

Abstract
Experimental evidence indicates that, under some circumstances, “surrogate” molecules may play the same role as cholesterol in ordering membrane lipids. The simplest molecule in this class is Ceramide. In this article, we describe atomic-level molecular dynamics simulations designed to shed light on this phenomenon. We run simulations of hydrated phosphoryl-oleoyl phosphatidylcholine (POPC) bilayers containing cholesterol, and containing ceramide, in concentrations ranging from 5% to 33%. We also perform a simulation of a pure POPC bilayer to verify the simulation force fields against experimental structural data for POPC. Our simulation data are in good agreement with experimental data for the partial molecular volumes, areas, form factors, and order parameters. These simulations suggest that ceramide and cholesterol have a very similar effect on the POPC bilayer, although ceramide is less effective in inducing order in the bilayer compared with cholesterol at the same concentrations.
INTRODUCTION
Eukaryotic cellular life has an undeniable dependence on cholesterol (1,2). It comprises ∼40 mol % of the lipid portion of the eukaryotic plasma membrane, and is generally responsible for the modulation of the physico-chemical properties required for viability and cell proliferation (1,2). It is known that cholesterol reduces the passive permeability of membranes, increases membrane mechanical strength, and modulates membrane enzymes (3). Cholesterol is involved in the formation of membrane rafts—domains in which cholesterol, saturated long-chained lipids, and specific proteins are concentrated (4,5), and consisting of saturated lipids like sphingomyelin (SM) and cholesterol. Numerous physical and thermodynamic studies over the past several decades have revealed details of lipid-cholesterol interactions (2,3). Three distinct types of lipid ordering are observed in mixtures of lipid and cholesterol: 1), gel-like ordering, in which lipid chains are highly ordered and lipid diffusion in two dimensions is very low; 2), fluid-like ordering, where lipid chains are disordered and lipid diffusion is much higher compared to gel phase; and 3), an intermediate state with gel-like chain order but fluid-like lateral lipid diffusion.
Since cholesterol is a critical constituent of bilayers, an important question arises: Is cholesterol the only membrane intercalator that can induce a state of intermediate order in lipid bilayers? In recent experiments by Lange et al. (6), Megha and London (7), and Zitzer et al. (8), the amount of cholesterol displaced by different membrane intercalators has been studied. Interestingly these experiments show that ceramides, diglycerides, amphipathic alcohols, and other sterols can act as cholesterol substitutes in membranes, including raft-like domains (7). These data suggest that smaller molecules that can induce condensation effect upon addition in lipid bilayer, can, like cholesterol, affect lateral organization in a lipid bilayer.
The simplest lipid in this group is ceramide. Ceramide (Cer), in addition to being an important signaling lipid, is a major lipid in the stratum corneum epidermal layer of many animals, including humans. Here, it participates in the formation of a highly ordered barrier to permeation, essential to the skin. The physical and thermodynamic properties of Cer bilayers have been studied by a number of groups (9–14). Of particular interest is C16:0 Cer. Shipley and co-workers synthesized and purified C16:0 Cer, and used differential scanning calorimetry and x-ray scattering to obtain structural and thermodynamic properties of this lipid in both hydrated and anhydrous dispersions (9). They found that hydrated C16:0 Cer is in an ordered lamellar phase up to ∼90°C, where it undergoes a chain-melting transition. Other than in the stratum corneum, Cer is found only as a relatively minor component in membranes. However, as discussed above, it has several important biological roles. For this reason, it is of interest to investigate the properties of mixed lipid bilayers containing phospholipids, sphingolipids, Cer, and cholesterol.
Massey (13) used fluorescence spectroscopy to examine the interactions of Cer with the phospholipids DPPC and POPC, and with bovine brain sphingomyelin (in the latter case both with and without 33% cholesterol). Bovine brain Cer, and synthetic C16:0, C18:0, C18:1, C24:1 Cer were used, along with bovine brain, bovine erythrocyte, and egg yolk sphingomyelin. In all cases the addition of Cer increased the phase transition temperature of the host phospholipid or sphingolipid. The magnitude of the effect ranged from 0.12°C per mol % Cer for C24:1 Cer in DPPC to 1.15°C per mol % Cer for bovine brain Cer in bovine erythrocyte sphingomyelin. The effect of Cer on DPPC bilayers above the chain-melting temperature for DPPC is similar to that observed for POPC.
In comprehensive deuterium NMR and calorimetry studies, Thewalt et al. (14) measured the order and phase properties of mixtures of POPC and C16:0 Cer over a range of temperatures and Cer concentrations. Their data suggests that POPC/Cer bilayers show gel phase immiscibility, and that the addition of Cer raises the chain-melting temperature of the bilayer above that of pure POPC. Based on their experiments they proposed a partial phase diagram for the C16:0 Cer-POPC system. At temperatures above ∼0°C and Cer concentrations above ∼5%, there are separate regions of gel (rich in Cer) and liquid crystalline (mainly POPC) lipid. Order parameters for both Cer and POPC chains were also measured. The order parameter values increase with the addition of Cer up to 15% Cer, the maximum concentration for which order parameters were measured. The quantitative data presented by Thewalt et al. (14) make the POPC–Cer system the best choice for simulation, to allow for the testing of simulation parameters.
Strong evidence for Cer-induced phase separations comes from experiments in which sphingomyelinase is introduced into mixed lipid systems consisting of PC and SM. Sphingomyelinase, which cleaves the phosphocholine from SM, producing Cer, has the effect of introducing Cer in controlled amounts into the system. It is found that sphingomyelinase induces separations in SM-PC lipid vesicles (15) and monolayers (16) into Cer-SM rich domains, which coexist with Cer poor domains. The addition of Cer to one side of a model membrane either directly or through the action of sphingomyelinase, induced transbilayer lipid redistribution, which may be related to the signaling properties of Cer (17).
In this article, we introduce molecular dynamics (MD) simulations designed to compare the effects of Cer and Chol on bilayers of POPC. Simulations are performed on mixtures of POPC-Chol and POPC-Cer at the concentrations studied in the NMR experiments of Hsueh et al. (18). In the following sections, we describe our simulations and the results.
METHOD
Molecular dynamics (MD) simulations were performed on a hydrated POPC bilayer and POPC mixtures with Cer and Chol at 10%, 20%, 25%, and 33%. These simulations were performed using the GROMACS package (19,20). Force-field parameters for the Chol and Cer molecules were taken from our previous work (21,22). Parameters for the phosphocholine polar groups were taken from our DPPC force field (23). The hydrocarbon chain parameters were taken from our earlier determination of these quantities by fitting to density and heat of vaporization data (24). The LINCS algorithm was used to constrain all bonds in the system (25) allowing an integration time step of 4 fs. Periodic boundary conditions were applied in all three dimensions and long-range electrostatics were calculated using the SPME algorithm (26) with a real-space cutoff of 9.5 Å. A cutoff of 18 Å was employed for van der Waals interactions. The temperature in all the simulations was maintained at 303 K, using the Nosé-Hoover scheme. The systems were equilibrated in an NPT ensemble using the Parrinello-Rahman pressure-coupling scheme (27,28) at a constant pressure of 1 atm.
Initial configurations for all the mixture systems were generated by random placement of 100 mixture molecules per leaflet in appropriate proportion such that the phosphorus atoms of the headgroups were at the z = 25 Å and z = − 25 Å planes and the hydrocarbon chain were pointing toward the z = 0 plane. Two slabs of 5000 SPCE waters were placed above and below the constructed bilayers. The systems were energy-minimized to remove bad contacts. A 200 ps MD simulation was performed on each system at 500 K. This was done to ensure proper disordering of the hydrocarbon chains. Then the temperature was brought down to 303 K in steps of 50 K. At each temperature step a small 100 ps MD simulation was performed on each system. Both the systems were simulated for 2 ns of MD with regeneration of velocities from a Maxwellian distribution at 303 K after every 100 ps. Then 3 ns of continuous MD simulations were performed on each system. At this point, the velocities were regenerated as before and continuous 20-ns simulations were performed on all the systems. Throughout the simulations, we monitored the dimensions of the simulation cells.
The hydrated POPC bilayer was constructed with 64 POPC molecules per leaflet and 28 waters per molecule. The bilayer was equilibrated for 10 ns followed by 60 ns continuous MD run at 303 K and 1 bar. All the other simulation parameters were exactly like the mixture simulations except for the time step, which was 2 fs.
RESULTS AND DISCUSSIONS
Hydrated POPC bilayer
Before studying the mixing properties of Cer and Chol in POPC, we validated force-field parameters for POPC by computing key structural properties such as volume and area per lipid. The area per lipid Al is given by
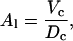 |
(1) |
where Vc is the volume of the hydrocarbon core and Dc is the thickness of the average molecular hydrocarbon core (29).
The hydrocarbon volume is calculated by the method proposed by Petrache et al. (30). In this method the system is divided into ns slabs and the partial specific volumes of the atom types (vi) in the system are obtained by imposing conservation of total volume for each slice element through minimization of the following function,
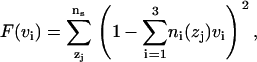 |
(2) |
over vi, where ni(zj) is the number density of ith type in zj slice. In these calculations, we divided the atoms into three distinct types: water, headgroup+glycerol backbone (HG), and hydrocarbon core (HC). The volume of each element is reported in Table 1. We note that the HC, HG, and total POPC volumes agree well with experimental volumes (31). The hydrocarbon thickness, Dc, is calculated by determining the half-value of the probability distribution of the hydrocarbon core (29). Fig. 1 shows this probability distribution of the hydrocarbon chains. This distribution is obtained by computing the number densities of the atoms from hydrocarbon, headgroup, and water region and normalizing it with the total number of atoms in the slice. Two vertical lines at 1.37 nm are the Dc boundaries. The hydrocarbon thickness determined by this method is in excellent agreement with the experimental values (see Table 1).
TABLE 1.
Structural results for hydrated POPC bilayer
| POPC (MD) | POPC (Experiment 31) | |
|---|---|---|
| Al (Å2) | 66.5 | 68.3 |
| VPOPC (Å3) | 1241.8 | 1256 |
| Vc (Å3) | 911.4 | 924.2 |
| VHG (Å3) | 330.4 | 331 |
| Vwater (Å3) | 30.54 | — |
| 2Dc (Å) | 27.4 | 27.1 |
| DHH (Å) | 36.6 | 37.0 |
FIGURE 1.

Probability distribution for the hydrocarbon region of pure POPC. Vertical axis is the normalized probability (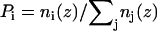 ) of locating a hydrocarbon chain, headgroup, and water. Horizontal axis is the distance from the bilayer center (z = 0). The vertical dashed lines are the Dc boundaries. The horizontal dashed line is the line of half probability. To precisely calculate the hydrocarbon thickness, the plot is not symmetrized across the two leaflets.
) of locating a hydrocarbon chain, headgroup, and water. Horizontal axis is the distance from the bilayer center (z = 0). The vertical dashed lines are the Dc boundaries. The horizontal dashed line is the line of half probability. To precisely calculate the hydrocarbon thickness, the plot is not symmetrized across the two leaflets.
A commonly studied structural property of the bilayer is its electron density profile. In experiments, the form factor of the bilayer is obtained from the x-ray diffraction pattern. Then the electron density profile is deduced from the form factor. In simulations, we directly calculate the electron density. We then calculate the form factors from the electron densities using
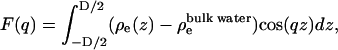 |
(3) |
where D is average length of the simulation cell in z direction, ρe(z) is symmetrized electron density of the system, and 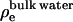 is the electron density of the bulk water. Fig. 2 a shows the calculated x-ray form factors for POPC, in comparison with experimental data from Kucerka et al. (31). We observe good agreement, down to the third lobe, where simulation data rises above the experimental data points. Fig. 2 b shows the electron density profiles, for both simulation and model-fitted electron densities from x-ray scattering. The data show good agreement in the hydrocarbon core. The simulated electron densities are slightly lower in the headgroup and bulk water region than the experiments. In these simulations the bulk density of the SPC/E water is 986 g/L as opposed to the real water density of 997 g/L at the same temperature. These numbers are consistent with a recent study by van der Spoel and Maaren (32) on bulk water densities for various water models. We hypothesize that the slightly lower electron density in bulk and headgroup regions of hydrated POPC bilayer is due to this lower water density.
is the electron density of the bulk water. Fig. 2 a shows the calculated x-ray form factors for POPC, in comparison with experimental data from Kucerka et al. (31). We observe good agreement, down to the third lobe, where simulation data rises above the experimental data points. Fig. 2 b shows the electron density profiles, for both simulation and model-fitted electron densities from x-ray scattering. The data show good agreement in the hydrocarbon core. The simulated electron densities are slightly lower in the headgroup and bulk water region than the experiments. In these simulations the bulk density of the SPC/E water is 986 g/L as opposed to the real water density of 997 g/L at the same temperature. These numbers are consistent with a recent study by van der Spoel and Maaren (32) on bulk water densities for various water models. We hypothesize that the slightly lower electron density in bulk and headgroup regions of hydrated POPC bilayer is due to this lower water density.
FIGURE 2.

Simulated and experimental x-ray structure data for pure POPC. (a) Form factors. (b) Electron density. The x-ray data are from Kucerka et al. (31). The electron density plot is symmetrized around the bilayer center by averaging densities over two leaflets because, in Eq. 3, a symmetric electron density is assumed.
Computing the area per POPC molecule from our simulated data by the method of Nagle and Tristram-Nagle (29) (see Eq. 1) gives a value of 66.5 Å2 (see Table 1) in close agreement with the current best experimental value of 68.3 ± 1.5 Å2 31. On the other hand, if we divide our simulated box cross-sectional area by the number of POPC molecules in one leaflet, we obtain an area per molecule, which we denote as “geometric area” (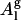 ), of 63 Å2, and is 6% lower. Since the molecular volume, the form factor, and the electron density obtained from the pure POPC simulations are in good agreement with experiments, it is appropriate to consider Al (Eq. 1) as the molecular area predicted by the simulation instead of
), of 63 Å2, and is 6% lower. Since the molecular volume, the form factor, and the electron density obtained from the pure POPC simulations are in good agreement with experiments, it is appropriate to consider Al (Eq. 1) as the molecular area predicted by the simulation instead of 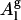 . There are two additional reasons for choosing Al as lipid area over
. There are two additional reasons for choosing Al as lipid area over 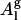 : 1), the experimentally reported area per lipid is Al (
: 1), the experimentally reported area per lipid is Al (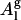 cannot be measured experimentally); and 2), the value of
cannot be measured experimentally); and 2), the value of 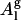 depends on the details of the pressure-coupling algorithm and the parameters used in the implementation of the pressure-coupling scheme. Further, if we use
depends on the details of the pressure-coupling algorithm and the parameters used in the implementation of the pressure-coupling scheme. Further, if we use  and the simulation hydrocarbon volume (Table 1) and calculate the hydrocarbon thickness, we obtain
and the simulation hydrocarbon volume (Table 1) and calculate the hydrocarbon thickness, we obtain 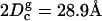 . This value is larger than the experimental and the simulation value reported in Table 1. We suspect that the discrepancy between the values of
. This value is larger than the experimental and the simulation value reported in Table 1. We suspect that the discrepancy between the values of 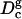 and Dc is due to slow moving water molecules that are trapped in the vicinity of the glycerol backbone and the hydrocarbon chains. A simulation at least an order-of-magnitude longer is needed to resolve this issue. Another possible cause of the discrepancy between the values of
and Dc is due to slow moving water molecules that are trapped in the vicinity of the glycerol backbone and the hydrocarbon chains. A simulation at least an order-of-magnitude longer is needed to resolve this issue. Another possible cause of the discrepancy between the values of 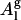 and Al may be undulations in the simulated bilayer. Undulations could introduce errors into the calculation of the projected area, thereby producing as lower value for the geometrical area. However, due to small size and periodic boundaries, such undulations are suppressed. Hence, we believe that it is more likely that the discrepancy is due to slow-moving trapped water molecules, in the vicinity of the glycerol backbone. So far, no phospholipid/water force field has been shown to match bilayer experiment to simulation in a completely consistent manner (33,34).
and Al may be undulations in the simulated bilayer. Undulations could introduce errors into the calculation of the projected area, thereby producing as lower value for the geometrical area. However, due to small size and periodic boundaries, such undulations are suppressed. Hence, we believe that it is more likely that the discrepancy is due to slow-moving trapped water molecules, in the vicinity of the glycerol backbone. So far, no phospholipid/water force field has been shown to match bilayer experiment to simulation in a completely consistent manner (33,34).
Mixtures of intercalators with POPC
With respect to the structural properties of mixed POPC-Cer and POPC-Chol bilayers, we first consider molecular volumes. Recent experimental data by Greenwood et al. (35) for partial molecular volumes of POPC–Chol mixtures indicate POPC volume to be 1256.2 Å3 and Chol volume as 622.5 ± 10 Å3. In our simulations, we computed these volumes by computing the volume of the simulation cell and fitting a straight line to the expression
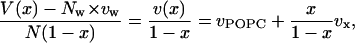 |
(4) |
where V is the volume of the simulation cell, Nw is the number of water molecules in the system, vw is the volume of a water molecule, N = NPOPC + Nx is the total number of lipids in the system, x = Nx/N, and vPOPC, vx are the volumes per molecule of POPC and the intercalator in the system, respectively. Fig. 3 shows plots of v(x)/(1 − x) as a function of x/(1 − x). The simulated points fall almost on a straight line for Cer as well as Chol simulations. The slopes of these lines give the specific molecular volume of Chol to be 604.76 ± 4.98 Å3 and of Cer to be 943.11 ± 3.72 Å3, and the y intercept gives the specific molecular volume of POPC as 1240.42 ± 1.46 Å3 in Chol mixture and 1239.69 ± 1.09 Å3 in Cer mixture. These numbers are in good agreement with the experimental volumes reported by Greenwood et al. (35).
FIGURE 3.

Plot of total volume per lipid molecule of the simulated systems as function of concentrations of cholesterol (solid line) and ceramide (dashed line). Lines are guides to the eye between data points.
Using this same method, we computed the areas per molecule for POPC and the intercalator molecules. The application of this method to area computation is discussed by Edholm and Nagle (36). Fig. 4 shows a(x)/(1 − x) (a(x) is the area of the simulation cell divided by the total number of molecules) as a function of x/(1 − x) for Chol as well as Cer simulations. Unlike the volume curves (Fig. 3), the area curves are not linear. In fact, below 10% Chol and Cer concentrations, the derivatives of the curves in Fig. 4 have negative slopes, implying negative partial molecular areas of Chol and Cer. Negative partial molecular areas are interpreted by Edholm and Nagle (36) as a manifestation of the condensation effect on the lipid molecules due to Cer and Chol, respectively. The curves in Fig. 4 are almost linear above 10% concentrations of Cer and Chol. The derivatives of the curves, i.e., the partial molecular area of the intercalator molecules, above 10% concentrations are ACer = 31.2 Å2 and AChol = 12.4 Å2. The partial molecular areas of the POPC, above x > 10%, are 61.6 Å2 and 61.0 Å2 in Chol and Cer mixtures, respectively.
FIGURE 4.

Plot of the total molecular area a(x) (the area of the simulation cell divided by the total number of molecules) of simulated system versus concentrations of cholesterol (solid line) and ceramide (dashed line). Lines are guides to the eye between data points. Light dashed lines represent linear fits to data for non-zero cholesterol and ceramide concentrations. The slope of the fitted lines correspond to the partial molecular areas of the intercalators and the y intercept gives the partial molecular area of POPC.
The area per Chol obtained from Fig. 4 is much smaller than the area per Chol observed in mixtures with saturated chain lipids such as DPPC (23,36). This implies that the Chol is better solvated in POPC than DPPC. Based on our previous work on mixtures of saturated, unsaturated lipids, and Chol (21) we propose that better solvation of Chol in POPC is due to availability of saturated as well as unsaturated chains in POPC. We hypothesize that the saturated chains of POPC entropically favor the (smooth) α-face of the Chol and the unsaturated chains favor the (rough) β-face. Much longer simulations are required to verify this claim. From Fig. 4, we also note that, despite a more flexible structure, Cer induces condensation in POPC bilayer comparable to, albeit weaker than, that induced by Chol. This suggests that Cer may, under some circumstances, act as a surrogate for Chol. The similarities we observe may explain the similarities observed in proposed Cer–PC and Chol–PC phase diagrams (37).
To further explore the similarities between Cer and Chol in POPC, we calculate electron densities for mixtures at the simulated concentrations. Fig. 5, a–d, show the electron densities of the system with Chol and Cer. Both Cer and Chol increase the thickness of the bilayer by approximately the same amount at each concentration. Further, both the intercalators introduce plateaus in electron density corresponding to more ordered chains. However, the effect due to Chol is much more prominent than that of Cer, especially at the higher concentrations.
FIGURE 5.

Plots of electron densities for POPC-Cer and POPC-Chol mixtures. (Dashed lines) POPC-Cer; (solid lines) POPC-Chol. The densities are not symmetrized across the bilayer.
The ordering of hydrocarbon tails is determined in NMR experiments by measuring the deuterium order parameters, and presents another structural property for comparison of Chol and Cer in POPC. The order parameter tensor, S, is defined as
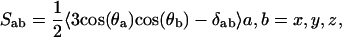 |
(5) |
where θa is the angle made by the ath molecular axis with the bilayer normal and δab is the Krönecker delta. In the simulations, with the united atom force field, the order parameter for saturated and unsaturated carbons SCD can be determined using the relations
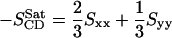 |
(6) |
and
 |
(7) |
respectively (see (38) for the derivation and assumptions behind these expressions). In united atom simulations, the component of order parameter tensors must be obtained from the relative positions of the neighboring carbons in hydrocarbon chain. Fig. 6, a–h, show the deuterium order parameter profile for POPC Sn–1 and Sn–2 chains in Chol and Cer mixtures. As expected, the Sn–2 chain of POPS shows characteristic drop in order parameter at the unsaturated bond in the chain. Furthermore, we note that at lower concentrations, both Cer and Chol increase the POPC chain order by the same amount for concentrations <33%. At the highest concentration, Chol-POPC chains are slightly more ordered than the Cer-POPC chains. Fig. 7 shows the order parameter profiles for acyl and sphingosine chains of Cer. The order parameters of Cer increase with increasing concentration of Cer, as expected, since pure Cer has a main transition temperature of over 90°C. At the 33% concentration level, Cer is more ordered and therefore should induce more order on neighboring POPC chains. However at this concentration, the order induced by Cer is less than that induced by Chol (Fig. 5). The 33% Cer-POPC bilayer still consists of 67% POPC, which has a very low melting temperature, so that Cer molecules likely retain considerable conformational flexibility, compared to a pure Cer bilayer at the simulation temperature.
FIGURE 6.

Plots of POPC SCD order parameter profiles for POPC-Cer and POPC-Chol mixtures. (Solid lines) POPC-Cer; (dashed lines) POPC-Chol. (Left column) Sn–1 chains. (Right column) Sn–2 chains with characteristic drop in order at the 9–10 double-bond location.
FIGURE 7.

Plots of Cer chain SCD order parameter profiles for acyl (top plot) and sphingosine (bottom plot) chains, for simulated POPC–Cer mixtures at various concentrations.
It is interesting to note the overall trend in Figs. 4–6, at least for concentrations of 25% or less, is that Cer can also induce changes similar to Chol. This suggests a generalized hypothesis that flexible molecules like Cer can have interactions with lipid bilayer similar to that of Chol, provided they have sufficiently high transition temperatures and are miscible in bilayers. Of course, this effect should diminish as one approaches transition temperature of the intercalator. Chol, on the other hand, by its rigid ring structure and asymmetric faces, induces chain ordering in lipid bilayers for a large range of temperatures and concentrations. A large number of simulations with different types of intercalators are required to establish these hypotheses. However, this idea forms a central point in our self-consistent mean field theory-based coarse-grain model for mixtures of various lipids and Chol.
In summary, the main conclusion of this article is that, at lower concentrations, 16:0 ceramide orders POPC chains at 300 K in a manner that is very similar, structurally, to the order induced by cholesterol at the same concentration and temperature. At higher concentration, however, the ordering effect of cholesterol is more prominent. Our current results for POPC-Chol give relatively small partial molecular area for Chol in POPC (12.4 Å2) compared to Chol in DPPC (24 Å2) (23). This suggests that Chol packs around POPC more tightly than it packs around DPPC, which in turn suggests that POPC-Chol complexes, or clusters, will have longer lifetimes than DPPC-Chol clusters.
While our simulations and the experimental data reveal details of lipid chain structure, they do not shed light directly on details of the lateral organization of the molecules in the bilayer. For a lipid cholesterol bilayer (DPPC-Chol), we have recently developed a modeling approach, based on self-consistent mean field theory (SCMFT) (39). The principal conclusion of SCMFT simulations is that the DPPC-cholesterol bilayer is monophasic; that is, cholesterol does not induce thermodynamic phase separations in DPPC (40). Regions that appear in the model mimic gel-like, fluid-like, and intermediate chain ordering. However, these regions do not resemble coexisting phases. While an SCMFT model has not yet been constructed for POPC-Chol or POPC-Cer, we suspect that, in the case of POPC-Chol, the system will also be monophasic. The situation is more complicated for Cer-POPC, because Cer by itself is highly ordered at the simulation temperature. Phase separations may indeed take place above some critical Cer concentration in this system. These questions will be investigated within the framework of SCMFT in the future.
Acknowledgments
We thank Prof. John Nagle for valuable discussion, and for providing us with his x-ray data on POPC bilayer.
S.AP. and A.Y.G. are supported by the National Science Foundation grant No. DMR 0427540. E.J. and H.L.S. are supported by National Institutes of Health grant No. UIUC/NIH 2006-139-1.
References
- 1.Miao, L., M. Nielsen, J. Thewalt, J. H. Ipsen, M. Bloom, M. J. Zuckermann, and O. G. Mouritsen. 2002. From lanosterol to cholesterol: structural evolution and differential effects on lipid bilayers. Biophys. J. 82:1429–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohvo-Rekilä, H., B. Ramstedt, P. Leppimäki, and J. P. Slotte. 2002. Cholesterol interactions with phospholipids in membranes. Prog. Lip. Res. 41:66–97. [DOI] [PubMed] [Google Scholar]
- 3.Yeagle, P. L. 1993. The biophysics and cell biology of cholesterol: an hypothesis for the essential role of cholesterol in mammalian cells. In Cholesterol in Membrane Models. L. Finegold, editor. CRC Press, Boca Raton, FL.
- 4.Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- 5.Xu, X., and E. London. 2000. The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochem. 39:843–849. [DOI] [PubMed] [Google Scholar]
- 6.Lange, Y., J. Ye, and T. L. Steck. 2005. Activation of membrane cholesterol by displacement from phospholipids. J. Biol. Chem. 280:36126–36131. [DOI] [PubMed] [Google Scholar]
- 7.Megha and E. London. 2004. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): implications for lipid raft structure and function. J. Biol. Chem. 279:9997–10004. [DOI] [PubMed] [Google Scholar]
- 8.Zitzer, A., R. Bittman, C. A. Verbicky, R. K. Erukulla, S. Bhakdi, S. Weis, A. Valeva, and M. Palmer. 2001. Coupling of cholesterol and cone-shaped lipids in bilayers augments membrane permeabilization by the cholesterol-specific toxins streptolysin O and Vibrio cholerae cytolysin. J. Biol. Chem. 276:14628–14633. [DOI] [PubMed] [Google Scholar]
- 9.Shah, J., J. M. Atienza, R. I. Duclos, A. V. Rawlings, Z. Dong, and G. G. Shipley. 1995. Structure and thermotropic properties of synthetic c16:0 (palmitoyl) ceramide: effect of hydration. J. Lip. Res. 36:1936–1944. [PubMed] [Google Scholar]
- 10.Shah, J., J. M. Atienza, A. V. Rawlings, and G. G. Shipley. 1995. Physical properties of ceramides: effect of fatty acid hydroxylation. J. Lip. Res. 36:1945–1955. [PubMed] [Google Scholar]
- 11.Moore, D. J., M. E. Rerek, and R. Mendelsohn. 1997. FTIR studies of the conformational order and phase behavior of ceramides. J. Chem. Phys. B. 101:8933–8940. [Google Scholar]
- 12.Saxena, K., P. Zimmermann, R. R. Schmidt, and G. G. Shipley. 2000. Bilayer properties of totally synthetic C6:0 lactosyl ceramide. Biophys. J. 78:306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massey, J. B. 2001. Interaction of ceramides with phosphatidylcholine, sphingomyelin, and sphingomyelin/cholesterol bilayers. Biochim. Biophys. Acta. 1510:167–184. [DOI] [PubMed] [Google Scholar]
- 14.Thewalt, J. L., and M. Bloom. 1992. Phosphatidylcholine: cholesterol phase diagram. Biophys. J. 63:1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holopainen, J. M., M. Subramaniam, and P. K. J. Kinnunen. 1998. Sphingomyelinase induces lipid microdomain formation in a fluid phosphatidylcholine/sphingomyelin membranes. Biochemistry. 37:17562–17570. [DOI] [PubMed] [Google Scholar]
- 16.Härtel, A., M. L. Fanani, and B. Maggio. 2005. Shape transitions and lattice structuring of ceramide-enriched domains generated by sphingomyelinase in lipid monolayers. Biophys. J. 88:287–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Contreras, F. X., G. Basanez, A. Alonso, A. Herrmann, and F. M. Goni. 2005. Asymmetric addition of ceramides but not dihydroceramides promotes transbilayer (flip-flop) lipid motion in membranes. Biophys. J. 88:348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsueh, Y.-W., R. Giles, N. Kitson, and J. Thewalt. 2002. The effect of ceramide on phosphatidylcholine membranes: a deuterium NMR study. Biophys. J. 82:3089–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berendsen, H., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comp. Phys. Comm. 91:43–56. [Google Scholar]
- 20.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Mod. 7:306–317. [Google Scholar]
- 21.Pandit, S. A., E. Jakobsson, and H. L. Scott. 2004. Simulation of the early stages of nano-domain formation in mixed bilayers of sphingomyelin, cholesterol, and dioleoylphosphatidylcholine. Biophys. J. 87:3312–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandit, S. A., and H. L. Scott. 2006. Molecular-dynamics simulation of a ceramide bilayer. J. Chem. Phys. 124:014708. [DOI] [PubMed] [Google Scholar]
- 23.Chiu, S.-W., M. M. Clark, E. Jakobsson, S. Subramaniam, and H. L. Scott. 1999. Application of a combined Monte Carlo and molecular dynamics method to the simulation of a dipalmitoylphosphatidylcholine lipid bilayer. J. Comp. Chem. 20:1153–1164. [Google Scholar]
- 24.Chiu, S.-W., M. M. Clark, E. Jakobsson, S. Subramaniam, and H. L. Scott. 1999. Optimization of hydrocarbon chain interaction parameters: application to the simulation of fluid phase lipid bilayers. J. Chem. Phys. B. 103:6323–6327. [Google Scholar]
- 25.Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaige. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comp. Chem. 18:1463–1472. [Google Scholar]
- 26.Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 27.Nosé, S., and M. L. Klein. 1983. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 50:1055–1076. [Google Scholar]
- 28.Parrinello, M., and A. Rahman. 1981. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52:182–7190. [Google Scholar]
- 29.Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrache, H. I., S. E. Feller, and J. F. Nagle. 1997. Determination of component volumes of lipid bilayers from simulations. Biophys. J. 70:2237–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kucerka, N., S. Tristram-Nagle, and J. F. Nagle. 2005. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 208:193–202. [DOI] [PubMed] [Google Scholar]
- 32.van der Spoel, D., and P. J. van Maaren. 2006. The origin of layer structure artifacts in simulations of liquid water. J. Chem. Theory Comput. 2:1–11. [DOI] [PubMed] [Google Scholar]
- 33.Klauda, J. B., N. Kucerka, B. R. Brooks, R. W. Pastor, and J. F. Nagle. 2006. Simulation-based methods for interpreting x-ray data from lipid bilayers. Biophys. J. 90:2796–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benz, R. W., F. Castro-Roma, D. J. Tobias, and S. H. White. 2005. Experimental validation of molecular dynamics simulations of lipid bilayers: A new approach. Biophys. J. 88:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenwood, A. I., S. Tristram-Nagle, and J. F. Nagle. 2006. Partial molecular volumes of lipids and cholesterol. Chem. Phys. Lipids. 143:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edholm, O., and J. F. Nagle. 2005. Areas of molecules in membranes consisting of mixtures. Biophys. J. 89:1827–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silva, L., R. F. M. de Almedia, A. Fedorov, A. P. A. Matos, and M. Prieto. 2006. Ceramide-platform formation and induced biophysical changes in a fluid phospholipid membrane. Mol. Membr. Biol. 23:137–148. [DOI] [PubMed] [Google Scholar]
- 38.Douliez, J.-P., A. Léonard, and E. J. Dufourc. 1995. Restatement of order parameters in biomembranes: calculation of C–C bond order parameters for C-D quadrupolar splittings. Biophys. J. 68:1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khelashvili, G. A., S. A. Pandit, and H. L. Scott. 2005. Self-consistent mean field model based on molecular dynamics: application to lipid-cholesterol bilayers. J. Chem. Phys. 123:034910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pandit, S. A., G. Khelashvili, E. Jakobsson, A. Grama, and H. L. Scott. Lateral organization in lipid-cholesterol mixed bilayers. Biophys. J. 92:440–447. [DOI] [PMC free article] [PubMed]