

Abstract
Model biological membranes consisting of peptide/lipid-bilayer complexes can nowadays be studied by classical molecular dynamics (MD) simulations at atomic detail. In most cases, the simulation starts with an assumed state of a peptide in a preformed bilayer, from which equilibrium configurations are difficult to obtain due to a relatively slow molecular diffusion. As an alternative, we propose an extension of reported work on the self-organization of unordered lipids into bilayers, consisting of including a peptide molecule in the initial random configuration to obtain a membrane-bound peptide simultaneous to the formation of the lipid bilayer. This strategy takes advantage of the fast reorganization of lipids, among themselves and around the peptide, in an aqueous environment. Model peptides of different hydrophobicity, CH3-CO-W2L18W2-NH2 (WL22) and CH3-CO-W2A18W2-NH2 (WA22), in dipalmitoyl-phosphatidylcholine (DPPC) are used as test cases. In the equilibrium states of the peptide/membrane complexes, achieved in time ranges of 50–100 ns, the two peptides behave as expected from experimental and theoretical studies. The strongly hydrophobic WL22 is inserted in a transmembrane configuration and the marginally apolar, alanine-based WA22 is found in two alternative states: transmembrane inserted or parallel to the membrane plane, embedded close to the bilayer interface, with similar stability. This shows that the spontaneous assembly of peptides and lipids is an unbiased and reliable strategy to produce and study models of equilibrated peptide/lipid complexes of unknown membrane-binding mode and topology.
INTRODUCTION
Biological membranes are highly dynamic supramolecular complexes composed mainly of weakly interacting lipid and protein molecules. Because of the intrinsic disorder of their biologically relevant liquid-crystalline state, experimental methods encounter severe limitations for obtaining models of membranes at atomic detail (1). In contrast, the fast molecular motions of bilayer lipids make these complex structures particularly amenable to molecular dynamics (MD) simulations (2–4). Such computational methods allow the description of the spatial organization and temporal dynamics of the system, also providing a useful framework to interpret experimental results.
Among the biomembrane systems for which MD simulations can be applied, membrane/protein complexes are of special interest, due to the importance of protein-lipid interactions in understanding numerous biological functions. Particularly relevant are the fundamental processes of protein insertion and folding in the lipid membrane and the assembly of transmembrane protein segments. These have been studied by MD using simplified models of hydrophobic or amphipathic peptides and lipid bilayers, and binding of peptides to the membrane/water interface, their insertion into the bilayer and packing of peptide α-helices have been investigated (as recent examples see (5–8)). Typical strategies involve the use of, often arbitrary, preformed membrane systems, where several lipids are removed from one or the two leaflets to accommodate the protein inclusion. However, such preassembled systems present important limitations: i), They are difficult to build without imparting unrealistic bilayer stress. ii), The initial position of the peptide with respect to the membrane, i.e., whether it is bound to the surface or inserted, its tilt angle or the depth of insertion is usually unknown and must be assumed for the starting configuration. iii), In the peptide/membrane complexes, the diffusion of the helix from its initial position is slow and limits the number of configurations and binding modes accessible during commonly used simulation times.
As an alternative to the preformed systems, the spontaneous aggregation of bilayer components provides a way to obtain unbiased lipid membrane systems (9). A self-aggregation strategy has been tried with success to generate micellar complexes, either of surfactant molecules alone (10) or in the presence of hydrophobic peptides or proteins (11–13). Similarly, the spontaneous formation of pure lipid bilayers by means of MD simulations has also been described (9,14). Guided by these studies, we have generated in this work self-assembled lipid-bilayer/peptide supramolecular complexes by allowing a number of randomly distributed dipalmitoyl-phosphatidylcholine (DPPC) lipids and an α-helical peptide to self-organize freely in an aqueous environment. We have chosen two model peptides flanked by Trp-anchoring residues and with different hydrophobicities, depending on the presence of a poly-Leu or a poly-Ala central sequence: CH3-CO-W2L18W2-NH2 (WL22) or CH3-CO-W2A18W2-NH2 (WA22), respectively, expected to be α-helical in a membrane environment. The molecules self-organize freely in the simulation box, giving characteristic peptide/membrane complexes. The assembly process occurs through a series of steps that are kinetically distinguishable but depends barely on the type of peptide or the initial or final peptide positions. In the final complexes, the two simulated peptides display characteristic behaviors with respect to their membrane-binding mode. Thus, although the very hydrophobic WL22 is always inserted across the lipid bilayer, the more polar WA22 is found in two alternative configurations: either transmembrane (TM) inserted or embedded close to the interface region, parallel to the membrane plane. These findings show that current MD simulations are able to predict the correct binding mode of characteristic model peptides depending on their sequence. Moreover, the strategy also reports the response of the lipid bilayer to the different ways of peptide binding.
METHODS
Software and simulation conditions
All simulations were performed using the GROMACS suite of programs, version 3.2 (15). The united atom lipid parameters were adapted from the work of Berger and co-workers (16) and the peptide used the GROMOS force field. For water, the single point charge (SPC) model (17) was used, which has been shown to behave well in lipid bilayer/water simulations (18). The simulations were carried out using periodic boundary conditions with constant pressure and temperature. A Berendsen thermostat (19), with a coupling constant of 0.1 ps, was used. The reference temperature was set to 323 K, well above the phase transition temperature of DPPC (315 K). Pressure coupling was applied anisotropically, also using the Berendsen scheme (19), with a coupling constant of 1.0 ps. The reference pressure was 1 bar in all directions. Lipids, solvent, and peptide were separately coupled to the temperature bath. Simulations were run with a 4-fs time step. Bond lengths were constrained using the LINCS algorithm (20). Short-range electrostatic and Lennard-Jones interactions were cut off at 1.0 nm, and long-range electrostatic interactions were calculated by using the particle mesh Ewald algorithm (21).
Setup protocol
Coordinates for the DPPC molecules were taken from http://moose.bio.ucalgary.ca/. DPPC has been extensively studied by MD methods using force fields and setups similar to ours (22–25). The coordinates for the peptides CH3-CO-W2L18W2-NH2 (WL22) and CH3-CO-W2A18W2-NH2 (WA22) in α-helical structures were generated using standard tools of the WHATIF software package (26).
The starting configurations were prepared through several steps: i), A peptide aligned with the z axis was placed in the center of a cubic box (8.0-nm edge length). ii), A total of 128 DPPC molecules were distributed in the box. For that, a lipid molecule was chosen randomly from a pool of different configurations and was randomly translated and rotated. The chosen lipid was placed in the box at any available space, using as many attempts as necessary. iii), The system was solvated with 6,000 SPC molecules. iv), Then, the system was energy minimized and an MD run with position restrained on the peptide backbone was performed. At this stage isotropic pressure coupling was used, shrinking the box to an edge length of 7.0 nm in ∼50 ps. v), Finally, the production run was performed by allowing the system to evolve freely under anisotropic pressure coupling. Similar protocols have been used before (27).
Analysis of trajectories
Trajectories were visualized with the help of the VMD program (28). Lipid clusters were defined using a general criterion based on the distance between the centers of mass of the lipids (or the lipids and the peptide). Two lipid molecules are defined to be in the same cluster if the distance between their centers of mass is smaller than a cutoff distance. The cutoff distance is chosen as that which detects two clusters (one per monolayer) along a trajectory of a simulated bilayer made of 128 DPPC lipids. We find acceptable cutoff values within the range 0.9 nm < cutoff < 1.2 nm. A value of 1.1 nm was used for the analysis, although the results do not depend significantly on the precise choice of the cutoff within the indicated range. A similar strategy has been applied to cluster dodecyl phosphatidylcholine (DPC) molecules while aggregating into a unique micelle (10).
The state of lipid organization throughout self-assembly at a given time is reflected by an orientational order parameter of the lipid acyl chains, Sl, defined as the average value of the instantaneous molecular order parameters, Smol(n), of each n segment of the chain (29). In turn, Smol(n) is calculated from Smol(n) = ½ 〈3cos2θn − 1〉, where θn is the angle between the nth segmental vector linking carbon atoms Cn−1 and Cn+1 in the acyl chain and the normal of the membrane (eventually formed during the assembly process), and the brackets denote an ensemble average calculated for each frame.
The tilt of lipid acyl chains is the angle 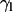 formed between the membrane normal and unit vectors pointing from the midpoint of C1 and C2 toward the midpoint of C15 and C16 (30). The peptide tilt
formed between the membrane normal and unit vectors pointing from the midpoint of C1 and C2 toward the midpoint of C15 and C16 (30). The peptide tilt 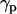 is the angle between the molecular axis of the peptide α-helix and the membrane normal.
is the angle between the molecular axis of the peptide α-helix and the membrane normal.
RESULTS
Self-assembling of peptide/membrane complexes
The starting system consisted of a cubic box with an 8-nm edge, filled with 128 DPPC lipids, an α-helical peptide (WL22 or WA22), and 6,000 water molecules. The lipids were placed with random conformations of their acyl chains, random orientations, and random rotations about their long axis (see Methods for more details). Because initially all three main axes are equivalent, the peptide helix was placed aligned with the z axis. We found that this initial alignment of the peptide influenced neither the direction eventually chosen by the system as the director axis (the bilayer normal) nor the final position occupied by the peptide with respect to the lipid membrane. A total of eight simulations of peptide/membrane complexes were performed, plus two control simulations with only lipids and water, all of them listed in Table 1.
TABLE 1.
List of simulations of this work
| System | Simulation No. | Simulated time (ns) | Pore closure time (ns) | Bilayer normal axis* | Peptide-binding mode | Lipids per monolayer† |
|---|---|---|---|---|---|---|
| WL22 | 1 | 50 | 37 | x | TM | 62/66 |
| 2 | 50 | 26 | z | TM | 65/63 | |
| 3 | 128 | 102 | z | TM | 65/63 | |
| WA22 | 4 | 90 | 85 | x | TM | 64/64 |
| 5 | 58 | 53 | z | TM | 62/66 | |
| 6 | 53 | 20 | y | TM | 65/63 | |
| 7 | 143 | 132 | y | Parallel | 61/67 | |
| 8 | 83 | 80 | z | Parallel | 67/61 | |
| Only lipids | 9 | 117 | 107 | z | – | 64/64 |
| 10 | 40 | 35 | y | – | 65/63 |
In all cases, the molecular axis of the peptide was aligned with the z axis in the initial configuration.
Number of lipids in each monolayer, separated by a slash (/) symbol, after pore closure.
For the sake of clarity, we base our description mainly on simulations 1 and 7, as they correspond to paradigmatic cases of TM-inserted WL22 and interface-bound WA22, respectively. The most important facts corresponding to each simulation are summarized in Table 1. Additionally, comparisons are made between the different cases when worth commenting. In all cases studied the self-assembling process evolves at an irregular pace, similar to that described for the self-formation of pure DPPC bilayers (9) and DPC micelles (10). We can distinguish during this process a number of characteristic stages, as follows:
Stage 1: initial clustering (0–200 ps)
In the starting random configuration of the lipids/peptide/water mixture (Figs. 1 a and 2, a and e), there is a large total hydrophobic surface exposed to the polar solvent, which drives the rapid reorganization of the system toward aggregation of the peptide and lipid molecules and exclusion of water. Thus, at the beginning of the simulation the exposed area of lipids is quickly reduced (Fig. 3 a). However, the accessible surface of the peptide is initially low and does not suffer important variations until the bilayer is completely formed (Fig. 3 b, see also below). This is a consequence of the crowdedness in the starting random system, where the peptide is closely surrounded by lipids facilitating it acting as an aggregation nucleus. The hydrophobic effect gives rise to the rapid formation of a first generation of small, irregular lipid groupings, together with a large cluster where the peptide recruits 20–40 lipids (Fig. 4 c). The number of clusters decreases slowly during this stage, and it appears that the initial reduction of the lipid apolar surface is mainly due to the reorganization of the first set of clusters, rather than to their fusion into larger ones (Fig. 4, a and b). It is interesting to note that despite the facilitated nucleation of a unique large cluster in the presence of the peptide, no important global kinetic variations were observed with respect to the aggregation of only lipids in control simulations (not shown).
FIGURE 1.

Snapshots of the spontaneous aggregation of a mixture of DPPC lipids, water, and the WL22 peptide (simulation 1 of Table 1). Headgroup atoms are depicted blue, atoms of the lipid tails are depicted light gray, and water molecules are drawn in green. The backbone of the peptide is shown in a simplified tube representation with dark gray color. The side chains of Trp residues are in red. For clarity, the figures do not exactly correspond to actual simulation boxes but show part of the system repeated in space. The initial random distribution of molecules (a) evolves into micelle-like clusters that concentrate in a distinct area, where water starts to be excluded ((b), 1.5 ns). Extensive fusion and ordering yields a metastable bilayer structure where the two leaflets are fused at the level of a transbilayer lipid pore, here shown at time 35 ns (c). The pore eventually closes (at 37 ns), and the membrane is further equilibrated up to 50 ns (d). During the aggregation process, the peptide accompanies the lipids through hydrophobic interactions, first as part of a big, micelle-like cluster (b). As the primordial bilayer forms, the peptide reorients (c) and keeps inserted across the membrane for the rest of the simulation (d).
FIGURE 2.

Density distribution of characteristic groups during the self-assembling of peptide/bilayer complexes, resolved along the direction normal to the membrane formed at the end of the process. Boxes a–d and e–h correspond to simulations 1 (complex with the WL22 peptide) and 7 (complex with the WA22 peptide), respectively. The solid line marks densities of lipid headgroup atoms, dotted lines are for lipid acyl tails, dashed lines for water molecules, and dotted-dashed lines for peptide atoms. Four different averages are shown, with labels corresponding to the same stages as in Fig. 1: (a and e), time interval 0–200 ps; (b and f), time 1–2 ns; (c and g), time 30–40 ns; and (d and h), final equilibrated complexes (time 45–50 ns and 135–140 ns, respectively). The positional order characteristic of a lipid bilayer can be seen in c, d, g, and h, although in the first two cases the water density across the membrane indicates the presence of a pore. The density of peptide atoms in the final stages shows clearly its position across the membrane (WL22, (d)) or bound parallel to the membrane reaching both the hydrocarbon and interface regions (WA22, (h)).
FIGURE 3.

Water accessible surface for hydrophobic groups in lipid (a) and WL22 peptide (b) molecules in simulation 1. (a) Lipid acyl tail accessibility: the exposed carbon tail surface is quickly lowered within the first nanosecond (inset, logarithmic timescale) and subsequent reduction attenuates. A final small step toward lower accessibility occurs after ∼15 ns, due to the ordering of lipids into a liquid crystal bilayer. (b) Hydrophobic peptide accessibility: From the beginning, the peptide is closely surrounded by nonordered lipids, which largely excludes water and keeps accessible area low and fairly constant during the first ∼30 ns. After this time, accessibility decreases to a lower rung, coinciding with a fluctuation of the peptide orientation (Fig. 6 b) just before the pore closes (37 ns).
FIGURE 4.

Evolution of the number of clusters throughout the self-assembly process for simulations 10 (no peptide, (a)) and 1 (with peptide WL22, (b and c)). The inset in b corresponds to the number of clusters of simulation 1 but calculated considering only lipids. Two lipids (or a peptide and a lipid) are defined to be in the same cluster if the distance between their center of mass is smaller than 1.1 nm. (a and b) The number of clusters decreases at an irregular pace. At the end of the process two clusters are formed, corresponding to the two monolayers (a and inset in b), which reduce to one cluster if the peptide is considered in the peptide/membrane complex (b). (c) Size of the cluster including the peptide. The peptide cluster size does not increase significantly until the first nanosecond, when small and irregular lipid-only clusters coalesce massively into it.
Stage 2: cluster fusion (200 ps–3 ns)
After the first 200 ps the fusion of clusters typically increases, reducing their total number (Fig. 4, a and b) and giving rise to bigger aggregates with micelle-like structure (Fig. 1 b). In parallel, all lipid groupings tend to concentrate in a centered region from which water is gradually excluded (Figs. 1 b and 2, b and f). In this lipid-rich area, fusion events are more probable and most lipid domains coalesce quickly into the biggest, peptide-containing cluster (Fig. 4, b and c), which in turn accelerates the aggregation process and accentuates the confinement of water and lipids to distinct regions of space. At ∼3 ns only a few lipids remain outside a central big cluster to which the peptide is bound (Fig. 4 c). Although in this aggregate there appears to be some bilayer-like patches, the micellar organization still dominates, as is witnessed by the low acyl-chain lipid order parameter, Sl (Fig. 5; see Methods for a definition of Sl). A higher level ordering of the phospholipids is a much slower process, which we distinguish as a separate stage.
FIGURE 5.

Formation of the liquid-crystal bilayer seen as the time evolution of an order parameter of lipid acyl tails (Sl, see Methods). The increase of Sl proceeds in a step-wise manner, with fast and slow phases overlapping with other important events of self-assembly (see text).
Stage 3: bilayer organization (3–30 ns)
In the big main cluster, the lipids tend to be confined to a centered layer of the box (Fig. 1 c), increasing intermolecular connectivities in the two directions of a plane and decreasing connectivities in the direction perpendicular to that plane. This corresponds to an improvement of the positional order of the lipids (Fig. 2, c and g) that occurs simultaneously to an increase of their orientational order (Fig. 5). As a consequence, lipid packing improves and the dimensions of the box readjust into prism shapes, allowed by the anisotropic pressure-coupling scheme used during the simulation (9,14).
The time evolution of Sl indicates formation of a liquid-crystal bilayer at ∼30 ns (Fig. 5, see a more detailed description below). This reorganization also affects the hydrophobic surface accessibility, which is further reduced during the same time window (15–25 ns, Fig. 3 a). At the end of this stage, the aggregated structure consists of a deformed lipid bilayer traversed by a water-filled lipid pore (Figs. 1 c and 2, c and g). The peptide molecule orders together with the lipids and in most simulations at this stage is already aligned close to its final position with respect to the membrane normal, whether TM inserted (WL22 in simulations 1–3, and WA22 in simulations 4–6) or lying flat in the bilayer plane (WA22 in simulations 7 and 8). Such orientations involved in some cases a large realignment of the peptide from its initial position (see below).
Stage 4: pore closure (>30 ns)
A bilayer with a water-filled lipid pore has been observed before as a characteristic intermediate state of the self-organization of lipid bilayers in MD simulations (9). It constitutes a metastable structure with variable lifetime and shape and a radius of 1–2 nm. For the pore to close, the lipids forming its wall must flip from their position and water must leave the volume corresponding to the center of the bilayer. This involves passing through a high energy state which makes the process of pore closure slow, being the limiting step for complete bilayer formation (9). Under these conditions, the pore may exist for a variable time, spanning from 40 to 100 ns. We did not observe significant differences in the mechanism of pore closure or in the pore lifetimes that can be attributed to the presence of a peptide during the self-assembly process (see Table 1). On the other hand, the metastable pore facilitates the equilibration of the primordial bilayer, sometimes deformed by an asymmetric number of lipids per monolayer. Although under normal circumstances the transbilayer redistribution of lipids is very slow, here it is facilitated by lateral diffusion at the pore wall. Such an equilibration is accompanied by fluctuations of the pore shape and size, rearrangements of the box dimensions, and a better organization of the bilayer lipids. Nevertheless, in most cases the pore closes before complete equilibration of the number of lipids, which suggests some tolerance for intermonolayer asymmetry (Table 1).
Evolution of structure during the aggregation process
Peptide structure
A number of poly-Ala-, poly-Leu-, and Leu-Ala-based peptides have been shown to adopt predominantly α-helical conformations in membrane environments (31–36). Thus, for both peptides, WL22 and WA22, we use α-helical rods as the starting structures. Although these should be barely stable in aqueous media, the peptides are well surrounded by lipids from the very beginning of the simulation, as we have seen above (Fig. 4 c). This helps maintain an organized secondary structure throughout the simulations, with typically only 1–2 unfolded residues at the N- or C-terminal parts. However, in some cases we observed a partial loss of helical content. For instance, in simulation 2, corresponding to WL22, a kinked peptide appears as a result of early interactions with two large micelle-like clusters, but the complete helix structure is recovered after a few nanoseconds. On the other hand, during complex formation in simulation 8, WA22 curves as it interacts interfacially at the pore wall in the primordial bilayer. This deformation persists after the pore closes and the peptide shifts to the interface of one monolayer, giving rise to a kinked helix in the final configuration (not shown).
Lipid and peptide alignment
The assembly of peptide/membrane complexes from randomly oriented molecules involves increasing positional (in plane) and orientational order to form a smectic liquid crystal lamellar phase. As described above, in our simulated systems ordering occurs mainly after coalescence of most lipid clusters into a single central big cluster. This can be seen after the increase of the order parameter of lipids from an initial value of Sl ∼ 0, corresponding to a random distribution, to a value of Sl ∼ 0.35 when the membrane is formed (Fig. 5). During the complete ordering process there is typically a characteristic fast phase at ∼20 ns coinciding with the collapse of micelle-like clusters to form the lipid bilayer.
One can also follow the evolution with time of the tilt of a molecular axis for lipids with respect to the director axis, taken as the normal of the bilayer that is formed at the end of the process. As a representative lipid molecular axis, we take that of the hydrocarbon tails, which makes an angle 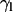 with the reference normal axis. Because this angle averages close to 0 throughout the simulation, we take the most populated value of the distribution of
with the reference normal axis. Because this angle averages close to 0 throughout the simulation, we take the most populated value of the distribution of 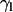 (
(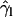 ) as a more informative magnitude (see Fig. 6 a for a typical distribution of lipid tilt angles). Illustrative examples are shown in Fig. 6, b and c, where we analyze the evolution of the lipid and peptide orientations in simulations 1 and 7, respectively. Starting from a random arrangement of lipids,
) as a more informative magnitude (see Fig. 6 a for a typical distribution of lipid tilt angles). Illustrative examples are shown in Fig. 6, b and c, where we analyze the evolution of the lipid and peptide orientations in simulations 1 and 7, respectively. Starting from a random arrangement of lipids, 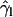 evolves slowly from ∼90° (averaged over 256 lipid tails) to ∼31°. This latter value is characteristic of an equilibrated lipid bilayer in all our simulations and is in agreement with other reported simulation data (30,37).
evolves slowly from ∼90° (averaged over 256 lipid tails) to ∼31°. This latter value is characteristic of an equilibrated lipid bilayer in all our simulations and is in agreement with other reported simulation data (30,37).
FIGURE 6.

Tilt angle of lipid chains (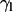 ) and peptide molecules (
) and peptide molecules (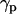 ) during self-assembly. (a) Distribution of
) during self-assembly. (a) Distribution of 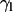 of sn1 and sn2 lipid acyl-chains in a 10-ns time window of the equilibrated WL22/membrane complex corresponding to simulation 1. Time evolution of
of sn1 and sn2 lipid acyl-chains in a 10-ns time window of the equilibrated WL22/membrane complex corresponding to simulation 1. Time evolution of 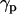 (black) and the mean value of the distribution of
(black) and the mean value of the distribution of 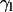 (gray) for simulations 1 (b) and 7 (c).
(gray) for simulations 1 (b) and 7 (c).
It is also interesting to analyze the evolution of the orientation of the helical peptides with respect to the bilayer normal, 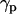 , along the simulation. In the case shown in Fig. 6 b corresponding to WL22, the bilayer forms in the y-z plane, with the normal being the x axis of the system. The peptide starts tilted 90° and, as the simulation proceeds, it rotates to accommodate itself in a TM fashion with ±10° fluctuations from the equilibrium
, along the simulation. In the case shown in Fig. 6 b corresponding to WL22, the bilayer forms in the y-z plane, with the normal being the x axis of the system. The peptide starts tilted 90° and, as the simulation proceeds, it rotates to accommodate itself in a TM fashion with ±10° fluctuations from the equilibrium 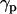 value. Comparable complexes, always with a TM peptide, are produced in two other simulations with WL22. However for WA22 we observe a variable behavior. Simulations 4–6 produce complexes with a TM-bound peptide in much the same way as for WL22. In contrast, simulations 7 and 8 give peptides bound parallel to the membrane plane in the final configurations. The time evolution of the peptide tilt for simulation 7 is shown in Fig. 6 c. In this case the bilayer normal corresponds to the y axis and the peptide is initially perpendicular to this direction. As soon as the lipids start to aggregate, the peptide rotates slightly, but it again positions perpendicular to y as the primordial lipid bilayer is formed (at time 15 ns in this case). During the rest of the simulation the peptide keeps essentially parallel to the membrane with ±10° fluctuations. Therefore, it appears that the alignment of peptides in the membrane is guided by the orientation of lipids but depends also on the peptide sequence.
value. Comparable complexes, always with a TM peptide, are produced in two other simulations with WL22. However for WA22 we observe a variable behavior. Simulations 4–6 produce complexes with a TM-bound peptide in much the same way as for WL22. In contrast, simulations 7 and 8 give peptides bound parallel to the membrane plane in the final configurations. The time evolution of the peptide tilt for simulation 7 is shown in Fig. 6 c. In this case the bilayer normal corresponds to the y axis and the peptide is initially perpendicular to this direction. As soon as the lipids start to aggregate, the peptide rotates slightly, but it again positions perpendicular to y as the primordial lipid bilayer is formed (at time 15 ns in this case). During the rest of the simulation the peptide keeps essentially parallel to the membrane with ±10° fluctuations. Therefore, it appears that the alignment of peptides in the membrane is guided by the orientation of lipids but depends also on the peptide sequence.
DISCUSSION
Atomistic simulations of self-assembly of surfactant molecules have proved successful for generating detailed and realistic models of micelles (10,38), protein/micelle complexes (11–13), and pure lipid bilayers (9,14). Here we apply an atomistic MD simulation approach to investigate the spontaneous formation of peptide/lipid bilayer complexes by allowing randomly distributed DPPC lipids and a peptide molecule to self-organize freely in an aqueous environment. Under the strong thermodynamic gradient imposed mainly by the hydrophobic effect, formation of peptide/membrane complexes proceeds fast and allows us to obtain equilibrium configurations in timescales of ∼100 ns. The autoorganized systems so produced are not biased by a chosen starting configuration. Instead, they are expected to depend only on objective factors, like the quality of the force field or details of the simulation methodology, and should be valid for selecting the preferred binding mode of peptides according to their physiochemical properties, codified in their sequence. An added value of this strategy is the inherent mechanistic information that can be obtained from the time evolution of the system during the formation of the complex, which helps us understand the molecular interactions defining each final configuration.
Bilayer self-assembly in the presence of a peptide
The simulated spontaneous formation of detergent micelles and liquid-crystal lipid bilayers follows similar mechanisms during the initial steps of the aggregation processes. However, the final organization is more complex for the lipid phases, often showing diverse structures and metastable intermediate states (9,14). Pure DPC and mixtures corresponding to the human bile aggregate into small micelles in ∼3 ns. At time ∼10 ns the small clusters coalesce to form large micelles of ∼50 molecules, which then reorganize internally and become spherical during time 10–20 ns (10,38). This has been extended to the self-assembly of surfactants together with hydrophobic proteins, with examples showing the formation of a sodium dodecyl sulfate micelle around dimeric glycophorin (11,12) or the β-barrel porins OmpF and OmpX (11,13). Similarly, in a series of landmark studies Marrink and co-workers have reported the spontaneous formation of fluid lipid bilayers. DPPC (9) or a mixture of dioleoylphosphatidylcholine (DOPC) and dioleoylphosphatidylethanolamine (DOPE) (27) form a large cluster within 2–3 ns, which then reorders internally to form a bilayer crossed by a metastable water-filed pore of variable size and stability. Eventually, the pore shrinks and collapses, completing bilayer formation within ∼50 ns from the starting random distribution of lipids in water (9).
For the work reported here we included a hydrophobic peptide in the initial random mixture of water and lipids. From the beginning, the peptide facilitates the formation of a unique large cluster (Fig. 4 c), which is not observed in the absence of peptides (control simulations 9 and 10, Table 1). Similar nucleation effects have been observed during the spontaneous formation of protein/micelle complexes (11,12). However, in our simulations of peptide/membrane complexes, the presence of the peptide does not alter significantly the global kinetics of self-assembling or the principal landmarks of this process, regardless of the type of peptide or the configuration finally achieved. Thus, as in the absence of peptides, after a rapid formation of the first generation of clusters, in ∼200 ps (Figs. 1 a and 2, b and f), coalescence into a big singular cluster is an order of magnitude slower (∼3 ns), and the increase of molecular order to form a primordial bilayer does not happen before ∼30 ns (Figs. 1 c and 5). This indicates that the process is limited by a number of slow phases, like diffusion of clusters, coalescence to form bigger clusters, and internal reorganizations within each cluster, including the final ordering of the bilayer, which appears to be independent of the presence of the peptide.
Again, similar to that reported in the absence of peptides, the bilayer organizes first as a metastable structure characterized by the presence of a lipidic pore, and pore closure is the rate-limiting step toward formation of a defect-free membrane (9). Previous studies have shown that the stability of the pore can be increased under specific stress conditions, up to a lifetime of more than 150 ns (39). Interestingly, we do not find significant variations of pore lifetime in the presence of WL22 or WA22 regardless of their binding mode. Both in the absence and in the presence of peptides, the pore seems to act as a mechanism for relaxing the stress that often builds up as the primordial bilayer organizes, coming from an asymmetric distribution of molecules between the two leaflets. However, in most cases the pore is closed before complete equilibration of the intermonolayer number of lipids, allowing maximum differences of three lipids in the absence of peptides, four lipids in complexes with a TM-bound peptide (WL22 or WA22), or six lipids for cases of a WA22 peptide bound parallel to the membrane (Table 1). For membranes with no peptide and with a TM peptide, this indicates a tolerance of up to 6% asymmetric area expansion. On the other hand, in the two cases where WA22 binds asymmetrically to only one monolayer (simulations 7 and 8), the defect of six lipids is found always at the side of the peptide. Interestingly, in these latter complexes, for an area per lipid of 0.65 nm2 calculated from the peptide-free monolayer, a defect of six lipids corresponds to 3.90 nm2, which fits well with an excess area of ∼3.96 nm2 contributed by one WA22 molecule at the opposite monolayer (estimated by assuming an ideal alanine-based α-helix of 22 residues).
System self-selection of the peptide-binding mode
One of our goals with this study was to test the atomistic self-assembling MD methods for the production of models of characteristic peptide/membrane complexes, sensitive to relevant physicochemical properties of the peptide, like hydrophobicity. To this aim, we chose two simple model membrane-binding α-helical peptides, WL22 and WA22. In both cases, the terminal Trp residues provide well-known interfacial anchoring of the peptides to the membrane (40–44). The core 18 Leu residues of WL22, compared to the 18 Ala residues of WA22, make the first peptide much more hydrophobic than the second (45), allowing us to test the influence of this property in the observed peptide-membrane-binding modes. To provide for a lipid environment, we chose DPPC for which the bilayer hydrophobic thickness (1) should match the hydrophobic length of our peptides.
Although leucine is clearly a very hydrophobic residue, hydrophobicity scales consider alanine from moderately apolar (46–48) to even slightly polar, if one accounts for the contribution of the hydrophilic peptide backbone (40,45). Such ambiguity of Ala is supported both experimentally (40,49–54) and from free energy calculations, which predict poly-Ala-based peptides in transmembrane- (TM-) and surface-bound configurations with similar probabilities (55). Thus, we take the poly-Leu-based WL22 as a paradigm for strongly interacting TM peptides and the poly-Ala-based WA22 as an ambivalent case with alternative binding modes. In agreement with such expectations, after simulated self-assembling, WL22 showed an absolute preference for TM binding. In contrast, WA22 was found in two distinct configurations: TM in three out of five simulations, and parallel to the membrane in two simulations (Table 1). Before discussing these facts in more detail, we elaborate briefly on their statistical significance. In principle it could be argued that chances are that all simulations, including cases of WL22 and WA22 (eight in total), are drawn from the same distribution. This would give a relatively high probability for a random sample of three measurements, as for WL22, to be exclusively TM (0.753 = 0.4). However, we find this latter possibility very unlikely, as any additional self-assembly simulation run for peptides having in common a central 18-Leu stretch (up to a total of six simulations; S. Estaban-Martin and J. Salgado, unpublished) gave a TM configuration. Thus, the cases of poly-Leu- and poly-Ala-based peptides should follow different distributions, and their differentiated behavior through the simulated self-assembly process appears to reflect the underlying thermodynamic driving forces relevant for their membrane-binding state.
The ratio of alternative states found here for WA22 is in agreement with theoretical predictions from free energy calculations (55) and with results obtained for similar poly-Ala-based peptides by solid-state 15N-NMR (52). The latter study shows an equilibrium between in-plane and TM states for K3A18K3 peptides, similar to our WA22 peptide, where the TM alignment can be stabilized by replacing a few Ala residues with Leu. Thus, it appears that the hydrophobicity of poly-Ala peptides is close to the threshold for TM insertion (52). Similarly, a recent investigation of the integration of GGPG-(LnA19−n)-GPGG peptides in endoplasmic reticulum membranes using the natural translocon machinery (56) yields a small, positive apparent free energy of insertion per Ala residue, 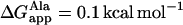 that can be counteracted by a few Leu residues in the peptide sequence (
that can be counteracted by a few Leu residues in the peptide sequence (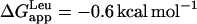 ). For the case of WA22 studied here, we should also consider the contribution of the flanking Trp residues to the free energy of insertion in the membrane. In agreement with the preference of these latter residues for interfaces, it has been shown that their contribution to a TM state is strongly dependent on their position with respect to the center of the helix, with maximum reduction of total ΔGapp for Trp-to-Trp separation ≥10 residues (56). Therefore, the flanking Trp residues in WA22 can effectively counteract the marginal polarity of the central Ala18 stretch, and the peptide can be stable both in TM and parallel to the membrane-binding modes, as shown by the simulations. It is interesting that despite the change in orientation of the peptide between the two states, the central Ala-stretch and flanking Trp residues are found in similar membrane regions in the two alternative configurations. Thus, even in the parallel-binding state, the peptide is immersed and most of its volume occupies the hydrocarbon region (Fig. 1 h), whereas the Trp residues always reside close to the membrane interface.
). For the case of WA22 studied here, we should also consider the contribution of the flanking Trp residues to the free energy of insertion in the membrane. In agreement with the preference of these latter residues for interfaces, it has been shown that their contribution to a TM state is strongly dependent on their position with respect to the center of the helix, with maximum reduction of total ΔGapp for Trp-to-Trp separation ≥10 residues (56). Therefore, the flanking Trp residues in WA22 can effectively counteract the marginal polarity of the central Ala18 stretch, and the peptide can be stable both in TM and parallel to the membrane-binding modes, as shown by the simulations. It is interesting that despite the change in orientation of the peptide between the two states, the central Ala-stretch and flanking Trp residues are found in similar membrane regions in the two alternative configurations. Thus, even in the parallel-binding state, the peptide is immersed and most of its volume occupies the hydrocarbon region (Fig. 1 h), whereas the Trp residues always reside close to the membrane interface.
In summary, self-assembly of peptide/membrane complexes by MD is able to distinguish the preferred binding mode of two model peptides depending on physicochemical properties codified by their sequence. Moreover, it correctly predicts available configurations for the WA22 peptide, which are almost equivalent in terms of their thermodynamic stability.
CONCLUSIONS
The spontaneous assembly, starting from random mixtures of DPPC lipids with a hydrophobic peptide in water by means of MD simulations, is an unbiased method for generating peptide/membrane complexes. The aggregation process proceeds through a number of distinct steps, characterized by the formation of lipid clusters of growing size and increasing positional and orientational order, until a lipid bilayer with a peptide bound to it is finally formed. The number of clusters change discontinuously in two main phases: a first one consisting of an increased packing of small micelle-like clusters, with an initial reduction of the accessible surface area due to the internal reorganization of existent clusters; and a second one characterized by the massive fusion of the clusters, accompanied by a further reduction of the hydrophobic lipid accessibility. After the main aggregate is formed, the reorganization of lipids to constitute a liquid-crystal bilayer and the closure of a lipid pore, which always accompanies bilayer formation, are slower processes and constitute rate-limiting steps.
The peptide binds strongly to the lipids from the beginning of the simulation, which facilitates aggregation of an initial big cluster. However, the mechanism and kinetics of formation of the lipid bilayer do not change significantly with respect to a system in the absence of the peptide. Likewise, the peptides do not seem to stabilize the intermediate metastable state characterized by a lipidic pore. The fact that the diffusion of lipids through the pore wall is faster than the lifetime of the pore itself allows for an efficient mechanism to compensate the tension induced by either an initial asymmetric random distribution of lipids, asymmetric inserted peptides, or peptides adsorbed at one monolayer interface. Particularly, when WA22 binds parallel to the membrane in only one monolayer, the surface occupied by the peptide is compensated by an equivalent reduction of the number of lipids.
During the self-assembly process the peptides are free to accommodate to their preferred configuration in the emerging bilayer, depending on their physiochemical properties, such as hydrophobicity, codified in their sequence. Thus, the very hydrophobic WL22 acquires a TM-inserted state, whereas the borderline apolar WA22 is found in TM-bound and parallel-bound states. Such a distinction of states with marginal differences of stability cannot be made starting from preformed bilayers, which makes the spontaneous assembly of peptides and lipids an unbiased strategy to produce reliable models of equilibrated peptide/lipid complexes. This opens the possibility of studying peptide/membrane complexes with peptides of unknown membrane-binding mode and topology, as well as systems where the equilibrium configuration depends on complex dynamic processes, like pore-forming peptides.
Acknowledgments
This work has been supported by grants from the Spanish Ministerio de Educación y Ciencia (CTQ2004-03444 and FPU fellowship (S.E.)), and Generalitat Valenciana (GVACOMP2006-107).
References
- 1.Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saiz, L., and M. L. Klein. 2002. Computer simulation studies of model biological membranes. Acc. Chem. Res. 35:482–489. [DOI] [PubMed] [Google Scholar]
- 3.Hansson, T., C. Oostenbrink, and W. F. van Gunsteren. 2002. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 12:190–196. [DOI] [PubMed] [Google Scholar]
- 4.Ash, W. L., M. R. Zlomislic, E. O. Oloo, and D. P. Tieleman. 2004. Computer simulations of membrane proteins. Biochim. Biophys. Acta. 1666:158–189. [DOI] [PubMed] [Google Scholar]
- 5.Nymeyer, H., T. B. Woolf, and A. E. Garcia. 2005. Folding is not required for bilayer insertion: replica exchange simulations of an alpha-helical peptide with an explicit lipid bilayer. Proteins. 59:783–790. [DOI] [PubMed] [Google Scholar]
- 6.Im, W., and C. L. Brooks. 2005. Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 102:6771–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stockner, T., W. L. Ash, J. L. MacCallum, and D. P. Tieleman. 2004. Direct simulation of transmembrane helix association: role of asparagines. Biophys. J. 87:1650–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sparr, E., W. L. Ash, P. V. Nazarov, D. T. Rijkers, M. A. Hemminga, D. P. Tieleman, and J. A. Killian. 2005. Self-association of transmembrane alpha-helices in model membranes: importance of helix orientation and role of hydrophobic mismatch. J. Biol. Chem. 280:39324–39331. [DOI] [PubMed] [Google Scholar]
- 9.Marrink, S. J., E. Lindahl, O. Edholm, and A. E. Mark. 2001. Simulation of the spontaneous aggregation of phospholipids into bilayers. J. Am. Chem. Soc. 123:8638–8639. [DOI] [PubMed] [Google Scholar]
- 10.Marrink, S. J., D. P. Tieleman, and A. E. Mark. 2000. Molecular dynamics simulation of the kinetics of spontaneous micelle formation. J. Phys. Chem. B. 104:12165–12173. [Google Scholar]
- 11.Bond, P. J., J. M. Cuthbertson, S. S. Deol, and M. S. Sansom. 2004. MD simulations of spontaneous membrane protein/detergent micelle formation. J. Am. Chem. Soc. 126:15948–15949. [DOI] [PubMed] [Google Scholar]
- 12.Braun, R., D. M. Engelman, and K. Schulten. 2004. Molecular dynamics simulations of micelle formation around dimeric Glycophorin A transmembrane helices. Biophys. J. 87:754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bockmann, R. A., and A. Caflisch. 2005. Spontaneous formation of detergent micelles around the outer membrane protein OmpX. Biophys. J. 88:3191–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel, R. Y., and P. V. Balaji. 2005. Effect of the choice of the pressure coupling method on the spontaneous aggregation of DPPC molecules. J. Phys. Chem. B. 109:14667–14674. [DOI] [PubMed] [Google Scholar]
- 15.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. (Online). 7:306–317. [Google Scholar]
- 16.Berger, O., O. Edholm, and F. Jahnig. 1997. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72:2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, and J. Hermans. (1981). Interaction models for water in relation to water hydration. In Intermolecular Forces. B. Pullman, editor. Reidel, Dordrecht, The Netherlands. 331–342.
- 18.Tieleman, D. P., and H. J. C. Berendsen. 1996. Molecular dynamics simulations of a fully hydrated dipalmitoyl phosphatidylcholine bilayer with different macroscopic boundary conditions and parameters. J. Chem. Phys. 105:4871–4880. [Google Scholar]
- 19.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. di Nola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 20.Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- 21.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 22.Anezo, C., A. H. de Vries, H. D. Holtje, D. P. Tieleman, and S. J. Marrink. 2003. Methodological issues in lipid bilayer simulations. J. Phys. Chem. B. 107:9424–9433. [Google Scholar]
- 23.Patra, M., M. Karttunen, M. T. Hyvonen, E. Falck, P. Lindqvist, and I. Vattulainen. 2003. Molecular dynamics simulations of lipid bilayers: major artifacts due to truncating electrostatic interactions. Biophys. J. 84:3636–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patra, M., M. Karttunen, M. T. Hyvonen, E. Falck, and I. Vattulainen. 2004. Lipid bilayers driven to a wrong lane in molecular dynamics simulations by subtle changes in long-range electrostatic interactions. J. Phys. Chem. B. 108:4485–4494. [Google Scholar]
- 25.Wohlert, J., and O. Edholm. 2004. The range and shielding of dipole-dipole interactions in phospholipid bilayers. Biophys. J. 87:2433–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vriend, G. 1990. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8:52–56. [DOI] [PubMed] [Google Scholar]
- 27.de Vries, A. H., A. E. Mark, and S. J. Marrink. 2004. The binary mixing behavior of phospholipids in a bilayer: a molecular dynamics study. J. Phys. Chem. B. 108:2454–2463. [Google Scholar]
- 28.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 29.Hubbell, W. L., and H. M. McConnell. 1971. Molecular motion in spin-labeled phospholipids and membranes. J. Am. Chem. Soc. 93:314–326. [DOI] [PubMed] [Google Scholar]
- 30.Takaoka, Y., M. Pasenkiewicz-Gierula, H. Miyagawa, K. Kitamura, Y. Tamura, and A. Kusumi. 2000. Molecular dynamics generation of nonarbitrary membrane models reveals lipid orientational correlations. Biophys. J. 79:3118–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oliver, A. E., and D. W. Deamer. 1994. Alpha-helical hydrophobic polypeptides form proton-selective channels in lipid bilayers. Biophys. J. 66:1364–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang, Y. P., R. N. Lewis, G. D. Henry, B. D. Sykes, R. S. Hodges, and R. N. McElhaney. 1995. Peptide models of helical hydrophobic transmembrane segments of membrane proteins. 1. Studies of the conformation, intrabilayer orientation, and amide hydrogen exchangeability of Ac-K2-(LA)12–K2-amide. Biochemistry. 34:2348–2361. [DOI] [PubMed] [Google Scholar]
- 33.Liu, L. P., S. C. Li, N. K. Goto, and C. M. Deber. 1996. Threshold hydrophobicity dictates helical conformations of peptides in membrane environments. Biopolymers. 39:465–470. [DOI] [PubMed] [Google Scholar]
- 34.Liu, L. P., and C. M. Deber. 1998. Uncoupling hydrophobicity and helicity in transmembrane segments. Alpha-helical propensities of the amino acids in non-polar environments. J. Biol. Chem. 273:23645–23648. [DOI] [PubMed] [Google Scholar]
- 35.Percot, A., X. X. Zhu, and M. Lafleur. 1999. Design and characterization of anchoring amphiphilic peptides and their interactions with lipid vesicles. Biopolymers. 50:647–655. [DOI] [PubMed] [Google Scholar]
- 36.Harzer, U., and B. Bechinger. 2000. Alignment of lysine-anchored membrane peptides under conditions of hydrophobic mismatch: a CD, 15N and 31P solid-state NMR spectroscopy investigation. Biochemistry. 39:13106–13114. [DOI] [PubMed] [Google Scholar]
- 37.Rog, T., K. Murzyn, R. Gurbiel, Y. Takaoka, A. Kusumi, and M. Pasenkiewicz-Gierula. 2004. Effects of phospholipid unsaturation on the bilayer nonpolar region: a molecular simulation study. J. Lipid Res. 45:326–336. [DOI] [PubMed] [Google Scholar]
- 38.Marrink, S. J., and A. E. Mark. 2002. Molecular dynamics simulations of mixed micelles modeling human bile. Biochemistry. 41:5375–5382. [DOI] [PubMed] [Google Scholar]
- 39.Leontiadou, H., A. E. Mark, and S. J. Marrink. 2004. Molecular dynamics simulations of hydrophilic pores in lipid bilayers. Biophys. J. 86:2156–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wimley, W. C., and S. H. White. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3:842–848. [DOI] [PubMed] [Google Scholar]
- 41.Killian, J. A., and G. von Heijne. 2000. How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 25:429–434. [DOI] [PubMed] [Google Scholar]
- 42.Ulmschneider, M. B., and M. S. P. Sansom. 2001. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta. 1512:1–14. [DOI] [PubMed] [Google Scholar]
- 43.Aliste, M. P., J. L. MacCallum, and D. P. Tieleman. 2003. Molecular dynamics simulations of pentapeptides at interfaces: salt bridge and cation-pi interactions. Biochemistry. 42:8976–8987. [DOI] [PubMed] [Google Scholar]
- 44.White, S. H., and G. von Heijne. 2005. Transmembrane helices before, during, and after insertion. Curr. Opin. Struct. Biol. 15:378–386. [DOI] [PubMed] [Google Scholar]
- 45.White, S. H., and W. C. Wimley. 1999. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28:319–365. [DOI] [PubMed] [Google Scholar]
- 46.von Heijne, G. 1981. Membrane proteins: the amino acid composition of membrane-penetrating segments. Eur. J. Biochem. 120:275–278. [DOI] [PubMed] [Google Scholar]
- 47.Kyte, J., and R. F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105–132. [DOI] [PubMed] [Google Scholar]
- 48.Engelman, D. M., T. A. Steitz, and A. Goldman. 1986. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 15:321–353. [DOI] [PubMed] [Google Scholar]
- 49.Chen, H., and D. A. Kendall. 1995. Artificial transmembrane segments. Requirements for stop transfer and polypeptide orientation. J. Biol. Chem. 270:14115–14122. [DOI] [PubMed] [Google Scholar]
- 50.Chung, L. A., and T. E. Thompson. 1996. Design of membrane-inserting peptides: spectroscopic characterization with and without lipid bilayers. Biochemistry. 35:11343–11354. [DOI] [PubMed] [Google Scholar]
- 51.Lu, L. P., and C. M. Deber. 1998. Guidelines for membrane protein engineering derived from de novo designed model peptides. Biopolymers. 47:41–62. [DOI] [PubMed] [Google Scholar]
- 52.Bechinger, B. 2001. Membrane insertion and orientation of polyalanine peptides: a 15N solid-state NMR spectroscopy investigation. Biophys. J. 81:2251–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis, R. N., Y. P. Zhang, R. S. Hodges, W. K. Subczynski, A. Kusumi, C. R. Flach, R. Mendelsohn, and R. N. McElhaney. 2001. A polyalanine-based peptide cannot form a stable transmembrane alpha-helix in fully hydrated phospholipid bilayers. Biochemistry. 40:12103–12111. [DOI] [PubMed] [Google Scholar]
- 54.de Planque, M. R., and J. A. Killian. 2003. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol. Membr. Biol. 20:271–284. [DOI] [PubMed] [Google Scholar]
- 55.Ben-Tal, N., A. Ben-Shaul, A. Nicholls, and B. Honig. 1996. Free-energy determinants of alpha-helix insertion into lipid bilayers. Biophys. J. 70:1803–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hessa, T., H. Kim, K. Bihlmaier, C. Lundin, J. Boekel, H. Andersson, I. Nilsson, S. H. White, and G. von Heijne. 2005. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 433:377–381. [DOI] [PubMed] [Google Scholar]