

Abstract
Fibroblast growth factor (FGF)1 is released from cells as a constituent of a complex that contains the small calcium binding protein S100A13, and the p40 kDa form of synaptotagmin (Syt)1, through an ER-Golgi-independent stress-induced pathway. FGF1 and the other components of its secretory complex are signal peptide-less proteins. We examined their capability to interact with lipid bilayers by studying protein-induced carboxyfluorescein release from liposomes of different phospholipid (pL) compositions. FGF1, p40 Syt1, and S100A13 induced destabilization of liposomes composed of acidic but not of zwitterionic pL. We produced mutants of FGF1 and p40 Syt1, in which specific basic amino acid residues in the regions that bind acidic pL were substituted. The ability of these mutants to induce liposomes destabilization was strongly attenuated, and they exhibited drastically diminished spontaneous and stress-induced release. Apparently, the non-classical release of FGF1 and p40 Syt1 involves destabilization of membranes containing acidic pL.
Keywords: FGF1, synaptotagmin 1, S100A13, non-classical release, membrane, phospholipid
Abbreviations: 5,6 carboxyfluorescein (CF); fibroblast growth factor (FGF); molten globule (MG); phospholipid (pL); phosphatidylinositol (pI); phosphatidylserine (pS); phosphatidylglycerol (pG); phosphatidylcholine (pC); synaptotagmin (Syt); wild type (WT)
INTRODUCTION
Fibroblast growth factor (FGF)1 regulates embryonic development of vertebrates [1] and plays important roles in angiogenesis, inflammation, wound healing, and as a neurotrophic factor [2, 3]. Similar to another ubiquitous and biologically important prototype member of the FGF family, FGF2, FGF1 belongs to a large group of proteins that lack a conventional signal sequence and gain access to the extracellular compartment independently of the endoplasmic reticulum (ER)-Golgi apparatus [4–13]. Indeed, FGF1 release is insensitive to Brefeldin A [14], which blocks ER-to-Golgi vesicular transport [15], and FGF1 does not appear to be present in the cytoplasmic vesicles [16]. Thus, FGF1 export through exocytotic fusion of secretory vesicles with the cell membrane is unlikely. FGF1 is secreted from cells upon stress stimulation such as heat shock [14], hypoxia [17], serum starvation [18], and treatment with oxidized LDL [19]. The availability of free intracellular copper ions is necessary for FGF1 release, and in vitro data suggest the formation of a copper- and stress-dependent multiprotein export complex [20], which contains the calcium-binding proteins, S100A13 and p40 Syt1 [21, 22]. p40 Syt1 is a non-transmembrane isoform of the integral component of secretory vesicles, synaptotagmin (Syt)1, that is involved in the fusion of exocytotic vesicles with the plasma membrane [23]. Our laboratory demonstrated that p40 Syt1 is produced by alternative initiation of translation of p65 Syt1 mRNA [24].
Both p40 Syt1 and S100A13 display a constitutive and stress-induced release from transfected NIH 3T3 cells. Their constitutive release is blocked when they are cotransfected into the cells along with FGF1; however upon stress, they are released in a complex with FGF1 [21, 22].
Although significant progress has been achieved in the study of non-classical protein release, the final step that allows these polypeptides to translocate across the cell membrane remains elusive. Confocal immunofluorescence microscopy studies demonstrated that in response to stress, cytosolic FGF1, S100A13, and p40 Syt1 traffic to the inner surface of the plasma membrane of the cells which co-express them [16]. It was suggested that the assembly of the FGF1 release complex may occur in this locale through the interaction of the individual polypeptide components with membrane phospholipids (pL). Indeed, it is known that: (i) FGF1 is able to bind acidic pL in a solid phase pL assay [25]; (ii) Syt1 can bind phosphatidylserine (pS) through its C2A domain and phosphorylated forms of phosphatidylinositol (pI) through its C2B domain [26]; and (iii) some members of the S100 family, such as S100A6 and S100A10, bind pL and they are important regulators of the functions of pL-binding proteins [27, 28]. However, the ability of S100A13 to interact with pL has not been evaluated.
FGF1 disrupts acidic pL-containing liposome integrity [29], is able to deform lipid bilayers [30], and it exhibits molten globule (MG) character at temperatures above 30°C [31, 32], at acidic pH and in the presence of acidic pL [29]. MG is a partially unfolded conformation that bestows more hydrophobic features on the protein, and therefore increases the capability to interact with lipids [33]. Given the pL binding capability and pL-induced MG state of FGF1 [25, 29], we investigated whether specific membrane pL play a role in FGF1 non-classical release. To this end, we studied the interaction of FGF1, S100A13, and p40 Syt1 with liposomes of various pL compositions. Our results demonstrated that the three members of the FGF1 release complex altered the integrity of membranes composed of acidic pL. Based on these data, we found that the mutations of specific basic amino acid residues in the C2B domain of p40 Syt1 and in the C-terminal part of FGF1 attenuated their ability to destabilize liposomes and blocked or drastically decreased their release from the cells.
MATERIALS AND METHODS
Materials
Dioleoylphosphatidylinositol (pI), dioleoylphosphatidylglycerol (pG), dioleoylphosphatidylserine (pS), and dioleoylphosphatidylcholine (pC) were purchased from Avanti Polar Lipid (Alabaster, AL). The fluorescent dye, 5,6-carboxyfluorescein (CF), was purchased from Molecular Probes, Inc (Eugene, OR).
Plasmids and recombinant proteins
The plasmids for eukaryotic expression of FGF1 (pXZ38) and p40 Syt1 (p40-Syt1:Myc in pMEX hygro) were prepared as previously described [14, 21, 22]. The K326,327,331Q mutant of p40 Syt1 was produced by mutagenesis from the p40 Syt1:Myc pMEX Hygro and GST-p40 Syt1-pGEX-KG original plasmids [22, 34] for eukaryotic and prokayotic expression, respectively. FGF1 K126,127A and FGF1 K114,115A mutants were produced by mutagenesis of the FGF1:HA pCR3.1 construct (gift of Andrew Baird, Human BioMolecular Research Institute, San Diego, CA) for eukaryotic expression, and the FGF1 pET3C construct [35] for prokaryotic expression. Recombinant FGF1, S100A13, and p40 Syt1 were produced and purified by HPLC as previously described [21, 34, 35].
Liposome preparation and fluorescence measurement
CF-loaded unilammelar liposomes were prepared as previously described [36]. The fluorescence of liposomes resuspended in 10 mM HEPES, 150 mM NaCl (pH 7.0) was monitored for 10 minutes by a Fluorolog-3 spectrofluorimeter (Jobin Yvon Horiba, Edison, NJ) at an excitation wavelength 470 nm and an emission wavelength 520 nm at 50°C, as previously described [29, 36]. Different concentrations of FGF1, S100A13, and p40 Syt1 recombinant proteins were added to the cuvette at the second minute of the experiment.
Cell culture, heat shock, conditioned media processing, and immunoblot analysis
NIH 3T3 cells were grown to 70% confluence in Dulbecco’s-modified Eagle’s medium (DMEM; HyClone) supplemented with 10% (v/v) bovine calf serum (BCS, HyClone) and 1X antibiotic/antimycotic mixture (Gibco ), on human fibronectin-coated (10 μg/cm2) 10 cm dishes (Corning). For transient transfections, p40 Syt1 WT, p40 K326,327,331Q Syt1, FGF1-HA WT, FGF1:HA K114,115A or FGF1:HA K126,127A DNA were used in combination with the JetPEI transfectant reagent according to the manufacturer’s instructions (Qbiogene Inc.).
Transiently transfected NIH 3T3 cells grown to 70–80% confluency were washed with DMEM containing 5 units/ml heparin (Sigma), and heat shock was performed as previously described [14] in DMEM containing 5 units/ml heparin for 110 minutes at 42°C; control cultures were incubated at 37°C in the same medium. Released p40 Syt1 was isolated using heparin Sepharose chromatography as described [22]. Released FGF1:HA was immunoprecipitated using anti-HA monoclonal antibodies (Covance) as described [21]. FGF1 and p40 Syt1 were resolved by SDS PAGE and detected by immunoblotting as described [14, 22].
Confocal microscopy analysis
NIH 3T3 cells were plated on fibronectin-coated glass coverslips in 6-well plates, grown to 70% confluence, and then transiently transfected with 1 μg per well of either p40 Syt1:Myc, p40 Syt1:Myc K326,327,331Q, FGF1:HA, FGF1:HA K114,115A, or FGF1:HA K126,127A DNA, as described above. After 24 hours, the culture medium was substituted with DMEM containing 5 units/ml heparin, and heat shock was performed as described above. The cells were fixed with 4% formaldehyde and immunostained as previously described [16] using a monoclonal anti-Myc antibody (Oncogene) or a monoclonal anti-HA antibody (Covance) followed by a fluorescein-conjugated anti–mouse IgG antibody and Hoechst 34580 (both from Molecular Probes). The cells were examined using a 100x objective of a LTCS-SP confocal system (Leica) at a 237 μM confocal pinhole.
RESULTS
Proteins of the FGF1 release complex induce liposome destabilization that is dependent on liposome composition and protein concentration
Liposomes loaded with fluorescent molecules represent an established model for studying the membrane destabilizing activity of proteins [37]. FGF1 destabilized mixed pG/pC liposomes [29]. To investigate the interaction of FGF1 with various plasma membrane pL, we compared the destabilizing effect of FGF1 on liposomes consisting of several acidic (pS, pI, pG) and a zwitterionic (pC) pL. We also evaluated whether other components of the FGF1 release pathway, i.e. S100A13 and p40 Syt1, induce liposome destabilization (Figure 1A, B, C). The recombinant forms of FGF1, S100A13, and p40 Syt1 were added individually to the pL liposome suspension in the cuvette, and the CF release was detected fluorimetrically. The release of CF from liposomes results in a dequenching of fluorescence due to a sharp decrease of CF concentration [29]. The final protein concentrations in the cuvette were 1 μM, 0.5 μM, 0.25 μM, 0.125 μM. The pL concentration was 2 μM. We used α-chymotrypsin (Sigma) at the maximal concentration employed for studied proteins as a standard negative control because it is unable to induce liposome leakage [29], and 0.1% Triton X-100 served as a positive control for complete liposome destabilization (maximal CF release).
Figure 1. The release of carboxyfluorescein (CF) from pL liposomes by FGF1, S100A13, and p40 Syt1.
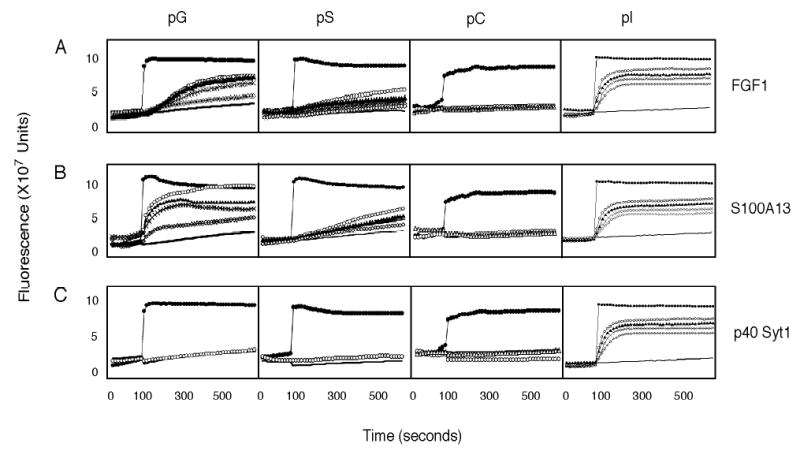
Liposomes were prepared using pS, pG, pI, or pC in the presence of CF. The release of CF after the addition of 1 mM (○-○), 500 nM (Δ-Δ), 250 nM (x-x), 125 nM (⋄-⋄) of recombinant FGF1, S100A13, and p40 Syt1 was continuously monitored using a fluorescence spectrophotometer (excitation:470 nm; emission:520 nm). The data are reported as a function of time in seconds. 1 mM α-chymotrypsin (—) or 0.1% Triton X100 (•-•) were used as respectively negative and positive controls of membrane destabiliization. The pL concentration in the cuvette was 2 μM.
We found that FGF1 induced destabilization of pI, pG, and pS liposomes, but not the pC liposomes. The dependence of the ability of FGF1 to induce liposome destabilization upon their pL composition may be represented by the following relationship: pI > pG > pS > pC (Figure 1A).
S100A13 exhibited destabilizing effects on liposomes with a similar relationship as FGF1: pG > pI > pS > pC (Figure 1B). Conversely, p40 Syt1 was not able to induce any CF release from pC, pS, and pG liposomes, but very efficiently destabilized pI liposomes (Figure 1C). It is important to stress that none of the proteins were able to induce CF release from zwitterionic (pC) liposomes (Figure 1A, B, C). The extent of CF release was proportional to protein concentration. Thus, S100A13 at 1 μM concentration induced CF release from pG liposomes almost equivalent to complete liposome destabilization induced by 0.1% Triton X-100 (Figure 1B).
We concluded that the proteins of the FGF1 release complex were able to induce destabilization of liposomes composed of acidic pL, but not zwitterionic pL, in a protein concentration-dependent manner. In addition, for p40 Syt1, this ability exhibited selectivity towards a specific acidic pL (pI).
p40 Syt1 K326,327,331Q mutant is not released by NIH 3T3 cells
The ability of the three members of the FGF1 release complex to destabilize acidic pL membranes prompted us to use mutagenesis to investigate whether this property is related to non-classical release. To that end, we first chose Syt1, whose interaction with membranes is extensively studied [26]. A loss of pI binding capability was described for a closely related protein Syt2 after substitution of three closely located lysine residues in the C2B domain [38]. Corresponding lysine residues of Syt1 bind PIP2, a pI derivate that is present at the plasma membrane [39]. We mutated these lysine residues (K326, 327 and 331) in the C2B domain of p40 Syt1, in order to evaluate the release of the resultant mutant during stress and at normal conditions. Heat shock experiments performed on NIH 3T3 cells transiently transfected with either the mutant or the WT form of p40 Syt1 demonstrated that, unlike the WT form, the p40 Syt1 K326,327,331Q mutant was not released either at 37º or 42ºC (Figure 2A). This suggests that the lysine residues at position 326, 327, 331 are required for the non-classical release of p40 Syt1 from cells.
Figure 2. A. The release of p40 Syt1 WT and p40 Syt1 K326,327,331Q from NIH 3T3 cells.
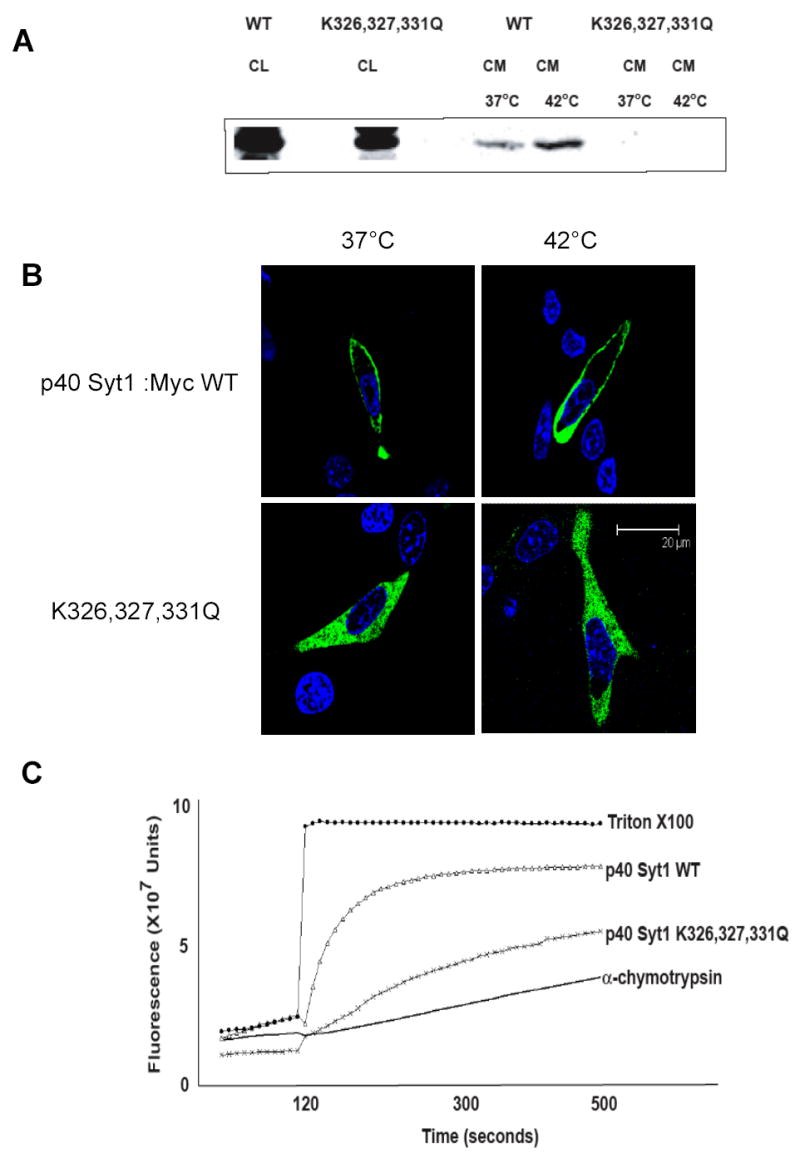
NIH 3T3 cells transfected with p40 Syt1 WT or p40 Syt1 K326,327,321Q were incubated for 2 hours at 37°C or 42°C. p40 Syt1 from conditioned medium was adsorbed to heparin-Sepharose and analyzed by immunoblotting as described [14]. CL- cell lysate; CM- conditioned medium. B. Confocal fluorescence microscopy analysis of p40 Syt1:Myc WT and p40 Syt1:Myc K326,327,331Q cellular localization at normal and stress conditions. NIH 3T3 cells transfected with either p40 Syt1:Myc WT or p40 Syt1:Myc K326,327,331Q were incubated for 2 hours at 37°C or 42°C. The cells were fixed with formaldehyde and immunofluorescently stained for Myc tag (green) as described in the “Materials and Methods”. The nuclei were stained with Hoechst 34580 (blue). The cells were studied using a confocal microscope with an X100 objective. Median horizontal confocal cell sections are presented. Bar- 20 μm. C. Analysis of the CF release from pI liposomes induced by p40 Syt1 WT and p40 Syt1 K326,327,331Q. The experiments on CF release from pI liposomes induced by 500 nM p40 Syt1 WT or p40 Syt1 K326,327,331Q were performed as described in Figure 1.
p40 Syt1 K326,327,331Q does not display enhanced perimembrane localization in transfected cell
Previously, we demonstrated that when the WT form of p40 Syt1 is co-expressed with FGF1, it migrates to the plasma membrane under stress conditions like other members of the FGF1 release complex [16]. Since p40 Syt1 K326,327,331Q is not released by NIH 3T3 cells, we evaluated its localization by confocal fluorescence microscopy during heat shock and at normal conditions in transiently transfected NIH 3T3 cells. Our study demonstrated that p40 Syt1 K326,327,331Q presented a diffuse cytosolic distribution pattern both at 37º and 42ºC (Figure 2B), which was unlike the WT form that displayed an enhanced localization in the vicinity of the cell membrane under both conditions (Figure 2B).
K326,327,331Q mutations in the p40 Syt1 C2B domain lead to a sharp decrease of pI liposome destabilization activity
The inability of p40 Syt1 K326,327,331Q to be released by NIH 3T3 cells and to exhibit preferential localization in the vicinity of the plasma membrane, prompted us to explore its liposome permeabilizing activity. The p40 Syt1 K326,327,331Q-induced CF release from pI liposomes was strongly reduced compared to WT p40 Syt1 (Figure 2C). These data suggest that the lysine residues at positions 326, 327, and 331 play a crucial role in the ability of p40 Syt1 to bind the plasma membrane, destabilize it, and exit to the extracellular compartment.
FGF1 K114,115A, and FGF1 K126,127A exhibit strongly attenuated release from NIH 3T3 cells
Since the C-terminal region of FGF1 is responsible for interaction with acidic pL [25] and we demonstrated that the substitution of a lysine cluster in p40 Syt1 results in the loss of the non-classical release of this protein from NIH 3T3 cells, we investigated the role of lysine clusters in the C-terminal region of FGF1 in FGF1 release, and its ability to destabilize acidic pL membranes. For this purpose, we produced two FGF1 mutants, FGF1 K114,115A and FGF1 K126,127A, and we tested their ability to be released by transiently transfected NIH 3T3 cells after heat shock. These experiments demonstrated that FGF1 K114,115A exhibited a drastically reduced release at 42°C as compared to the WT FGF1, while FGF1 K126,127 was not released at this temperature (Figure 3A). These data suggest that the clusters of lysine residues in the FGF1 C-terminal domain are important for stress-induced release of this growth factor from cells.
Figure 3. A. The release of FGF1 WT, FGF1 K114,115A, and FGF1 K126,127A from NIH 3T3 cells.
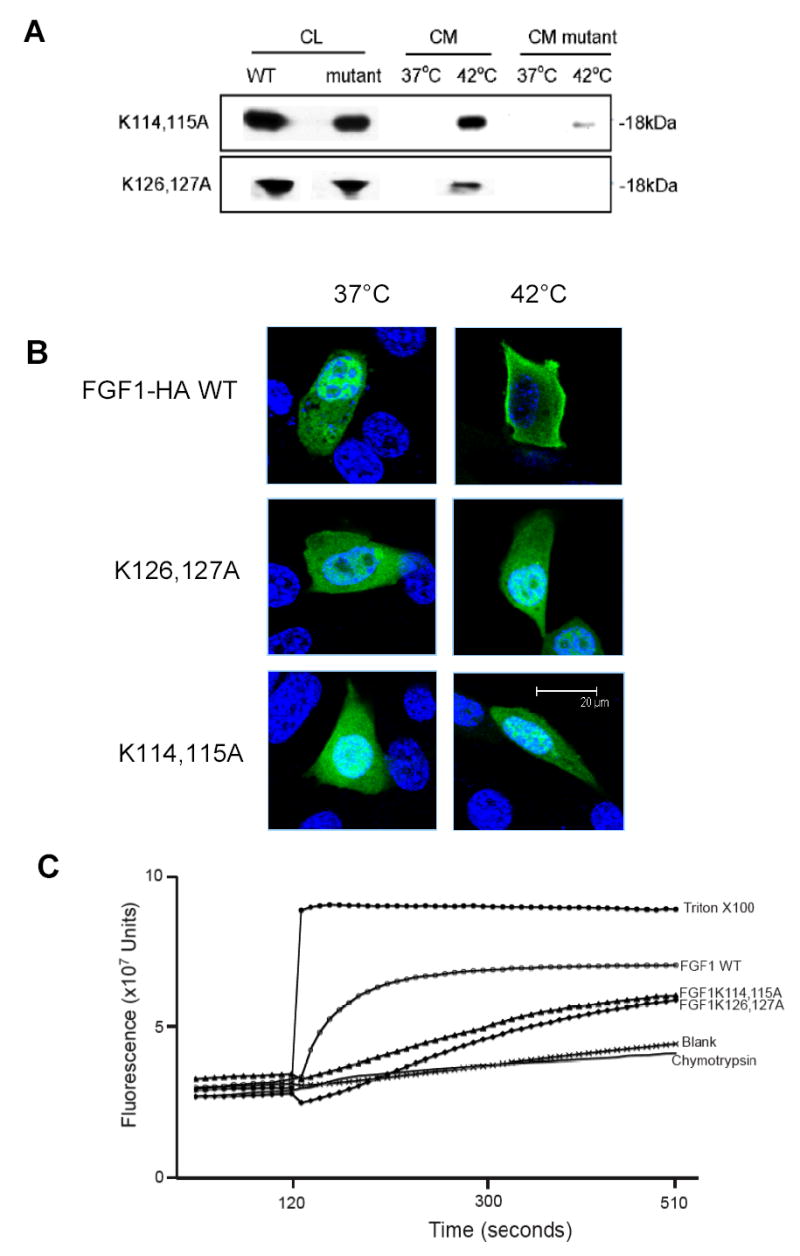
NIH 3T3 cells transfected with FGF1:HA WT, FGF1:HA K114,115A or FGF1:HA K126,127A were incubated for 2 hours at 37°C or 42°C. FGF1:HA from conditioned medium was immunoprecipitated and analyzed by immunoblotting as described in the Materials and Methods. CL- cell lysate; CM- conditioned medium. B. Confocal fluorescence microscopy analysis of FGF1:HA WT, FGF1:HA K114,115A and FGF1:HA K126,127A cellular localization at normal and stress conditions. NIH 3T3 cells transfected with either FGF1:HA WT, FGF1:HA K114,115A, or FGF1:HA K126,127A were incubated for 2 hours at 37°C or 42°C. Cells were fixed with formaldehyde and immunofluorescently stained for HA tag (green) as described in the Materials and Methods. Nuclei were stained with Hoechst 34580 (blue). The cells were studied using a confocal microscope with a X100 objective. Median horizontal confocal cell sections are presented. Bar- 20 μm. C. Analysis of the CF release from pG liposomes induced by FGF1 WT, FGF1 K114,115A, or FGF1 K126,127A. The experiments on CF release from pG liposomes induced by 500 nM FGF1 WT, FGF1 K114,115A, or FGF1 K126,127A were performed as described in Figure 1.
FGF1 K114,115A and FGF1 K126,127A do not display perimembrane redistribution at heat shock
We previously demonstrated that FGF1 and the components of its secretory pathway migrate toward the plasma membrane prior to release [16]. Since FGF1:HA K114,115A and FGF1:HA K126,127A, like p40 Syt1 K326,327,331Q, are not released from NIH 3T3 cells, we studied their intracellular localization under normal and stress conditions in order to define whether their impaired release was related to a defect in the association with the cell membrane. After transfecting NIH 3T3 cell cultures with either the mutants or the WT form of FGF1 and submitting them to heat shock, we performed immunofluorescence staining for the HA tag and studied the cells using confocal microscopy. We found (Figure 3B) that contrary to FGF1 WT, neither FGF1:HA K114,115A nor FGF1:HA K126,127A exhibited enhanced perimembrane localization after heat shock.
K114,115A or K126,127A mutations in the C-terminal region of FGF1 lead to the decrease of its ability to destabilize liposomes
To determine whether the attenuated secretion of FGF1:HA K114,115A and FGF1:HA K126,127A correlates with the decreased ability to destabilize acidic pL liposomes, we produced recombinant forms of FGF1 K114,115A and FGF1 K126,127A mutants and tested their effects on CF-loaded acidic pL liposomes. Recombinant FGF1 K114,115A and FGF1 K126,127A (Figure 3C) demonstrated a strong decrease of destabilizing activity towards pG liposomes as compared to destabilizing activity of the recombinant WT FGF1, suggesting that lysine clusters at the C-terminal domain of FGF1 are critical for both membrane destabilization and non-classical protein release.
DISCUSSION
Despite the absence of a classical signal sequence in its structure, FGF1 exhibits stress-induced release [14]. However, the mechanism used by FGF1 to cross the cell membrane is not known. Previously it was demonstrated that FGF1 binds acidic pL [25], and is able to deform lipid bilayers [30]. Moreover, FGF1 destabilizes liposomes containing acidic pL and assumes a partially unfolded MG-like conformation in the presence of acidic pL [29]. In the MG-like state conformation, FGF1 retains most of its secondary structure but its tertiary structure is almost completely disrupted. The solvent-exposed non-polar surface(s) in the MG-like state probably facilitates the interaction of FGF1 with the lipid membrane. Subsequently, the bound FGF1 appears to disorganize the lipid membrane, due to acquired hydrophobic characteristics that may facilitate its solubility in membranes and perturb their integrity [29, 33]. However, since FGF1 is released in association with S100A13 and p40 Syt1 [21, 22], the question arised whether the two latter proteins could also display membrane destabilizing properties. Similarly to FGF1 [25], Syt1 is able to associate with acidic pL, particularly pS and pI [26]. In addition, some S100 family members bind pL or pL-binding proteins [27, 28]. These previous observations prompted us to undertake a comparative study of the interaction of FGF1, p40 Syt1, and S100A13 with artificial membranes composed of different pL, to determine whether individual members of the FGF1 release complex exhibit preferential membrane destabilizing abilities dependent upon specific pL membrane composition. We chose pC, a zwitterionic pL, and three acidic pL - pI , pG, and pS. All of these pL are present in the plasma membrane [40–42], and acidic pL are preferentially localized in the plasma membrane inner leaflet [40]. We observed that the three proteins destabilized lipid membranes composed of acidic pL but not of pC, which is a zwitterionic pL. While FGF1 and S100A13 exhibited destabilizing activity towards pG, pS, and pI membranes, p40 Syt1 induced selective destabilization of only the pI liposomes.
Since we previously demonstrated that when the components of the FGF1 release complex are co-expressed, they localize at the plasma membrane in response to stress [16], the observation that FGF1 release complex members destabilize acidic pL liposomes suggests a critical role for plasma membrane pL in the assembly and export of the FGF1 release complex [12]. Particularly, the preference exhibited by p40 Syt1 in permeabilizing pI liposomes indicates the existence of sites of specific pL compositions that recruit different constituents of the FGF1 release complex at the inner leaflet of the plasma membrane. Therefore, it is likely that specific pL function as anchors for individual proteins, promoting the formation of the multiprotein FGF1 release complex at the inner leaflet of the cell membrane. Additionally, we should consider the interaction between acidic pL and the components of the FGF1 secretion pathway in terms of reciprocal structural changes: acidic pL might be responsible for inducing tertiary structure modifications that allow the proteins to become more hydrophobic [29]; on the other hand, the proteins might be responsible of membrane deformation [30]. Thus, both of these mechanisms might ultimately contribute to protein release. This might be the case of p40 Syt1, which binds pS and pI respectively through the C2A and C2B domains [26], but exhibits destabilizing activity only towards pI liposomes. It is also important to mention that the ability of some pL, i.e. pS, to flip from the inner to the outer face of the plasma membrane in response to stress [43, 44] might also be involved in the protein translocation process.
The calcium-binding protein, S1001A13, which is released from cells in association with FGF1 [21], and similarly to FGF1 exhibits destabilizing activity upon acidic pL liposomes (Figure 2), is a member of the S100 proteins family. S100 proteins regulate the functions of their protein partners through induced conformational changes. In particular, S100A10 regulates the process of insertion of annexin II into the plasma membrane [45]. Similarly to S100A10, S100A13 combined with acidic pL might promote FGF1 and p40 Syt1 conformational changes, and translocation of the FGF1 release complex across the plasma membrane.
The establishment of membrane destabilizing properties of the FGF1 release complex components prompted us to use mutational analysis to identify the membrane destabilizing domains of p40 Syt1 and FGF1. To this purpose, we used available information regarding the acidic pL-binding domains of synaptotagmins [38, 39] and FGF1 [25]. We found that mutation of lysines 326, 327, and 331 to glutamines resulted in the inability of p40 Syt1 to be released from NIH 3T3 cells both at 37º and at 42ºC, as well as a strong decrease of pI liposome destabilization. Additionally, whereas p40 Syt1 WT exhibited perimembrane localization, p40 Syt1 K326,327,331Q was distributed diffusely in the cytoplasm of transfected cells. Similarly, substitution of lysine residues in the C-terminal domain of FGF1 abolished its capability to be released from NIH 3T3 cells, to localize at the plasma membrane under stress conditions, and to perturb acidic pL bilayer integrity. Thus, clusters of lysine residues (i) are responsible for the interaction of p40 Syt1 and FGF1 with the plasma membrane, (ii) are required for their non-classical release, (iii) are at least partially responsible for the destabilization of acidic pL membranes.
In conclusion, this work demonstrated that the members of non-classical FGF1 release complex exhibited the ability to destabilize artificial membranes composed of acidic pL, which are preferentially localized in the inner leaflet of the cell membrane [46, Micheva, 2001 #3641]. Moreover, the membrane destabilizing ability of p40 Syt1 and FGF1 correlated with their non-classical release from NIH 3T3 cells. Additionally, we previously reported that interleukin 1α, a protein exported through a non-classical release pathway similar to FGF1, also efficiently permeabilizes pG liposomes [36]. Further identification of the membrane destabilizing domains of S100A13 and structural characterization of the interaction of the FGF1 release complex components with lipid membranes is important for the understanding of the mechanism of non-classical protein release, which may be applied to the rational design of therapeutic compounds that interfere with this process.
Acknowledgments
This work was supported in part by NIH grants, HL 32348, HL 35627, and P20 RR 15555 (Project # 4) to IP and by the NSF grant 0132384 to DN. It was also partially supported by the grants to TKSK: NIH NCRR COBRE (P20 RR15569), Department of Energy (DE-FG02-01ER15161), and to CY: The Arkansas Bioscience Institute and the National Science Council, Taiwan (NSC 94-2320-B007-005 and NSC-94-2113-M007-036).
The authors wish to thank the laboratory of R. Middaugh at the University of Kansas for their help in the design of the liposome studies, and Andrew Baird from the Human BioMolecular Research Institute, San Diego, CA for the FGF1:HA construct.
The work was performed by IG and MD in partial fulfillment of the requirements for Ph.D. degree from the Department of Critical Care Medicine and Surgery, Gerontology and Geriatrics Unit, University of Florence, Florence, Italy, and from the Life and Health Science University of Minho in Braga, Portugal, respectively.
Footnotes
The article is dedicated to the memory of Tom Maciag, friend, scientist, and mentor.
References
- 1.Bottcher RT, Niehrs C. Fibroblast growth factor signaling during early vertebrate development. Endocr Rev. 2005;26:63–77. doi: 10.1210/er.2003-0040. [DOI] [PubMed] [Google Scholar]
- 2.Friesel R, Maciag T. Fibroblast growth factor prototype release and fibroblast growth factor receptor signaling. Thromb Haemost. 1999;82:748–754. [PubMed] [Google Scholar]
- 3.Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16:233–247. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Nickel W. Unconventional secretory routes: direct protein export across the plasma membrane of mammalian cells. Traffic. 2005;6:607–614. doi: 10.1111/j.1600-0854.2005.00302.x. [DOI] [PubMed] [Google Scholar]
- 5.Florkiewicz RZ, Majack RA, Buechler RD, Florkiewicz E. Quantitative export of FGF-2 occurs through an alternative, energy- dependent, non-ER/Golgi pathway. J Cell Physiol. 1995;162:388–399. doi: 10.1002/jcp.1041620311. [DOI] [PubMed] [Google Scholar]
- 6.Mignatti P, Morimoto T, Rifkin DB. Basic fibroblast growth factor, a protein devoid of secretory signal sequence, is released by cells via a pathway independent of the endoplasmic reticulum-Golgi complex. J Cell Physiol. 1992;151:81–93. doi: 10.1002/jcp.1041510113. [DOI] [PubMed] [Google Scholar]
- 7.Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol Biol Cell. 1999;10:1463–1475. doi: 10.1091/mbc.10.5.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrei C, Margiocco P, Poggi A, Lotti LV, Torrisi MR, Rubartelli A. Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: Implications for inflammatory processes. Proc Natl Acad Sci U S A. 2004;101:9745–9750. doi: 10.1073/pnas.0308558101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunter-Lavin C, Davies EL, Bacelar MM, Marshall MJ, Andrew SM, Williams JH. Hsp70 release from peripheral blood mononuclear cells. Biochem Biophys Res Commun. 2004;324:511–517. doi: 10.1016/j.bbrc.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 10.Jeske NA, Glucksman MJ, Roberts JL. Metalloendopeptidase EC3.4.24.15 is constitutively released from the exofacial leaflet of lipid rafts in GT1-7 cells. J Neurochem. 2004;90:819–828. doi: 10.1111/j.1471-4159.2004.02557.x. [DOI] [PubMed] [Google Scholar]
- 11.Lee SJ, Saravanan RS, Damasceno CM, Yamane H, Kim BD, Rose JK. Digging deeper into the plant cell wall proteome. Plant Physiol Biochem. 2004;42:979–988. doi: 10.1016/j.plaphy.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 12.Prudovsky I, Mandinova A, Soldi R, Bagala C, Graziani I, Landriscina M, Tarantini F, Duarte M, Bellum S, Doherty H, Maciag T. The non-classical export routes: FGF1 and IL-1alpha point the way. J Cell Sci. 2003;116:4871–4881. doi: 10.1242/jcs.00872. [DOI] [PubMed] [Google Scholar]
- 13.Jaye M, Howk R, Burgess W, Ricca GA, Chiu IM, Ravera MW, O’Brien SJ, Modi WS, Maciag T, Drohan WN. Human endothelial cell growth factor: cloning, nucleotide sequence, and chromosome localization. Science. 1986;233:541–545. doi: 10.1126/science.3523756. [DOI] [PubMed] [Google Scholar]
- 14.Jackson A, Friedman S, Zhan X, Engleka KA, Forough R, Maciag T. Heat shock induces the release of fibroblast growth factor 1 from NIH 3T3 cells. Proc Natl Acad Sci U S A. 1992;89:10691–10695. doi: 10.1073/pnas.89.22.10691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J Biol Chem. 1986;261:11398–11403. [PubMed] [Google Scholar]
- 16.Prudovsky I, Bagala C, Tarantini F, Mandinova A, Soldi R, Bellum S, Maciag T. The intracellular translocation of the components of the fibroblast growth factor 1 release complex precedes their assembly prior to export. J Cell Biol. 2002;158:201–208. doi: 10.1083/jcb.200203084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carreira C Mouta, Landriscina M, Bellum S, Prudovsky I, Maciag T. The comparative release of FGF1 by hypoxia and temperature stress. Growth Factors. 2001;18:277–285. doi: 10.3109/08977190109029116. [DOI] [PubMed] [Google Scholar]
- 18.Shin JT, Opalenik SR, Wehby JN, Mahesh VK, Jackson A, Tarantini F, Maciag T, Thompson JA. Serum-starvation induces the extracellular appearance of FGF-1. Biochim Biophys Acta. 1996;1312:27–38. doi: 10.1016/0167-4889(96)00013-4. [DOI] [PubMed] [Google Scholar]
- 19.Ananyeva NM, Tijurmin AV, Berliner JA, Chisolm GM, Liau G, Winkles JA, Haudenschild CC. Oxidized LDL mediates the release of fibroblast growth factor-1. Arterioscler Thromb Vasc Biol. 1997;17:445–453. doi: 10.1161/01.atv.17.3.445. [DOI] [PubMed] [Google Scholar]
- 20.Landriscina M, Bagala C, Mandinova A, Soldi R, Micucci I, Bellum S, Prudovsky I, Maciag T. Copper induces the assembly of a multiprotein aggregate implicated in the release of fibroblast growth factor 1 in response to stress. J Biol Chem. 2001;276:25549–25557. doi: 10.1074/jbc.M102925200. [DOI] [PubMed] [Google Scholar]
- 21.Landriscina M, Soldi R, Bagala C, Micucci I, Bellum S, Tarantini F, Prudovsky I, Maciag T. S100A13 participates in the release of fibroblast growth factor 1 in response to heat shock in vitro. J Biol Chem. 2001;276:22544–22552. doi: 10.1074/jbc.M100546200. [DOI] [PubMed] [Google Scholar]
- 22.LaVallee TM, Tarantini F, Gamble S, Carreira CM, Jackson A, Maciag T. Synaptotagmin-1 is required for fibroblast growth factor-1 release. J Biol Chem. 1998;273:22217–22223. doi: 10.1074/jbc.273.35.22217. [DOI] [PubMed] [Google Scholar]
- 23.Yoshihara M, Montana ES. The synaptotagmins: calcium sensors for vesicular trafficking. Neuroscientist. 2004;10:566–574. doi: 10.1177/1073858404268770. [DOI] [PubMed] [Google Scholar]
- 24.Bagala C, Kolev V, Mandinova A, Soldi R, Mouta C, Graziani I, Prudovsky I, Maciag T. The alternative translation of synaptotagmin 1 mediates the non-classical release of FGF1. Biochem Biophys Res Commun. 2003;310:1041–1047. doi: 10.1016/j.bbrc.2003.09.119. [DOI] [PubMed] [Google Scholar]
- 25.Tarantini F, Gamble S, Jackson A, Maciag T. The cysteine residue responsible for the release of fibroblast growth factor-1 residues in a domain independent of the domain for phosphatidylserine binding. J Biol Chem. 1995;270:29039–29042. doi: 10.1074/jbc.270.49.29039. [DOI] [PubMed] [Google Scholar]
- 26.Bai J, Chapman ER. The C2 domains of synaptotagmin--partners in exocytosis. Trends Biochem Sci. 2004;29:143–151. doi: 10.1016/j.tibs.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Rescher U, Gerke V. Annexins--unique membrane binding proteins with diverse functions. J Cell Sci. 2004;117:2631–2639. doi: 10.1242/jcs.01245. [DOI] [PubMed] [Google Scholar]
- 28.Nacken W, Sorg C, Kerkhoff C. The myeloid expressed EF-hand proteins display a diverse pattern of lipid raft association. FEBS Lett. 2004;572:289–293. doi: 10.1016/j.febslet.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 29.Mach H, Middaugh CR. Interaction of partially structured states of acidic fibroblast growth factor with phospholipid membranes. Biochemistry. 1995;34:9913–9920. doi: 10.1021/bi00031a013. [DOI] [PubMed] [Google Scholar]
- 30.Doyle AW, Fick J, Himmelhaus M, Eck W, Graziani I, Prudovsky I, Grunze M, Maciag T, Neivandt D. Protein deformation of lipid hybrid bilayer membranes studied by Sum Frequency Generation Vibrational Spectroscopy (SFS) Langmuir. 2004;20:8961–8965. doi: 10.1021/la0484220. [DOI] [PubMed] [Google Scholar]
- 31.Copeland RA, Ji H, Halfpenny AJ, Williams RW, Thompson KC, Herber WK, Thomas KA, Bruner MW, Ryan JA, Marquis-Omer D, et al. The structure of human acidic fibroblast growth factor and its interaction with heparin. Arch Biochem Biophys. 1991;289:53–61. doi: 10.1016/0003-9861(91)90441-k. [DOI] [PubMed] [Google Scholar]
- 32.Dabora JM, Sanyal G, Middaugh CR. Effect of polyanions on the refolding of human acidic fibroblast growth factor. J Biol Chem. 1991;266:23637–23640. [PubMed] [Google Scholar]
- 33.Bychkova VE, Pain RH, Ptitsyn OB. The ‘molten globule’ state is involved in the translocation of proteins across membranes? FEBS Lett. 1988;238:231–234. doi: 10.1016/0014-5793(88)80485-x. [DOI] [PubMed] [Google Scholar]
- 34.Tarantini F, LaVallee T, Jackson A, Gamble S, Carreira C Mouta, Garfinkel S, Burgess WH, Maciag T. The extravesicular domain of synaptotagmin-1 is released with the latent fibroblast growth factor-1 homodimer in response to heat shock. J Biol Chem. 1998;273:22209–22216. doi: 10.1074/jbc.273.35.22209. [DOI] [PubMed] [Google Scholar]
- 35.Engleka KA, Maciag T. Inactivation of human fibroblast growth factor-1 (FGF-1) activity by interaction with copper ions involves FGF-1 dimer formation induced by copper-catalyzed oxidation. J Biol Chem. 1992;267:11307–11315. [PubMed] [Google Scholar]
- 36.Mandinova A, Soldi R, Graziani I, Bagala C, Bellum S, Landriscina M, Tarantini F, Prudovsky I, Maciag T. S100A13 mediates the copper-dependent stress-induced release of IL-1{alpha} from both human U937 and murine NIH 3T3 cells. J Cell Sci. 2003;116:2687–2696. doi: 10.1242/jcs.00471. [DOI] [PubMed] [Google Scholar]
- 37.Matsuzaki K, Murase O, Tokuda H, Funakoshi S, Fujii N, Miyajima K. Orientational and aggregational states of magainin 2 in phospholipid bilayers. Biochemistry. 1994;33:3342–3349. doi: 10.1021/bi00177a027. [DOI] [PubMed] [Google Scholar]
- 38.Kida Y, Sakaguchi M, Fukuda M, Mikoshiba K, Mihara K. Amino acid residues before the hydrophobic region which are critical for membrane translocation of the N-terminal domain of synaptotagmin II. FEBS Lett. 2001;507:341–345. doi: 10.1016/s0014-5793(01)03000-9. [DOI] [PubMed] [Google Scholar]
- 39.Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- 40.McLaughlin S, Hangyas-Mihalyne G, Zaitseva I, Golebiewska U. Reversible - through calmodulin - electrostatic interactions between basic residues on proteins and acidic lipids in the plasma membrane. Biochem Soc Symp. 2005:189–198. doi: 10.1042/bss0720189. [DOI] [PubMed] [Google Scholar]
- 41.Momchilova-Pankova AB, Markovska TT, Koumanov KS. Acyl-CoA synthetase activity depends on the phospholipid composition of rat liver plasma membranes. J Lipid Mediat Cell Signal. 1995;11:13–23. doi: 10.1016/0929-7855(94)00024-7. [DOI] [PubMed] [Google Scholar]
- 42.Schroeder F, Fontaine RN, Feller DJ, Weston KG. Drug-induced surface membrane phospholipid composition in murine fibroblasts. Biochim Biophys Acta. 1981;643:76–88. doi: 10.1016/0005-2736(81)90220-0. [DOI] [PubMed] [Google Scholar]
- 43.Kuypers FA, de Jong K. The role of phosphatidylserine in recognition and removal of erythrocytes. Cell Mol Biol (Noisy-le-grand) 2004;50:147–158. [PubMed] [Google Scholar]
- 44.Kagan VE, Borisenko GG, Serinkan BF, Tyurina YY, Tyurin VA, Jiang J, Liu SX, Shvedova AA, Fabisiak JP, Uthaisang W, Fadeel B. Appetizing rancidity of apoptotic cells for macrophages: oxidation, externalization, and recognition of phosphatidylserine. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1–17. doi: 10.1152/ajplung.00365.2002. [DOI] [PubMed] [Google Scholar]
- 45.Donato R. Intracellular and extracellular roles of S100 proteins. Microsc Res Tech. 2003;60:540–551. doi: 10.1002/jemt.10296. [DOI] [PubMed] [Google Scholar]
- 46.Holz RW, Hlubek MD, Sorensen SD, Fisher SK, Balla T, Ozaki S, Prestwich GD, Stuenkel EL, Bittner MA. A pleckstrin homology domain specific for phosphatidylinositol 4, 5-bisphosphate (PtdIns-4,5-P2) and fused to green fluorescent protein identifies plasma membrane PtdIns-4,5-P2 as being important in exocytosis. J Biol Chem. 2000;275:17878–17885. doi: 10.1074/jbc.M000925200. [DOI] [PubMed] [Google Scholar]