial homologue of Na+/Cl− dependent neurotransmitter transporters. Nature. 437:215–223. [DOI] [PubMed] [Google Scholar]
- 61.Gouaux, E., and R. Mackinnon. 2005. Principles of selective ion transport in channels and pumps. Science. 310:1461–1465. [DOI] [PubMed] [Google Scholar]
- 62.Toyoshima, C., and H. Nomura. 2002. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 418:605–611. [DOI] [PubMed] [Google Scholar]
- 63.Wu, J., and H. R. Kaback. 1997. Helix proximity and ligand-induced conformational changes in the lactose permease of Escherichia coli determined by site-directed chemical crosslinking. J. Mol. Biol. 270:285–293. [DOI] [PubMed] [Google Scholar]
- 64.Wu, J., D. Hardy, and H. R. Kaback. 1998. Tilting of helix I and ligand-induced changes in the lactose permease determined by site-directed chemical cross-linking in situ. Biochemistry. 37:15785–15790. [DOI] [PubMed] [Google Scholar]
- 65.Wu, J., D. Hardy, and H. R. Kaback. 1998. Transmembrane helix tilting and ligand-induced conformational changes in the lactose permease determined by site-directed chemical crosslinking in situ. J. Mol. Biol. 282:959–967. [DOI] [PubMed] [Google Scholar]
- 66.Zhang, W., L. Guan, and H. R. Kaback. 2002. Helices VII and X in the lactose permease of Escherichia coli: proximity and ligand-induced distance changes. J. Mol. Biol. 315:53–62. [DOI] [PubMed] [Google Scholar]
- 67.Jung, H. 2002. The sodium/substrate symporter family: structural and functional features. FEBS Lett. 529:73–77. [DOI] [PubMed] [Google Scholar]
- 68.von Germar, F., A. Galan, O. Llorca, J. L. Carrascosa, J. M. Valpuesta, W. Mäntele, and A. Muga. 1999. Conformational changes generated in GroEL during ATP hydrolysis as seen by time-resolved infrared spectroscopy. J. Biol. Chem. 274:5508–5513. [DOI] [PubMed] [Google Scholar]
- 69.Guan, L., and H. R. Kaback. 2005. Lessons from lactose permease. Annu. Rev. Biophys. Biomol. Struct. 35:67–91. [DOI] [PMC free article] [PubMed] [Google Scholar]