

Abstract
Gradient sensing, polarization, and chemotaxis of motile cells involve the actin cytoskeleton, and regulatory modules, including the phosphoinositides (PIs), their kinases/phosphatases, and small GTPases (Rho proteins). Here we model their individual components (PIP1, PIP2, PIP3; PTEN, PI3K, PI5K; Cdc42, Rac, Rho; Arp2/3, and actin), their interconversions, interactions, and modular functions in the context of a one-dimensional dynamic model for protrusive cell motility, with parameter values derived from in vitro and in vivo studies. In response to a spatially graded stimulus, the model produces stable amplified internal profiles of regulatory components, and initiates persistent motility (consistent with experimental observations). By connecting the modules, we find that Rho GTPases work as a spatial switch, and that the PIs filter noise, and define the front versus back. Relatively fast PI diffusion also leads to selection of a unique pattern of Rho distribution from a collection of possible patterns. We use the model to explore the importance of specific hypothesized interactions, to explore mutant phenotypes, and to study the role of actin polymerization in the maintenance of the PI asymmetry. We also suggest a mechanism to explain the spatial exclusion of Cdc42 and PTEN and the inability of cells lacking active Cdc42 to properly detect chemoattractant gradients.
INTRODUCTION
The aim of this article is to develop a model of the events underlying initial steps in cell motility and chemotaxis, from redistribution of signaling components to the reorganization and protrusion of the lamellipodial cytoskeleton. We investigate three major modules involved in coordinating directed movement (Fig. 1, a and b), and study how their interactions lead to characteristic behaviors of normal and mutant cells.
FIGURE 1.

(a) Simplified schematic diagram of proposed interactions between the phosphoinositides PI, PIP1, PIP2, and PIP3, the Rho proteins Cdc42, Rac, and Rho, and actin dynamics considered in this article. Interconversion of the PIs is indicated by dotted lines. Inhibition is denoted by ⊣ and activation by →. Arrows are based on the following references: 1, (82,84,131); 2, (83,84); 3, (85,110); 4, (8,84,95); 5, (90); 6, (78–80); 7, (119,132); 8, (4,53); and 9, (62,66). (b) Schematic representation of components included in the model with their spatial localization: membrane-bound PIs and active (GTP-bound) Rho proteins, cytoplasmic PI3K, PTEN, inactive (GDP-bound) Rho proteins, actin, and Arp2/3.
Chemotactic stimulation of motile cells (e.g., neutrophils) initiates an intricate chain of events. First, the cell polarizes internally, producing intracellular gradients in response to stimuli. Then, after reorganizing the actin cytoskeleton and undergoing changes in shape, the cell extends protrusions, and starts to move in the direction of the stimulus (1). Cells can detect gradients as shallow as 1–2% (2–5), by transducing and amplifying external signals into clear, persistent intracellular maps. These internal maps correspond to differences in levels of enzymes, lipids, and proteins at an emergent front and back of the cell. Notable among the first to redistribute are the kinase PI3K (front), and phosphatase PTEN (back), followed by phosphoinositides, and small GTPases of the Rho protein family (1,6–8).
The interactions and crosstalk of these signaling components at many levels form an important organizing principle, and the subject of this article. A key aspect of the downstream effect of these signaling components is their effect on the actin cytoskeleton. By regulating the initiation of new growth sites (i.e., by nucleation of new actin filament barbed ends), and by releasing inhibition of growth at some sites (i.e., by inhibition of barbed end capping), these regulatory pathways lead to the directional protrusion of the cytoskeleton, formation of a leading edge, and eventual initiation of cell motion (Fig. 1 b).
Aside from amplification of weak stimuli, chemotactic cells display a panoply of characteristic behaviors. The term “amplification” denotes the fact that internal gradients are macroscopic, with similar magnitudes in response to strong or weak stimuli (4,9). The term “adaptation” denotes the fact that some cells (e.g., Dictyostelium) return to rest after transient responses to spatially uniform chemoattractant distributions (see Fig. 3 in (10)). Other cells (e.g., neutrophils) randomly choose a direction and initiate directed motion (11,12). Normal motile cells move up gradients of attractants, but remain sensitive to new or changing stimuli from other directions (6,13). Finally, some cell types appear to initiate directed movement in the absence of spatial cues (14). In this article, we explore how detection of an external stimulus can lead to directed movement.
FIGURE 3.

(a) Growth and translocation of barbed ends as the cell initiates motion in response to graded 10-s stimulus (kPI3K gradient: zero at the rear to baseline at the front edge, and opposite for kPTEN; see Graded Stimuli). Barbed ends are moving to the right, shown initially at 2–4 s intervals, then at 10-s intervals to show steady-state motion. When the stimulus is applied, there is an overshoot in the number of barbed ends near the leading edge, leading to a faster membrane speed during stimulus application. (b) Membrane speeds resulting from different stimulus application time-lengths. Dotted line corresponds to simulation of barbed ends shown in panel a. Stimuli at t = 20, as in panel a, but for durations 0.01 s (dash-dash-dot line), 0.1 s (dot-dash line), 1 s (dashed line), 10 s (dotted line), and 40 s (solid line). Exposure to a brief stimulus results in directed movement with a time delay while stimuli applied for a longer amount of time rapidly initiate movement. Similar results are seen for weak stimuli, but with longer delays (data not shown).
Signaling cascades that impinge on the actin cytoskeleton and on cell motility are gradually being deciphered. Their parts list and wiring diagrams (e.g., KEGG, at www.genome.jp/kegg/pathway/hsa/hsa04810.html) are composites that emanate from studies of many different cell types, with diverse experimental treatments, and technologies (1). Some difficulties with the current state of knowledge is that these diagrams are difficult to comprehend, some key components are missing or not well-characterized, the plethora of those included sometimes obscures which are central and which peripheral, and the static diagrams cannot indicate how these components work together in real time.
In this article, our aim is first, to investigate three important “modules” that act in concert to initiate cell motility, second, to understand their essential dynamical functions, and third, to show how they work together in real time to produce cellular polarization and initiate motility (both normally, and in knockout or mutant cells). By a “module”, we mean a set of interrelated proteins or lipids that can be identified as a unit with specific dynamical functions (amplifying, switching, filtering, etc.). Based on our experience, we focus on the Rho family of small GTPases, the phosphoinositides, and the actin cytoskeleton, as shown in Fig. 1, a and b. We explore their components, crosstalk, and interactions based on the experimental literature.
Previous theoretical work has addressed cell polarization and motility phenomena. Some studies focus on putative activators, inhibitors, etc., with overall appropriate dynamics (15–18). Many rely on a local excitation/global inhibition (LEGI) module to produce specific behaviors such as gradient amplification and adaptation (9,19). Others have investigated signaling networks (20–25). Excellent recent reviews of both theoretical and experimental approaches can be found in the literature (26,27). We compare our model to others in Table 1 (listing components parts included and indicating behaviors that each model could account for). To our knowledge, ours is the first attempt to link together the above three biochemical modules in a model for the polarization and initiation of cell motility. In this work, our guiding principles have been to base assumptions, where possible, on what is known, to assume the simplest mechanisms where knowledge is lacking, and to explore hypotheses for parts of the system that are uncertain. We restrict attention to a one-dimensional “motile cell” (see Geometry of the Model) to enable us to understand the dynamical behavior in the simplest possible geometry, before attempting to move to more computationally challenging or intensive two- or three-dimensional versions (but see (28), for initial steps in simulating a two-dimensional moving cell).
TABLE 1.
Major components considered and resulting behavior obtained in previous models of gradient sensing, cell polarization, and cell motility
| Reference | Actin | CRρ | PIs | Other | GS | Amp | Adapt | Pol | SC | Mot | Sens | Pers |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Meinhardt (15) | Activator/inhibitor | × | × | × | × | × | ||||||
| Narang et al. (16) | × | Activator/inhibitor; receptor | × | × | × | × | × | |||||
| Subramanian and Narang (17) | × | Activator/inhibitor | × | × | × | × | × | |||||
| Narang (18) | Activator/inhibitor; front/back | × | × | ? | × | |||||||
| Levchenko and Iglesias (9) | general | × | LEGI | × | × | × | × | × | ||||
| Ma et al. (19) | × | LEGI | × | × | × | × | × | |||||
| Krishnan and Iglesias (20) | × | × | × | × | × | × | ||||||
| Postma and v. Haastert (109) | Receptor, 2nd messenger, effector | × | × | × | × | |||||||
| Haugh and Schneider (21) | × | × | × | × | ||||||||
| Schneider and Haugh (127) | × | PDGF gradient sensing | × | × | × | |||||||
| Maly et al. (22) | EGFR, MAPK cascade | × | × | × | ? | ? | ||||||
| Sakumura et al. (23) | × | GAPs | × | × | × | |||||||
| Gamba et al. (24) | × | Receptor binding | × | × | × | × | ||||||
| Skupsky et al. (25) | general | × | Four variants | × | × | × | × | × | × | |||
| Herrmann et al. (128) | × | × | ||||||||||
| Gracheva and Othmer (96) | × | Myosin | × | |||||||||
| Mogilner and E-Keshet (45) | × | Arp2/3, thymosin, profilin, etc. | × | |||||||||
| Grimm et al. (129) | × | × | × | |||||||||
| Rubinstein et al. (47) | × | Myosin | × | × | ||||||||
| Dawes et al. (98) | × | Arp2/3 | × | |||||||||
| Marée et al. (28) | × | × | Arp2/3, myosin | × | × | × | × | × | × | × | ||
| Civelekoglu-Scholey et al. (130) | × | × | Alignment of stress fibers | × | × | |||||||
| This article | × | × | × | Arp2/3 | × | × | × | × | × | × |
Actin, Rho proteins, Cdc42, Rac Rho (CRρ), and phosphoinositides (PIs), as well as other components included in these models are listed in the first four columns. The remaining columns list behaviors observed in the models. GS, gradient sensing; Amp, amplification; Adapt, adaptation; Pol, polarization; SC, shape change; Mot, motility; Sens, sensitivity to change in stimulus location; and Pers, persistence of polarization when stimulus removed.
BIOLOGICAL BACKGROUND
We briefly describe some essential biological background needed for understanding the model and its assumptions. Much of this background has been reviewed elsewhere. We label key (well-established) biological facts (B1, B2, etc.) for later use in justifying model assumptions. Readers not interested in the detailed justification can refer to Fig. 1 and proceed to Questions We Address.
Regulation of actin polymerization in motile cells
To initiate motility, cells reorganize their actin cytoskeleton into a thin protruding sheet (the lamellipod), with filaments oriented toward the membrane (29–31), impinging on and pushing the leading edge forward. In motile cells, active polymerization occurs almost exclusively at the leading edge (29,32–34). The extension of actin filaments (at their fast growing “barbed ends”) is tightly regulated by many factors that nucleate, cap, and depolymerize them (35,36). Arp2/3 is essential for cell motility (37). It is activated close to the membrane by WASp or N-WASp (34,38,39), associates with, and causes, side-branching off actin filaments, thereby nucleating new growing barbed ends (40,41). Arp2/3 is incorporated into the actin network, and is recycled when filaments depolymerize, at the back of the lamellipod. The literature on theoretical approaches to actin cytoskeletal growth is extensive, and includes Mogilner and Oster (42,43), Carlsson (44), Mogilner and Edelstein-Keshet (45), Carlsson et al. (46), and Rubinstein et al. (47).
Rho proteins
The best-studied members of the Rho subfamily, Cdc42Hs, Rac1, and RhoA (henceforth Cdc42, Rac, and Rho) are expressed by many cell types (e.g., fibroblasts, neutrophils, neurons) and are crucial for cell motility (48–50), and organization of the actin cytoskeleton (51,52). In a resting cell, the Rho proteins are evenly distributed, but when a cell is stimulated by a spatially graded signal, active Rho proteins (bound to the membrane and GTP) reorganize into spatially graded distributions (Cdc42 and Rac high at the leading edge and Rho high at the rear) (13,53–56). Inactive (GDP bound) Rho proteins transit between the membrane and cytosol, where they diffuse more rapidly. Interconversion of these forms is accelerated by the activating guanine-nucleotide exchange factors (GEFs) and inactivating proteins (GAPs) (57–60). Rho family proteins interact with one another via crosstalk, although the detailed mechanism of that crosstalk is not yet known. It has been demonstrated in many cell types that Cdc42 activates Rac and Rac activates Rho (61–63). Whether mutual Rac-Rho inhibition (51,63–65) or mutual Cdc42-Rho inhibition (62,66) are the rule, is less clear. In theoretical work, Sakumura et al. (23) analyzed several variants. In our recent work (28,67,68) we adopted the crosstalk proposed by Giniger (66) (see also Fig. 1 a), with mutual inhibition of Cdc42 and Rho. We investigate the effect of inhibitory arrow 9 in Fig. 1 a with our numerical experiments in Cdc42 Spatially Excludes PTEN by Inhibiting Activation of Rho. A well-recognized actin-related role of the Rho family GTPases includes the following:
B1. Cdc42 interacts with WASp or N-WASp, membrane-associated proteins, to increase the activation of Arp2/3, causing filament branching and new barbed end formation at the leading edge (52,69,70). In the models developed here, we incorporate Cdc42 activation of Arp2/3 (arrow 6 of Fig. 1 a) and investigate the role Cdc42 plays in gradient sensing and polarization.
Phosphoinositides
Excellent reviews of the structure, function, and interconversions of these membrane lipids are given in the literature (71–74). In this article, we focus on PI(4)P, PI(4,5)P, and PI(3,4,5)P (henceforth PIP1, PIP2, and PIP3, respectively). The kinases PI5K and PI3K add phosphates to the 5- and 3-positions, respectively, while the phosphatase PTEN removes phosphates from the 3-position. Together, these inter-convert phosphoinositides (PIs), as shown in Fig. 1 a. PI3K, PTEN, and PI5K can diffuse in the cytoplasm. When a cell is exposed to a chemoattractant gradient, PTEN is released from the membrane at the front of the cell, allowing PI3K to associate with the membrane. (PTEN remains bound to the sides and the back of the stimulated cell.) This spatial redistribution of PI3K and PTEN causes PIP3 to be elevated at the leading edge (75–77). PIs diffuse on the membrane at the same rate or faster than Rho proteins. We use the two biological facts:
B2. The phosphoinositide PIP2 inhibits the association of capping protein with barbed ends of actin filaments (50,69). We include this effect in our model (arrow 7 in Fig. 1 a).
B3. According to Higgs and Pollard (78) and Rohatgi et al. (79), PIP2, (not Cdc42) is required to activate WASp and N-WASp, which then activate Arp2/3. In the presence of both PIP2 and Cdc42, activation of Arp2/3 is enhanced (80), suggesting that Cdc42 amplifies this effect (arrow 6 in Fig. 1 a).
Interconnection of the modules
It has been shown experimentally that PIP3 is required for the activation of Cdc42 and Rac (4,8,53,81). Indeed, using RNA interference, it was determined that PIP3 activates both Cdc42-specific (PIXα, (82)) and Rac-specific (P-REX1, (83)) GEFs, as well as shared GEFs such as Vav2 and Vav3 (84). Based on this, we explore the hypothesis that:
B4. PIP3 upregulates the active forms of Cdc42 and Rac (arrows 1 and 2 of Fig. 1 a). In Activation of Cdc42, Not Rac, by PIP3 Required for Proper Gradient Sensing, we explore the consequence of these interactions.
Evidence that Rac enhances the activity of the kinases PI5K and PI3K comes from several sources. In platelets and neutrophils, it was shown that Rac can directly activate PI5K (85,86). Observations of the fluorescent distribution of PIP3 and active Rac in neutrophils demonstrated that Rac (and not Cdc42) is required to enhance activity of PI3K (53). Experiments in neuron-like cells suggested that Cdc42 and Rac can interact with the GEFs Vav2, Vav3 to enhance PI3K activity (84), and further, in vitro, active Rac and Cdc42 can bind directly to PI3K (87–89). Based on this evidence, we assume that:
B5. Rac can directly activate PI5K, as well as enhance the activity of PI3K by an unknown mechanism. These assumptions lead to arrows 3 and 4 in Fig. 1 a. We use the model to investigate each of these feedbacks in Feedback from Rac to PI5K or PI3K Required to Maintain PI Asymmetry.
In epithelial cells and neutrophils, Rho activates Rho kinase, which directly binds and activates PTEN by phosphorylation (82,90). Based on this we assume that
B6. Active Rho upregulates the activity of PTEN (arrow 5 of Fig. 1 a).
Moreover, we show that this fact has important consequences for the spatial exclusion of PTEN from areas with high concentration of PI3K and Cdc42 (see Cdc42 Spatially Excludes PTEN by Inhibiting Activation of Rho).
We consider feedback from the actin cytoskeleton to its upstream signaling components. Blocking actin polymerization in motile cells results in a loss of asymmetry in the PIs, but not in the Rho proteins. When cells are exposed to the actin-sequestering latrunculin, and then to chemoattractant, their PIP3 level increases transiently, while Rac is persistently elevated at the leading edge (53). (Latrunculin treatment halts polymerization and prevents the cell from initiating movement.) Cells treated with jasplakinolide (which halts actin filament turnover), stop moving within 1 min and lose PIP3 at their membrane (4). This leads us to explore the hypothesis that:
B7. Actin polymerization (and not merely F-actin or free barbed ends) is necessary to maintain PIP3 accumulation at the leading edge. (See arrows 8 of Fig. 1 a). We explore this hypothesis in Polymerization-Dependent Rac Activation of PI5K and PI3K Causes Loss of PI Asymmetry, But Not Rho Protein Asymmetry.
QUESTIONS WE ADDRESS
Our work is geared toward investigating a number of general questions previously outlined. Chiefly, we ask how the three modules described above work together in real-time to generate prototypical polarization, and initiation of motility in response to a variety of chemotactic stimuli (graded or random). We are also interested in understanding specific observations or phenomena that have been noted, but not yet fully explained in the experimental literature. We list these questions below.
Q1: (a) What accounts for the typical profiles of proteins and PI lipids in a polarized cell? Why are PIP2, PIP3, Cdc42, and Rac high at the front, while Rho is high at the back of a polarized cell? (b) What accounts for the formation and maintenance of a steep gradient of PIP3 and the localization of active Rac (91)? (c) What prevents PI gradients from dissipating by diffusion along the membrane, i.e., what preserves that gradient (91)? (d) If such internal gradients are persistent, how does a cell turn in response to a new distinctly oriented signal, i.e., what prevents the internal gradient from becoming “frozen” (15,92)?
Q2: How can cell polarization and motion be persistent, even after the stimulus is removed (4,12,93)?
Q3: (a) How can cells respond consistently to weak stimuli (1,4,5,12)? (b) Why do neutrophils exposed to a uniform concentration of chemoattractant polarize in a random direction and move (14,93)?
Q4: What is the relative importance of feedbacks from PIP3 to Cdc42 versus to Rac?
Q5: How do we explain the fact that directional sensing is not obligatory for motility (12)?
Q6: Why are both cell polarity and cell motility blocked if PI3K is inhibited (4,53)?
Q7: Why are cells lacking active Cdc42 able to move but not to detect a gradient (53,94)?
Q8: What is the relative importance of feedback from Rac to PI5K versus PI3K (53,91,95)?
Q9: How do we explain the fact that PTEN and Cdc42 tend to spatially exclude one another in neutrophils (82,90)?
Q10: What prevents Rho proteins from forming arbitrary distributions of high activity in the cell in response to irregular or noisy stimuli?
Q11: How can we explain the fact that in latrunculin treated cells, Rac, but not PIP3, is persistently elevated at the leading edge in response to chemoattractant (53)?
Q12: (a) What is the main dynamic functional role of the PI module? (b) How is this related to the dynamic functional role of the Rho family GTPases module?
We present our model in the next section and then describe detailed numerical experiments that shed light on these questions.
MODEL OF PIs, RHO PROTEIN, AND ACTIN DYNAMICS
Our model incorporates the actin cytoskeleton, the Rho family of small GTPases, and the phosphoinositides (Fig. 1, a and b). We describe the geometry and essential ingredients of each module below, and present detailed equations in Appendix A and parameter estimates in Appendix B and in Table 2.
TABLE 2.
Parameter estimates relevant to actin dynamics, Rho-proteins, and phosphoinositide dynamics, with cited sources
| Parameter | Definition | Value | Source |
|---|---|---|---|
| Actin dynamics | |||
| μP | PIP2-dependent Arp2/3 activation rate. | 0.011 s−1 | (28) |
| nP | Hill coefficient of PIP2-mediated Arp2/3 activation. | 5 | (28) |
| P2half | Threshold concentration of PIP2 for Arp2/3 activation. | 50 μM | (28) |
| DA | Diffusion coefficient of Arp2/3. | 1 μm2 s−1 | (98) |
| η | Arp2/3 nucleation rate. | 0.06 μM s−1 | (98) |
| Km | Saturation constant for Arp2/3 nucleation. | 2 μM | (28) |
| l | Scale factor (converts units of F to concentration). | 0.255 μM | (28) |
| k | Scale factor (converts concentration to units of B). | 106 μm−1μM−1 | (28) |
| v | Actin filament growth rate (free polymerization). | 0.5 μm s−1 | (118,124) |
| γ | Actin filament turnover rate. | 0.03 s−1 | (125,45) |
| kmax | Barbed end capping rate. | 2.8 s−1 | (118,119) |
| kP2 | Maximum reduction of capping by PIP2. | 2.1 s−1 | (28) |
| w | Energy ratio in Eq. 7. | 5 μm−1 | (45,98) |
| Rho proteins | |||
| Cb, Rb, ρb | Typical levels of active Cdc42, Rac, Rho. | 1, 3, 1.25 μM | (28,67) |
| Ctot, Rtot, Ptot | Total levels of Cdc42, Rac, Rho. | 2.4, 7.5, 3.1 μM | (28,67,101) |
| IC, IR, Iρ | Cdc42, Rac, Rho activation input rates. | 3.4, 0.5, 3.3 μM s−1 | (28,67) |
| a1 | Rho level for half-maximum inhibition of Cdc42. | 1.25 μM | (28,67) |
| a2 | Cdc42 level for half-maximum inhibition of Rho. | 1 μM | (28,67) |
| n | Hill coefficient of Cdc42-Rho mutual inhibition. | 3 | (28,67) |
| α | Cdc42-dependent Rac activation rate. | 4.5 s−1 | (28,67) |
| β | Rac-dependent Rho activation rate. | 0.3 s−1 | (28,67) |
| dC,dR,dp | Decay rates of activated Rho-proteins. | 1 s−1 | (121,122) |
| Dm, Dmc | Diffusion coefficient of active, inactive Rho-proteins. | 0.1, 10 μm2 s−1 | (99,109) |
| Phosphoinositide dynamics | |||
| IP1 | PIP1 input rate. | 105 μM s−1 | (102) |
| δP1 | PIP1 decay rate. | 2.1 s−1 | (102) |
| kPI5K | PIP1 to PIP2 baseline conversion rate (by PI5K). | 0.84 s−1 | (102) |
| k21 | PIP2 to PIP1 conversion rate. | 1.4 s−1 | (102) |
| kPI3K | PIP2 to PIP3 baseline conversion rate (by PI3K). | 0.0072 s−1 | (102) |
| kPTEN | PIP3 to PIP2 baseline conversion rate (by PTEN). | 4.3 s−1 | (102) |
| DP | PI diffusion rate. | 0.5–5 μm2 s−1 | (99,100) |
| P1b, P2b, P3b | Typical levels of PIP1, PIP2, PIP3. | 50, 30, 0.05 μM | (9,126) |
Geometry of the model
The geometry of the model is illustrated in Fig. 2. A resting (unpolarized) or motile (polarized) cell with lamellipod is represented by a thin transect through its diameter. We assume implicitly that thickness and width of the transect are both small and constant (ignoring, for example, the bulging nucleus) relative to its length, making the one-dimensional model a reasonable approximation.
FIGURE 2.

Geometry of the model, showing the cell (stationary in panel a, and later motile in panel b) with a one-dimensional transect (shaded bar), as viewed from the top. (c) (Enlarged side view) The transect contains intermediates such as Rho proteins in the membrane (solid disks) and cytosol (open circles) treated as effective concentrations in vertical cross-sections (d), rather than in distinct compartments. Membrane proteins diffuse laterally more slowly than their cytosolic counterparts. As a result, the gradient in cytosolic proteins is much shallower than in the membrane fraction (thin versus thick lines in panel e). The axis indicates direction of motion, position of cell edge, and length of the cell. Other intermediates (actin filaments, etc.) are not shown in this schematic. The thickness of the transect has been greatly enlarged for visual clarity, and is neglected in the model.
Certain important effectors (e.g., Rho proteins) exist in both cytosolic and membrane-bound forms, but rather than assigning distinct compartments, we merely consider the “effective mean concentrations” of each within a vertical column though the cell. (Instead of resolving membrane versus cytosol, we consider the domain as a two-phase composite, with intermediates exchanging between phases (28,68).) The key and important distinction between membrane-versus-cytosolic forms is the rate of transverse diffusion along the one-dimensional axis of the model, and this is preserved by appropriate choices of diffusion coefficients. With this geometry, neither membrane nor cytosolic compartments are homogeneous along the one-dimensional domain, but since diffusion is much faster in the cytoplasm, gradients along the domain are much shallower in the cytoplasm.
Basic scheme
As a general rule, we restrict variables to known biochemical entities, and avoid including hypothetical inhibitors or activators. Activation/inactivation of a given substance, X (in a well-mixed system), subject to the influence of Y are described by the basic scheme
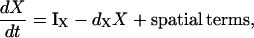 |
(1a) |
where
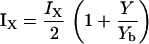 |
(1b) |
In Eq. 1a, the notation IX denotes a rate of activation (e.g., dependent on feedback from Y), whereas dX is the rate of inactivation (or turnover) of X. In Eq. 1b, we assume a linear activation, i.e., that a fractional increase of Y above some constant basal level, Yb, leads to a proportional increase in the activity of X. (As discussed later, we also tested Michaelis-Menten activation rates, and found qualitatively similar results.)
Spatial terms in Eq. 1a include diffusion and transport. The speed of the one-dimensional moving-cell depends on the cytoskeleton-mediated protrusion forces. We assume that both cytoplasmic and membrane-associated small molecules diffuse in this domain, and that they are also transported by a bulk flow, vbulk, when the cell edge protrudes forward.
Model of actin dynamics
The model of actin dynamics keeps track of actin filament density (F), density of growing barbed ends (B), and concentration of activated Arp2/3 (A) available to nucleate new barbed ends. Filaments are assumed to be essentially immobile due to crosslinking and attachment to adhesion sites (retrograde flow is here ignored). The growth of barbed ends deposits new filament density, and filaments turn over at some average constant rate, γ. The motion of the cell is modeled as protrusion-limited, with barbed ends pushing the membrane at the leading edge (x = xedge). We omit adhesion-contraction mechanics (but see (96)). We approximate the effect of barbed ends on protrusion speed at the leading edge, with a thermal ratchet relationship (42,43,97). In a previous article, we explored the relationship between Arp2/3-mediated branching of actin filaments and cell speed resulting from the formation of new barbed ends at the membrane (98). Here, we are concerned with the regulation of Arp2/3 activation that creates the surge of barbed ends in response to a stimulus.
Based on biological facts B1 and B3, Arp2/3 activation depends on Cdc42 (C) and PIP2 (P2). However, a fully linear relationship implies that even small elevations in PIP2 induce cell motion, which is unrealistic. Hence, we assumed that activation occurs once PIP2 exceeds some threshold,
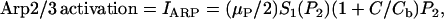 |
(2) |
where S1(P2) = 0 for low PIP2 levels, and S1(P2) = 1 beyond a low threshold value of PIP2 (see Appendix A for details). We also assumed (based on B2) that barbed end capping decreases in the presence of PIP2,
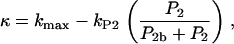 |
(3) |
where P2b is the baseline concentration of PIP2. In later tests of the model, we also incorporated feedback from newly synthesized actin to upstream signaling components.
Model of Rho protein dynamics
The Rho protein module forms the central component of the regulatory pathway in our model. Our treatment of this module is related to the previous work by Sakumura et al. (23) with the following essential differences: based on the experimental observations of spatial segregation of these proteins and their mutual crosstalk, we predict that this module is a switch between multiple equilibria (rather than an oscillator, as in (23)). We model crosstalk through GEF-mediated activation (rather than GAP-mediated inhibition): the actual mechanism is not yet known definitively. Inactive Rho proteins distribute to the cytosol where diffusion is much more rapid. This means that they can rapidly transmit “global information” (in the sense of (1,26,93), see also Discussion, this article). We use the simplest crosstalk scheme (66), to describe this module.
In our previous treatment of the Rho protein module (28,67,68), we showed that a minimal module that contains the essential features described above can be constructed from the following basic scheme: Crosstalk between the Rho proteins has to be of the type that allows for multiple coexisting steady states (i.e., high Cdc42 with low Rho and vice versa), as in the literature (23,66). Mutual inhibition between Cdc42 and Rho is a requirement of this scheme, and so is nonlinearity higher than Michaelian kinetics (i.e., some degree of cooperativity). These assumptions are essential for ensuring the existence of multiple steady states needed to account for observations. Finally, the rapidly diffusing inactive forms of the Rho proteins are important for stabilizing the polarized wavefront, and preventing one or another homogeneous steady state from sweeping through and taking over the whole domain (68). The basic scheme we arrived at has the form
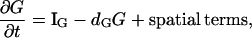 |
(4a) |
 |
(4b) |
for G = C, R, ρ active, Gi = Ci, Ri, ρi, inactive forms of Cdc42, Rac, and Rho, respectively, and dG an inactivation rate. The activation rate, IG, is some function of the form
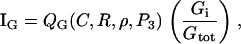 |
(5) |
with QG a GEF-mediated rate of activation of the inactive fraction, Gi/Gtot, of a given G protein. Crosstalk and effects of the phosphoinositides are depicted by QC, QR, Qρ. Based on assumed GEF-mediated crosstalk, QC decreases with Rho (ρ), Qρ decreases with Cdc42 (C), QR increases with C, and Qρ increases with Rac (R). (Based on B4, QC and QR increase with PIP3 (P3), a feature not explored in our previous work.) See Appendix A for details.
The spatial terms in Eqs. 4 and 5 include advection and diffusion. This leads to six equations for the three active and three inactive forms of these proteins. The motivation for specific choices of activation functions and other details are described in the literature (28,67,68). As shown in these background articles, with these assumptions, the Rho module dynamics are consistent with multiple distinct equilibria for a large range of parameter values. The module accounts for spatial polarization in response to graded or noisy inputs (e.g., in IC), producing a stable polarized distribution. Furthermore, in the preliminary two-dimensional cell motility simulation (28), we have shown that, once polarized, this module retains sensitivity to stimuli along new directions.
Model of PI dynamics
We do not model the full temporal dynamics of PI3K, PTEN, etc. Rather, we use equations based on the general strategy of Eq. 1, and Rho protein effects (B5, B6) to formulate a quasi-steady-state (QSS) assumption for these. This leads to a general form for the level φ = PTEN, PI3K, PI5K of the kinase/phosphatase that is then incorporated into the PI dynamics. To simulate the observed spatial asymmetry of PI3K and PTEN in response to a spatially graded external signal, we impose a gradient in the parameter Iφ (or kφ) across the one-dimensional domain (see Appendix A for details). Eqs. 13 tracks the dynamics of PIP1, PIP2, and PIP3 across the cell. These forms are interconverted, under the influences of PTEN, PI3K, and PI5K, mentioned above. Further, based on B5, Rac enhances the conversion of PIP1 to PIP2 (via PI5K) and the conversion of PIP2 to PIP3 via PI3K. Based on B6, Rho enhances the conversion of PIP3 to PIP2 via PTEN. We assume all PIs diffuse in the membrane at the same rate, DP, and undergo bulk convection as described above.
ESTIMATING PARAMETERS AND SIMULATING THE MODEL
We based parameter estimates, wherever possible, on experimental data. We used steady-state levels and turnover times to estimate rates of activation and inactivation, where available. Diffusion coefficients are based on previous studies, or on the size and location of the components (e.g., membrane forms diffuse slowly relative to cytosolic forms). Many parameter values collected in our previous articles are briefly summarized, with details provided in Appendix B, and default values gathered in Table 2. Some controversy exists about the rate of diffusion, DP of the PIs. Estimates for this diffusion coefficient range between 0.5 and 5 μm2/s in sources such as Postma et al. (99) and Schneider and Haugh (100). To deal with this discrepancy we studied the behavior of the model for a wide range (several orders of magnitude) of values of DP, as described in Robustness of the Model.
The full model equations (Eqs. 6, 8, 9, and 13 based on the pathways of Fig. 1 a) were simulated in a one-dimensional domain of length L, where x = xedge and x = xedge – L represent the two edges of the cell (eventually, the front and back, but initially not so specified). In some cases, we studied chemical polarization in the static domain xedge – L ≤ x ≤ xedge to investigate dynamics upstream of motility. Where motility was simulated, we allowed xedge(t) to evolve with time. Figures show some results in stationary lab-frame coordinates (e.g., Fig. 3), and others in a coordinate system moving with the cell (e.g., Fig. 4).
FIGURE 4.

(Left column) Spatial profiles of PIs (from top to bottom: PIP1, PIP2, PIP3); (middle column) Active Rho proteins (Cdc42, Rac, Rho); and (right column) actin (barbed ends, Arp2/3, and filament density) in response to the stimulus described in Fig. 3 a. The spatial profiles are consistent with experimental observations, with PIP2, PIP3, Cdc42, and Rac elevated at the front of the cell and Rho elevated at the rear. The dotted line shows the homogeneous prestimulus distribution (i.e., before a gradient is applied to kPI3K and kPTEN), while the solid line shows the post-stimulus steady-state distribution. The leading edge of the cell is on the right in each frame.
Time is scaled by seconds. The simulation was run for 20 s in the absence of a stimulus to initiate baseline (flat) distributions for all components. At t = 20 we applied different stimuli for various time durations. (All stimuli were imposed as internal redistributions of PI3K and PTEN, as we do not model upstream events.) We examined transients, as well as persistent polarization and motility. Steady-state profiles of PIs and Rho proteins were plotted at t = 100 s (well past the stage when transients were eliminated).
Polarization was examined by plotting the distributions of signaling components and actin across the one-dimensional domain (e.g., Fig. 4). Motility was assayed by relating the number of actin filament barbed ends at x = xedge to protrusion speed in the positive x direction. (We did not simulate motility of left-moving cells.) Details of the simulations and of the protocol used to run specific tests are provided in Appendix C.
RESULTS
One-dimensional cell polarizes, and initiates persistent motion in response to a spatially graded signal
Our first step was to run the full model with default parameter values. We examined the behavior of the model in response to a variety of graded stimuli. As explained, we incorporated these directly into graded profiles of PI3K and PTEN (see Graded Stimuli). We varied the steepness, magnitude, and duration of the stimuli as described further on. Fig. 3 a shows the growth and translocation of actin filament barbed ends (in lab coordinates) as a result of a strong stimulus. There is a brief time lag of 5–15 s while internal biochemistry reorganizes (Fig. 4). During this lag, the spatial asymmetries in the PIs and Rho proteins are established, and only then does Arp2/3 nucleate barbed ends. A large population of barbed ends are created, the edge is pushed outwards, and motion is initiated. The directed motion and spatial profiles are stable, and continue even after the stimulus is removed. Corresponding cell speed is shown by the dotted line of Fig. 3 b. The overshoot in speed or barbed ends does not occur if the stimulus is weaker.
The profiles of internal signaling components and actin cytoskeleton are shown in a coordinate system moving with the protruding membrane in Fig. 4. By this time (t = 100 s), the magnitudes of the steady-state spatial profiles are independent of the stimulus strength. PIP2 and PIP3 are highest at the front of the cell (left column, Fig. 4), consistent with observations (2,8,91). Note that the scales for phosphoinositides in Fig. 4 differ by two orders of magnitude: PIP3 occurs at very low levels relative to other forms (consistent with (9,71)). Cdc42, Rac, and Rho (center column, Fig. 4) also segregate spatially, with Cdc42 and Rac high at the front, and Rho high at the back. This is consistent with fluorescence imaging in live motile cells (13,54,55,101). Similar profiles of these proteins were obtained by us previously in Jilkine (67) and Jilkine et al. (68).
The profiles of actin filament density, barbed ends, and Arp2/3 are shown on the left column of the same figure. Arp2/3 is highest at the leading edge, followed by a peak of barbed ends and further back, a peak of actin filament density. Similar profiles that we previously obtained in an actin-only one-dimensional model (98) were compared to experimental profiles observed by (32,34,39). Related results for actin with Rho proteins were also discussed in Dawes (102) and in a two-dimensional motile cell (28).
As shown by this “control” simulation, the model produces appropriate one-dimensional spatial profiles of signaling components, and initiates protrusive motility in response to a strong graded stimulus. Thus, the interactions shown in Fig. 1 (the default model) suffice to account for basic phenomena to be explained (see Q1(a)). The persistence of polarization and motion is in agreement with the literature (4,12,93), and addresses question Q2.
Niggli (91) noted that the mechanism for formation of a steep gradient of PIP3 was uncertain, and that the localization of active Rac was also unclear. We can understand both phenomena from the scheme of Fig. 1: A weak stimulus that sets up a shallow gradient in the PIs feeds onto the Rho protein module, tripping the GTPase-switch and setting up a spatially segregated profile of these Rho proteins. The asymmetric gradient of the Rho proteins in turn feeds back onto the PI module, reinforcing and amplifying that PI gradient. When PIP3 is thereby concentrated at the front, it further reinforces and elevates the activity of Rac (arrow 2, Fig. 1). This set of interactions then maintains the asymmetric profile of the PIs after the stimulus is removed. This result addresses question Q1(b).
Before carrying out numerical experiments, we first investigated robustness of the default model to parameter values, kinetic terms, and types of stimuli. We afterwards modified the default model by one or another numerical experiments to investigate the effects of knockouts and mutants, or to understand the roles of specific pathway components or interactions.
Robustness of the model
The behavior described above constitutes a default behavior that was obtained in one realization of the model. We asked whether this behavior was robust to variations in the values of parameters, choices of kinetic terms, and applied stimuli.
We varied each kinetic parameter by 10% and found the same qualitative behavior (see Appendix B for further details). Many parameters could be varied on a much greater range. For example, we varied the rate of PI diffusion (Dp) in the range of 0.5–5 μm2/s and found no qualitative effect and little quantitative effect on the model. Thus the model cell is robust to parameter variations, with one exception: lowering the Hill coefficient in the Rho protein module leads to loss of bistability and polarization.
We explored whether assumed linearity versus nonlinearity of terms in the model creates artifacts in behavior. As previously discussed, nonlinearity in the Rho module is essential for the type of spatial bistability observed experimentally. Changing all linear terms in the PI and Rho protein equations to saturating (Michaelis-Menten) kinetics makes the slope of the internal steady-state gradients shallower, but does not qualitatively change the behavior of the model. In both linear and Michaelian cases, the model cell polarizes persistently in response to a graded stimulus.
The response of the model was also tested using stimuli superimposed on various baseline levels. (See protocol discussed in Appendix C.) We found that the steady-state response of the cell was identical. Based on these findings, we adopted the default model, parameter values, and stimuli as the basic protocol, from which further numerical experiments were done.
Weak stimuli cause a delay in initiation of motion
To determine the response of the model-cell and its sensitivity to weak graded stimuli, we varied the steepness, strength, and duration of graded stimuli (see protocol in Graded Stimuli). Stimuli applied for a short period of time (Fig. 3 b) resulted in directed motion, but only after some delay (a similar result was found for weak stimuli). A graded stimulus was applied for periods of time ranging from 0.01 s to the full length of the simulation. Membrane speed was computed as the simulation progressed, and plotted. The onset of motion is shown in Fig. 3 b. Stimuli applied for a short period of time (for example 0.1 s) cause directed motion but only after a time delay of ∼15 s. Longer or stronger stimuli organized the polarization and motion more rapidly, and caused a slight overshoot in the membrane speed before steady-state motion was established. (See also the overshoot previously described in Fig. 3 a.) Similar results are seen for shallower (i.e., weaker) graded stimuli applied for the same period of time (not shown).
These simulation results suggest that the pathways of Fig. 1 a suffice to account for the cell's response to weak or short-lived stimuli, addressing question Q3(a). However, we also found that the weaker the stimuli, the longer the time lag. The delay stems from the time taken to assemble the internal map. Our model is constructed so that this occurs even for very weak or short-lived stimuli, but the weaker/shorter the stimulus, the longer it takes the cell to polarize. This delay in initiation of motion (for a reasonable range of stimulus strengths) is one of our testable predictions. In a real cell, there are likely further mechanisms (not included in our model) to prevent polarization in response to extremely weak stimuli.
Activation of Cdc42, not Rac, by PIP3 required for proper gradient sensing
As noted in B4, PIP3 activates both Cdc42 and Rac by enhancing the activity of specific GEFs (82–84). However, the relative importance or roles of these feedbacks has not previously been examined. We investigated this question (Q4) by selectively abolishing either arrows 1 or 2 in Fig. 1 a, and simulating the model as previously described.
We first cut only arrow 2 of Fig. 1 a, from PIP3 to Rac. We obtained proper spatial localization of PIs and Rho proteins, with high Cdc42, Rac, PIP2, and PIP3 at the front (Fig. 5, solid lines). Once the cell polarizes, it begins moving to the right (the leading edge is formed in the area of the cell with high Cdc42, Rac, PIP2, and PIP3).
FIGURE 5.

Cells lacking active Cdc42 (dashed lines) cannot properly detect the direction of a stimulus but are able to initiate movement. The dotted line indicates the prestimulus distribution, when kPI3K and kPTEN, and all other intermediates are at their constant baseline values (as in Table 2) everywhere in the domain. Spatial profiles of PIs (left column) and Rho proteins (right column) in response to a transient graded stimulus in kPI3K and kPTEN when PIP3 enhances activation of only Cdc42 (solid line, note consistency with full model) or only Rac (dashed line, note polarity of profiles are reversed). This demonstrates the importance of arrow 1 (from PIP3 to Cdc42) in Fig 1 a.
We next cut only arrow 1 in Fig. 1 a from PIP3 to Cdc42. Surprisingly, we found that opposite spatial profiles were obtained: Rho is high at the front of the cell and PIP2, PIP3, Cdc42, and Rac are elevated at the back (dashed lines in Fig. 5). This type of cell would move in the opposite direction (motility not simulated). The result of this numerical experiment is consistent with the observation by Parent and Devreotes (12) that directional sensing is not obligatory for motility: mutants lacking the pathway represented by arrow 1 would still polarize and move, but may not correctly detect and align with the gradient. (This addresses aspects of question Q5.)
The reverse polarization can be understood in the context of Fig. 1: in a control-cell, a graded signal in PIs activates Cdc42, which both elevates Rac and depresses Rho at the front. In the mutant-cell lacking arrow 1, Cdc42 is bypassed: the PIs activate Rac, which activates Rho at the front. Due to mutual (Cdc42-Rho) inhibition, Rho takes over the front, Cdc42 is relegated to the back, and the polarity is reversed. GTPase and PI segregation can still occur, but Cdc42 cannot establish dominance at the front. Thus, the cell could still develop a polarity and move, but may not detect the gradient correctly or move in the correct direction.
Finally, we cut both arrows 1 and 2. The graded stimulus was applied to Cdc42 by imposing a linear gradient in the activation rate, Ic (since gradients in PIs no longer affect the Rho proteins). In this case (results not shown), the cell takes longer to polarize and initiate movement and the steady-state speed is much slower compared to the full model. This stems from a lack of positive feedback from the PIs to the Rho proteins. The Rho module polarizes first, and then, through arrows 3, 4, and 5, leads to PI polarization. The polarization and movement of the model cell are persistent.
We can also understand other observations in the context of these results. According to the literature (91,95,103), Rac activation depends somewhat on activation of PI3K. Moreover, Wang et al. (4,53) noted that if PI3K were entirely inhibited, both cell polarity and cell motility would be blocked. Since PI3K activation creates PIP3, which, in turn, feeds forward to the Rho module, this observation makes sense, and addresses question Q6. It is consistent with arrows 1 and 2 in Fig. 1, both of which lead to enhanced activation of Rac.
Further, we can put our numerical results into the context of experiments demonstrating that cells lacking active Cdc42 are able to move but cannot properly detect the direction of the gradient or the source of the stimulus (53,94) (see Q7). Absence of Cdc42 is analogous to severing arrows 1 and 9 of Fig. 1 a. First, this destroys the spatial polarizability of the Rho protein module, which depends on the double negative feedbacks of Cdc42 and Rho, and second, it prevents the spatial PI asymmetry from properly biasing the Rho proteins. This means that an external gradient may lead to internal restructuring, but not to the correct corresponding internal gradient. In summary, these results indicate that PIP3 activation of Cdc42 is required for proper polarization, not PIP3 activation of Rac.
Feedback from Rac to PI5K or PI3K required to maintain PI asymmetry
It has been shown experimentally (see B5) that Rac directly interacts with and activates PI5K (85,110) and is important for PI3K activity (53,91,95). To explore the relative importance of Rac feedback onto PI5K and PI3K (see Q8), we conducted numerical tests in which arrows 3 and 4 in Fig. 1 a were cut (see protocol in Dissecting the Pathways). As above, a graded stimulus (100% for 10 s) was applied after 20 s and the final data was collected at 100 s.
We first removed both feedbacks. This resulted in complete loss of polarity in PIP3 (but not Rho proteins) as soon as the graded stimulus was removed. In this case, PIP2 did not build up sufficiently to activate Arp2/3, and not enough barbed ends were nucleated to initiate movement. As shown in Fig. 6, restoring either feedback (Rac activation of PI5K or PI3K) leads to an asymmetric distribution of PIP3. When Rac enhances only PI3K (i.e., cut arrow 3), there is no asymmetric distribution of PIP1 or PIP2, only of PIP3, while Rac activation of PI5K (i.e., cutting arrow 4) leads to asymmetric distributions of all three PIs.
FIGURE 6.

If Rac enhances only PI5K activity (dashed line), all PIs develop spatially graded profiles. If Rac enhances only PI3K activity (solid line), only PIP3 develops a spatially graded profile. Variables same as in Fig. 5. Prestimulus distribution (dotted line). The spatial profiles that result from the transient stimulus are stable. The concentration of PIP3 at the front of the cell is higher when PI3K activity is enhanced, suggesting that conversion of PIP2 to PIP3 is more important than increased availability of the substrate PIP2. The conversion of PIs has little effect on the spatial profiles of the small G proteins.
The concentration of PIP3 at the front of the cell is higher when Rac enhances PI3K rather than PI5K activity, suggesting that increasing the conversion rate from PIP2 to PIP3 provides greater accumulation of PIP3 than simply increasing the concentration of the substrate, PIP2. These simulation results suggest that some feedback from Rac onto the PIs through kinase activity is required for the maintenance of asymmetric profiles in the PIs after the removal of the stimulus. Further, it shows that, under assumptions of our model, enhancing the rate of PIP2 to PIP3 conversion provides the greatest accumulation of PIP3 at the leading edge.
Cdc42 spatially excludes PTEN by inhibiting activation of Rho
While PI3K is essential for gradient sensing, the role of PTEN (and even its localization) is still controversial. In many motile cell types, PTEN establishes a spatial gradient reciprocal to that of PI3K. (However, there have been reports that PTEN is strictly cytosolic in migrating neutrophils; e.g., see (104).) It has been suggested that PTEN and active Cdc42 do not spatially colocalize (82,90), but the underlying reason for this has not been clear (see Q9). In this section we discuss a possible reason for this spatial exclusion. (Note, however, that the model cell is able to properly detect a gradient and initiate movement even when PTEN activity is constant everywhere on the domain; not shown.) In our default model runs, this phenomenon is observed indirectly: The activity of PTEN is assumed to depend positively on the activity of Rho. However, as previously discussed, mutual exclusion of Cdc42 and Rho is essential in our Rho protein module for spatial polarization to occur. (New evidence for an inhibitory role of Rho appears in Ohta et al. (65).) This inhibitory interaction then also implies mutual exclusion of Cdc42 and PTEN in the model predictions.
We simulated a variant of the model in which arrow 9 of Fig. 1 a was cut (i.e., omitting the inhibitory effect of Cdc42 in the dynamics of Rho). Using the usual protocol (see Dissecting the Pathways), we ran the model to its steady state and determined resulting profiles.
Whereas in simulations of the full model (Eqs. 8, 9, and 13), PTEN activity is low in areas where active Cdc42 is high, if Cdc42 does not inhibit Rho, the stable spatial asymmetry of the PIs and small G proteins is lost and PTEN activity is higher than baseline everywhere in the cell (not shown). These results suggest that the spatial exclusion of PTEN from areas with active Cdc42 may be due to the inhibition of Rho by Cdc42, addressing question Q9. Since Rho has been shown experimentally to activate PTEN (82,90), PTEN activity will be high only in those areas where active Rho is high and Cdc42 low.
This sheds light on experimental observations that Cdc42 activation plays a role in excluding PTEN from leukocyte protrusions (1,105). Ridley (1) postulated the existence of a “network of feedback loops” that conspire to produce and sustain motile cell polarity. We have shown here that the loops in Fig. 1 a suffice to explain this result, but that when loop 9 is cut, this spatial exclusion of PTEN is not observed. In a related comment, Niggli (91) remarked that it is not clear what mechanisms are responsible for retaining the gradient in PI molecules that forms in response to graded stimuli, i.e., what prevents these molecules from diffusing laterally along the membrane (see Q1(c)). While our model does not address the two- or three-dimensional localization, we have shown that the polarization of the Rho module in turn reinforces and maintains the polarity of the lipids, preventing the smearing of the PIs and loss of that internal gradient. Finally, these results are in concert with the statement in Kimmel and Parent (105) that PI3K-PTEN pathways are essential in chemotaxis, and that elevation of PIP3 in the front of the cell results from their reciprocal distributions. Our predictions here can be tested experimentally by microinjecting resting cells with active Cdc42 and observing the resulting spatial distribution of Cdc42, Rho, and PTEN.
Competing random stimuli cause the cell to polarize and initiate movement
We investigated the response of the one-dimensional model cell to a 10-s spatially irregular, but temporally fixed input, representing a superposition of competing random stimuli (protocol in Competing (Random) Stimuli).
Without PIs, Rho proteins can form multiple domains of high Cdc42
We first examined the model's response in the absence of the PIs (i.e., with only Rho proteins and actin). To do so, we modified the model so that PIP2 activation of Arp2/3 (arrow 6 in Fig. 1) is replaced by Cdc42 activation of Arp2/3, and PIP2 suppression of capping (arrow 7) is replaced by Rac inhibition of capping to preserve downstream effects on the cytoskeleton (as in (28)). We then ran the simulation with the stimulus applied directly to the Cdc42 activation rate, IC.
If the input is graded, no multiple peaks occur. Further, for weak random stimuli, transient multiple peaks coalesce into one, exactly as reported by Wong et al. (56). A strong random stimulus, however, leads to multiple stable domains with high Cdc42 and Rac, and reciprocal Rho as shown in Fig. 7. The profile of barbed ends, Arp2/3, and filament density reflect the spatial profile of the Rho proteins, i.e., produce a distribution of growing barbed ends that is inappropriate for efficient motility. In our one-dimensional model, this means that the cell slows down (speed not shown), since not as many barbed ends localize to the leading edge. In a more realistic two-dimensional version, it is easier to interpret multiple peaks of Cdc42 as multiple sites where nascent competing lamellipodia would form. Indeed, the appearance of multiple lamellipodia was noted by us in two-dimensional simulations of the motile cell in which PIs were not yet included (28). We can understand these results by noting that barbed ends do not persist outside areas with high levels of active Rac due to rapid capping. Barbed ends are nucleated in areas within the domain with high Cdc42 and Rac, but even if they grow toward the leading edge, they are mostly eliminated by capping before they reach the cell edge. This simulation suggests that in the absence of the PIs, a variety of putative Rho protein distributions can be manifested in response to competing or contradictory signals (see Q10).
FIGURE 7.

When the PI module is absent, multiple domains of Rho proteins and actin density are generated in response to a random signal. Barbed ends are generated in areas with high Cdc42 and Rac, but are capped before they can reach the membrane. (Left column) Cdc42 (top), Rac (middle), and Rho (bottom). (Right column) Barbed ends (top), Arp2/3 (middle), and filament density (bottom). The spatial profiles are shown before (dotted line), during (dashed line), and 50 s after (solid line) application of the stimulus.
With the help of PIs, Rho proteins form a defined front and back
We next investigated the effect of the PIs in a similar setting, i.e., with the same strong irregular stimulus, but in a simulation of the full model. Interestingly (as shown in Fig. 8), the inclusion of PIs abolishes the irregular profiles, and reestablishes the appropriate graded internal distributions (solid lines) of both PIs and Rho proteins after some transient (dashed lines). PIs rapidly distribute in a relatively shallow, smooth profile (dashed lines, left column of Fig. 8) This then damps the irregularity of the Cdc42, Rac, and Rho distribution. The actin cytoskeleton (not shown) also organizes as usual, and motion is restored.
FIGURE 8.

PIs act to smooth out Rho proteins and prevent establishment of multiple domains of high Cdc42 in response to a random stimulus (compare with Fig. 7). Spatial profiles are shown before (dotted line), during (dashed line), and 50 s after (solid line) application of the stimulus. Upstream regulation of the Rho proteins by PIs ensures the establishment of a single domain with high Cdc42 at the front of the cell when the cell is exposed to a random stimulus (in contrast to Fig. 7).
We asked what could account for this remarkable smoothing effect, and whether it stems from the relative size of the domain versus PI diffusion length. To explore this, we ran several simulations (not shown):
We repeated the same simulations, in a much larger domain (100 μm, instead of 10 μm). We found, as suspected, that the full model leads to a number of persistent peaks of components inside that domain (larger than typical cell size).
We ran similar simulations in a regular domain size, but with PI-like components whose diffusion coefficient was assumed to be smaller than in reality (D = 0.001, 0.01, 0.1 μm2 s−1).
For the first two cases, two peaks of Rho protein levels occurred in a 10-μm domain, whereas for the latter case, Rho proteins returned to their typical graded profile, as in the default runs. Multiple peaks in response to a random stimulus do not persist for Dp ≥ 0.05 μm2/s. Increasing or decreasing (by a factor of 10) other parameter values regulating the PIs does not significantly change the simulation results.
The results above can be used to draw the following conclusions: First, even with the topology of small GTPase crosstalk in our model, a variety of steady-state spatial patterns of Rho proteins could occur. When PIs are included, only the spatially polarized pattern of Rho proteins emerges; we infer that one role of the PIs could be to select a unique (polarized) pattern of Rho proteins from the larger class of possible patterns that could, in theory, exist. Second, and related to the above, PIs play a role in spatially smoothing out noise. Most graded and uniform signals are noisy, presenting conflicting information to directional selection of a cell. The PIs act as a filter, enabling an appropriate direction to be chosen even when multiple stimuli compete. Finally, these observations suggest that the activity of the PIs helps to establish a front and back of the cell, by ensuring that one domain of high Cdc42 and Rac is formed in response to a stimulus, random or otherwise. These observations help to address the global questions Q10 and Q12.
Cells polarize and move in response to near-uniform stimuli
Neutrophils exposed to a uniform concentration of chemoattractant often polarize in a random direction and begin to move (14,93). How this occurs has been the subject of debate and speculation, but still remains largely unknown (12,91) (Q3(b)).
We here interpret a “uniform stimulus” to mean a nongraded (but noisy) distribution. We ran the default model in a simulation with superimposed low amplitude noise. The method is similar to that of the previous sections, but with noise of much smaller amplitude, applied for shorter duration, see Competing (Random) Stimuli. We examined the internal distributions of PI and Rho proteins that resulted (not shown).
Applying a static random stimulus that deviates from baseline by 1%, for a duration of 0.1 s results in polarization of the PI and Rho proteins (same result with stimuli that impinged on either Cdc42 or PI3K/PTEN, and with noise that was temporally static or temporally fluctuating). Both cases lead to polarization with similarly short, weak stimuli. Thus, the model cell is highly sensitive to noise and the cell polarizes rapidly in response to very weak and short-lived stimuli. These observations are consistent with reports that cells such as neutrophils can establish and maintain localization of PIP3, and become polarized even in homogeneous chemoattractant levels (12,91), addressing question Q3(b).
Asymmetric spatial profiles of PIs and Rho proteins can be reversed in response to a second stimulus
Neutrophils respond quickly to a change in the position of a chemoattractant source by reorienting and moving in a new direction. We investigate the sensitivity of our one-dimensional model cell by determining whether the cell can reverse polarity in response to a new stimulus (see Q1(d)). To simulate a change in the position of the signal source, we first ran the default model with a graded stimulus. After transients are gone (at 80 s), we imposed a second gradient with the same strength but opposite slope for various time durations. The simulation was halted after 200 s.
We found that it is possible to reverse the gradients of the PIs and Rho proteins (representing repolarization and reorientation), provided the newly imposed gradient is sufficiently strong, and sufficiently long. For instance, in the one-dimensional model cell, a 100% gradient from front to back must be applied for ∼15 s before the spatial profiles will reverse completely. If the stimulus is applied for a shorter period of time, the spatial profiles recover their original directionality, after some lag, and the cell will continue moving in its original direction.
We can understand these numerical observations as follows: If the stimulus is sufficiently strong, the levels of PIP2, PIP3, Cdc42, and Rac decay from the front and grow in the back, whereas Rho activation grows at the front. If the stimulus is removed prematurely, this change is not sufficiently strong to create a new polarity. If, however, the stimulus duration is long enough to lead to appropriate accumulation, a new polarity is created and the direction of motion of the cell can reverse. This addresses question Q1(d). Note that in two-dimensional, a cell can turn to follow a new gradient at some acute angle to the original stimulus direction by gradually shifting its internal map (i.e., incrementally realigning the internal gradients). This turning behavior (and exquisite sensitivity to new weak stimuli) was already seen in our preliminary two-dimensional motile cell (28) in which PIs were not yet considered. In the one-dimensional geometry here discussed, this incremental realignment cannot be replicated. Hence a stronger and longer signal is needed to affect a full reversal in this one-dimensional model cell.
Polymerization-dependent Rac activation of PI5K and PI3K causes loss of PI asymmetry, but not Rho protein asymmetry
Despite occasional speculation (106–108), the feedback of the actin cytoskeleton on upstream signaling pathways, and therefore on the organization of cell polarity and motility remains mysterious. Several sources (4,53,91) have suggested that actin stabilizes the polarization of PIP3, for example, in neutrophils (see Biological Background, item B7).
To test the hypothesis that actin polymerization is involved in maintaining the PI asymmetry in motile cells, we modified the terms regulating Rac activation of PI5K, kPI5K and PI3K, kPI3K to include a dependence on active polymerization. In our model, polymerization is restricted to barbed ends, and occurs essentially at rate Bv, where v is polymerization velocity. Thus, our modification, described in Appendix C, consists in introducing a dependence on this term in the appropriate rates of activation.
We modified the PI equations so that the activity of PI5K and PI3K is enhanced by Rac only when active polymerization (even at a low level) is taking place. In the absence of barbed ends or free actin monomers (which prevents polymerization), Rac would be prevented from influencing the activity of PI5K or PI3K.
After so modifying the model, we simulated both the latrunculin and jasplakinolide experiments by adjusting the rate of polymerization in treated cells, vt. For latrunculin-treated cells, we assumed vt = 0 while for jasplakinolide-treated cells, we used vt = exp(– Kt) to simulate the gradual loss of actin monomers from continued polymerization. As shown in Fig. 9 (simulations of latrunculin-treated cells), a slight asymmetry in PIP3 (but not PIP1 or PIP2) is possible during the application of the stimulus (dashed line), but the asymmetry in PIP3 is not maintained after the stimulus is removed (solid line).
FIGURE 9.

Spatial asymmetries in PIs and directed motion are not maintained in latrunculin-treated cells if PI5K and PI3K activation relies on actin polymerization. Variables shown are the same as in Fig. 4, but with much lower magnitudes. Prestimulus distribution (dotted line). During stimulus application (dashed line), PIP3 and the Rho proteins, but not PIP1 or PIP2, become polarized. After the stimulus is removed (solid line), all PIs return to a uniform distribution but the asymmetry in the Rho proteins is maintained. Due to low levels of PIP2, Arp2/3 is not activated, causing barbed ends and filament density to remain at a low levels.
In contrast, an asymmetry in the Rho proteins is established and maintained. The simulated cell could not initiate motion since the low PIP2 concentration results in very low levels of Arp2/3, barbed ends, and filaments in the model cell. (Note scales in Fig. 9, right column, and compare with corresponding scales in Fig. 4.) Similar results are achieved in simulations of jasplakinolide-treated cells (not shown).
These results are consistent with suggestions in the literature that interaction of PIP3 with the newly formed actin network can lead to polarity of signaling components, and hence organize the polarization and directed motility of the cell (4,53,56). They further point to the idea that the actin cytoskeleton provides a feedback loop that plays a role in stabilizing and maintaining the PIP3 accumulation at the leading edge of locomoting neutrophils (4,53), addressing question Q11. It is not yet clear what precise molecular mechanisms could mediate such feedback, and we avoid speculating on this matter here.
DISCUSSION
While many of the individual components involved in cell polarization have already been well understood, it has not been at all clear how these interact to lead to cell polarity (8), how they are integrated temporally or spatially in a motile cell (1), and what sets up the initial symmetry breaking that defines a “front” and “back” and leads to the resulting self-organization (14). The importance of feedback loops (1,4,8,14), and of inhibitory and excitatory signaling (12,105) has been indicated in the past. Many investigators have stressed the importance of dissecting and understanding such loops (8), and of identifying putative inhibitors or activators proposed in theoretical models (9,15,18,109). The central issue of spatio-temporal regulation of the actin cytoskeleton has been noted before (105), and so has the importance of PIs and small GTPases of the Rho family (9).
In this article, we have developed an integrated model consisting of PIs and Rho proteins, i.e., biological signaling modules (with identified components), and the actin cytoskeleton. The model allows us to address broad questions (Q1(a–d)) about polarization and initiation of cell motility (e.g., what accounts for the intracellular segregation of components, how can a cell respond to such weak gradients), as well as specific experimental observations that have not previously been well understood (Q6, Q9, etc.). In this and previous work (67,68,98), we have shown that all three of the subsystems (actin, Rho GTPases, and PIs) produce distinct behaviors in isolation. Here we have demonstrated that, by their interaction as a single unified system, they conspire to reinforce internal spatial profiles of individual components, and to select a unique polarization, initiate motility, and ensure persistent movement even when a chemotactic stimulus is removed.
In this context, each module plays an important role. PIs transduce the initial signal, causing their Rho GTPase targets to establish asymmetric spatial profiles. The Rho protein crosstalk leads to spatial polarization, forming a stable internal gradient in response to graded inputs. The distribution of Rho proteins amplifies weak stimuli into a macroscopic internal map. This polarity persists even when the stimulus is removed, but it can be changed in response to new stimuli that lead to a reorganization of the polarity. In the context of the small GTPase map, PIs act to filter out noise, select a direction, and prevent multiple Rho GTPase domains from forming, hence acting as an internal compass (93,110,111). On the one hand, the PIs damp out noise and smear-out competing peaks of Rho proteins to prevent multiple protrusions from forming. On the other hand, the polarization of the Rho proteins prevents the polarized PI gradient from smearing-out altogether by membrane diffusion. Finally, the graded profiles of PI and Rho GTPases enhance actin polymerization by locally activating Arp2/3 and inhibiting capping. There, extension of the cytoskeleton forms a leading edge, and protrusive motility is initiated. Because Rho proteins (and possibly newly polymerized actin, though still controversial) feed-back onto the PIs, persistent polarity and motion can be maintained. Together, this addresses question Q12.
Numerous previous models of cell polarization have been formulated from an underlying Turing mechanism (15–17) or a local excitation/global inhibition (LEGI) mechanism (9,19,20). Hence it is useful to compare our model to these traditional formulations. Turing (112) reasoned that an activator and inhibitor that coexist stably in a well-mixed system could be destabilized by virtue of different spatial scales on which they act (i.e., distinct rates of diffusion). However, in our model, the basic pattern-forming mechanism resides in the Rho module, which acts as central polarity switch. There, instability emanates from the presence of multiple equilibria in the well-mixed system of three activated Rho components (Cdc42, Rac, Rho), even when they act on their own (68). These three proteins have equal sizes and hence equal rates of diffusion along the membrane. Separately, we have shown (68) that the system of three active Rho proteins alone cannot admit a Turing diffusion-driven instability. Including their inactive forms (for a total of six partial differential equations) leads to a higher dimensional system where a generalized Turing instability can occur; however, the segregation and spatial polarization of the GTPases is possible in a much larger parameter regime, even under conditions where Turing instability per se is absent.
Previous models have also mandated rapidly diffusing cytosolic inhibitors (for global inhibition in LEGI models (9,19,20)) that lead to fast communication between front and back of the cell. In our model, the rapidly diffusing cytosolic inactive Rho proteins facilitate this communication. However, they do not act as inhibitors, and they are not essential in the initial instability that leads to spatial polarization: we showed previously (68,28) that their role is to freeze the wave-front. If they are held at fixed homogeneous levels, the zone of transition between high Cdc42 and high Rho would sweep through the cell as a traveling wave, and eventually either Cdc42 or Rho would take over dominance of the entire domain.
Focusing on specific interactions, we find that PIP3 activation of Cdc42 (but not Rac) is required to establish asymmetric Rho protein gradients oriented in the proper (stimulus gradient) direction and that Rac activation of PI5K or PI3K is required to maintain an asymmetric distribution of PIP3. Within the Rho protein module, mutual inhibition of Cdc42 and Rho is needed to maintain the persistent internal graded pattern of the Rho proteins and, also, by feedback to the PIs, persistent spatial gradients of PIs and actin density. Finally, PIP2 localizes actin dynamics to the leading edge by locally enhancing Arp2/3 activation and inhibiting capping. Thus, simulating the full model with all three interacting modules has provided further motivation for the inhibitory crosstalk between Cdc42 and Rho that we conjectured to be important in our previous work (28,67,68).
Our model is robust to parameter values, and to changing the details of kinetic functions (e.g., from linear to saturating Michaelian kinetics). We also find that symmetry breaking occurs for a large range of stimuli strengths, durations, and functional forms (gradients or random stimuli both with and without superimposed constant backgrounds). Once the cell is moving, it is still sensitive to new stimuli, though in a one-dimensional model these have to be strong enough for full reversal.
The models presented here provide explanations for a variety of experimental observations, such as defects in gradient detection in mutants lacking Cdc42, and proper directed motion in mutants lacking PTEN. The models can also be used to suggest future experiments: For example, we hypothesized that mutual inhibition of Cdc42 and Rho could account for spatial exclusion of Cdc42 and PTEN. Thus, we predict that experimentally microinjecting active Cdc42 into a resting cell should lead to spatial exclusion of both Rho and PTEN. We showed that the strength and duration of a stimulus is correlated with the time it takes for the cell to polarize and initiate motion. This can be tested by varying the magnitudes and/or time durations of chemoattractant gradient stimuli. We proposed a mechanism that would prevent Rac-mediated activation of PI5K and PI3K in the absence of de novo actin polymerization. To test the role of barbed end polymerization in PI5K and PI3K activation, we suggest introducing capping protein into motile cells, either by permeabilization or by microinjection. This experiment would not affect availability of monomers but would prevent polymerization by blocking barbed ends. This would test our prediction that PIP3 asymmetry would not be maintained in a treated cell exposed to a chemoattractant gradient.
The model has limitations that make it only one first step in the quest to understand cell motility signaling.
First, this model, like many aspects of the current picture, is a composite, based on many types of cells, experiments, and information from many disparate sources. This problem is encountered by all biologists (and theorists) hoping to draw global conclusions from experiences with multiple distinct realizations of a given physiological process (1): details are bound to vary greatly from one cell type to another. (For example, D. discoideum has no small G proteins analogous to Cdc42 or Rho (113,114)). Nevertheless, as suggested by Weiner et al. (8), the PI and Rho protein system, with its emergent properties, could be an evolutionarily conserved interlinked unit that explains similar prototypical behavior shared by many disparate cell types.
Second, we simplified aspects of the pathways (leaving out many important intermediates) and assumed fairly elementary interactions (linear or Michaelian) in all but the essentially nonlinear Rho module. In the Rho module, our model (described and motivated in greater detail in (68)) is one reasonable (but as yet unproven) instantiation of bistable kinetics that sets up spatial segregation. This dynamic attribute is the key requirement for this central module, but the details of the way we chose to model it may deviate from reality; to date, no better information is available to refine the details of that module, though our predictions stem not from those details, but from the dynamics and bifurcations that it exhibits as a unit.
Third, as our framework is that of continuous differential equations, we cannot deal with stochastic effects of molecules with low copy-numbers (24). Such effects could be important for substances that are not very abundant. Finally, our description of a moving cell (using one-dimensional geometry and thermal ratchet protrusive velocity) is quite elementary, omitting the important effects of adhesion, contraction, and other processes known to be vital in a full description of motility.
Despite such limitations, our work fits into the collective efforts to clarify the molecular mechanisms responsible for symmetry breaking in cell polarity and motility, and to decipher the dynamic coupling between signaling components, a goal proposed in Wedlich-Soldner and Li (14). By basing our model on known molecular entities, and (as far as possible) known interactions and rates, we also come close to making quantitatively reasonable predictions. As new details emerge about these (or other) signaling modules, such models can be refined, extended, or combined to investigate further aspects of cell behavior. In this sense, a model of this type should be considered as an additional tool, helping to probe dynamics of cellular events that are below the resolution of light microscope (1), or helping to place the many observations into a common framework.
Acknowledgments
We are grateful to Alexandra Jilkine and Athanasius F. M. Marée for helpful discussions.
We have been supported by a subcontract (to L.E.K.) from the National Science Foundation grant (No. DMS-0240770) to Anders Carlsson, Washington University, St. Louis, MO, and by the Mathematics of Information Technology and Complex Systems (Canada). L.E.K. is supported by a Natural Sciences and Engineering Research Council discovery grant.
APPENDIX A: MODEL OF PI, RHO PROTEIN, AND ACTIN DYNAMICS
Actin dynamics
Model variables are defined in Table 3. Following Dawes et al. (98) and Marée et al. (28), barbed ends (B(x, t)), Arp2/3 (A(x, t)) and actin filament density (F(x, t)), dynamics are
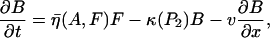 |
(6a) |
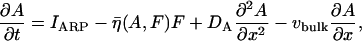 |
(6b) |
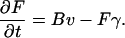 |
(6c) |
TABLE 3.
Definitions of variables that appear in the model
| Variable | Meaning | Units |
|---|---|---|
| F(x, t) | Actin filament length density. | μm/μm |
| B(x, t) | Density of actively growing barbed ends. | #/μm |
| A(x, t) | Arp2/3 concentration. | μM |
| C, R, ρ(x, t) | Concentration of active Cdc42, Rac, and Rho. | μM |
| Ci, Ri, ρi(x, t) | Concentration of inactive Cdc42, Rac, and Rho. | μM |
| P1, P2, P3(x, t) | Concentration of PIP1, PIP2, and PIP3. | μM |
| xedge | Position of leading edge. | μm |
The value v is free polymerization speed of barbed ends, DA is Arp2/3 diffusion coefficient, and IARP is Arp2/3 activation rate, given by Eq. 2, with the switch function 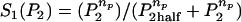 . The value P2half acts as the threshold value of PIP2 at which the response is triggered for large np. The capping rate, κ(P2), is given by the PIP2 dependent form in Eq. 3. We also include a bulk transport rate, vbulk = x′edge, which describes forward flow of the cytoplasm caused by membrane protrusion.
. The value P2half acts as the threshold value of PIP2 at which the response is triggered for large np. The capping rate, κ(P2), is given by the PIP2 dependent form in Eq. 3. We also include a bulk transport rate, vbulk = x′edge, which describes forward flow of the cytoplasm caused by membrane protrusion.
Nucleation of barbed ends by Arp2/3 (by side-branching of parent filaments), is proportional to filament density. A saturating function prevents unlimited formation of new barbed ends when there is a large amount of Arp2/3 or F-actin:
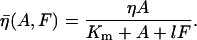 |
As in Dawes et al. (98), we assume that the membrane protrusion speed is determined by the number of barbed ends pushing on the membrane at the leading edge. Then the thermal ratchet (42) leads to the following approximate expression for forward speed,
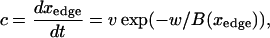 |
(7) |
where w = φBδ/kBT (φB is the load force per filament tip, δ a length increment due to one monomer, kB Boltzmann's constant, and T temperature). The expressions in Eqs. 6 are solved in a coordinate system moving with the cell edge (i.e., transformed so that xedge = 0).
Model of Rho protein dynamics
The model of Rho protein crosstalk is based on Marée et al. (28) and Jilkine et al. (68), modified to include PIs. Eq. 4a and Eq. 5 together with transport terms lead to
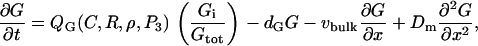 |
(8) |
for the active Rho proteins, where Dm is their diffusion rate (on the membrane). Experimental evidence suggests that membrane-associated molecules also experience an advective flow, vbulk at the speed of membrane protrusion (115).
The inactive forms obey Eq. 4b but their effective diffusion coefficient, Dmc > Dm, is faster, as these forms are partly cytosolic. In Jilkine et al. (68), we showed that
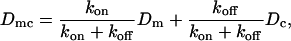 |
where kon, koff are rates of binding-unbinding of the inactive forms from the cell membrane, assumed to take place on a fast timescale. Hence, inactive Rho proteins are described by
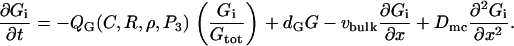 |
(9) |
Nonlinearities capturing crosstalk in Fig. 1 a and leading to appropriate behavior were chosen as follows (for reasons justified in the literature (28,67,68), and here using PIs, based on B4; see above),
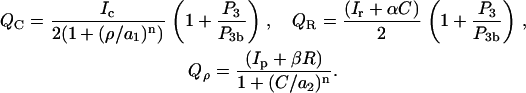 |
(10) |
The values Ic, Ir, and Ip are the baseline activation rates. The values a1 and a2 are the Rho and Cdc42 concentrations that elicit a half-maximal drop of Cdc42 and Rho activation, respectively. The value α determines the rate of Cdc42-enhanced activation of Rac and β determines the rate of Rac-enhanced Rho activation. The value P3b is the baseline concentration of PIP3 found in a resting cell. The values Ctot, Rtot, and Ptot are the total concentrations of Cdc42, Rac, and Rho. The details of the functions are less important than their nonlinear sigmoidal shape, with n ≥ 2. Michaelian (saturating) terms for P3 were also tested in Eqs. 10 and found to make little qualitative difference.
Model of PI dynamics
Quasi steady-state approximation of PI5K, PI3K, and PTEN
To keep the model as simple as possible, and to take into account the activity of a general Rho protein, G, on the activation of kinase or phosphatase φ, we assume that
 |
This expression is used in the model to simulate the effect of the Rho proteins (G = C, R, ρ) on the activity of PI5K, PI3K and PTEN. For example, based on arrow 5 of Fig. 1 a, the QSS level of PTEN is assumed to be
 |
(11) |
whereas, based on arrows 3 and 4, the activities of PI5K and PI3K are
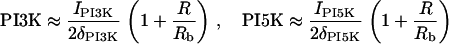 |
(12) |
To ensure that linear terms in Eqs. 11 and 12 were not introducing artifactual behavior, we also ran a variant of the model with Michaelis-Menten kinetics, i.e., terms of the form
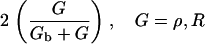 |
replacing linear terms in these equations. As discussed in the text, this had little or no qualitative effect. Since the kinase or phosphatase, φ, converts one form of PI to another, the equation for each PI will contain a term of the form
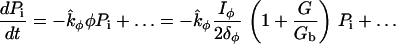 |
(or its Michaelian equivalent). To avoid carrying these expressions, we will use shorthand notation by defining
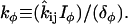 |
A gradient stimulus is represented in our model by a graded input of PI3K and/or PTEN, or, equivalently, a graded value of the parameters kPI3K, kPTEN.
PI equations
We do not keep track of the abundant unphosphorylated PI and instead assume that the conversion to PIP1 occurs at a constant rate, IP1. PIP1 decays back to the unphosphorylated pool at a rate δP1. Otherwise, the PIs convert between different phosphorylation states, where we assume only one phosphate can be added or removed at a time. We assume all PIs diffuse in the membrane at the same rate, DP. The PI equations are
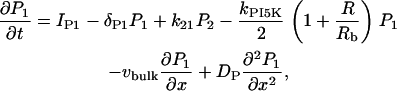 |
(13a) |
 |
(13b) |
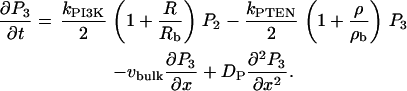 |
(13c) |
In the above, we have used the basic strategy of Eq. 1b in several places. We have assumed that Rac enhances the conversion of P1 to P2 (via PI5K) and the conversion of P2 to P3 via PI3K. Similarly, Rho enhances the conversion of P3 to P2 via PTEN. As discussed in the text, replacing terms in braces by Michaelian counterparts does not change the qualitative behavior of the model. Parameter values used to simulate the PI equations are given in Table 2.
Boundary conditions
We assume that there are no actively polymerizing actin barbed ends far from the leading edge, B(– ∞) = 0 (or for simulations, B(xedge – L) = 0). We impose no-flux boundary conditions on Arp2/3, all Rho proteins, and all phosphoinositides, i.e., Ax = Gx = Pix = 0, etc., at x = xedge, xedge – L, where xedge is the membrane position and L is the length of the lamellipod.
APPENDIX B: PARAMETER VALUES
Estimating values
Parameters used to simulate Eqs. 6, 8, 9, and 13 are discussed in detail in the literature (28,68,102). Many can be estimated directly from reported biological experiments. Others can be approximated or inferred by combining known basal (steady-state) concentrations with rough estimates for timescales of down or upregulation in response to a signal. We briefly describe our procedure for estimating the model parameters in the following sections, and we concentrate all default values in Table 2.
The rate of free actin polymerization was determined by electron microscope observations of polymerizing filaments (116–118). The rate of barbed end capping was obtained from in vitro studies of capping protein (118,119). The rate of Arp2/3-mediated nucleation of new barbed ends was estimated from average spacing between branch points and barbed end free polymerization rate (98,102). The rate of Arp2/3 activation was estimated by fitting the maximum barbed end edge density in the model to that observed in experiments (28,98). The rate of filament decay was based on fluorescence speckle microscopy (118,120).
The total concentration of Rho proteins was determined by immunoblotting (101). Rho protein activation rates were estimated using decay rates and steady-state concentrations (28,68), while the decay rates were estimated from in vitro observations of phosphorylation activity (121,122). Diffusion rates of Arp2/3, Rho proteins, and PIs were determined using fluorescence recovery after photobleaching (FRAP) (99,100,123).
There is little quantitative data about PIs in motile cells since most experiments are concerned with the spatial localization of the PIs. However, time-course data that tracks the increase in fluorescence indicate that PIs reach their peak concentration ∼10 s after a stimulus is applied (71,77). Given the approximate baseline concentration of PIs in unstimulated cells (Table 2) (9,71), and using the expressions in Eq. 13, we can determine the relationships between the biochemical parameters and the steady-state concentrations of the PIs in a resting cell (when all the Rho proteins are at their baseline concentrations, G = Gb). At baseline, the concentration of the three forms are
 |
(14) |
We simulated the PIs in the absence of the Rho proteins to estimate biochemical rates. In the simulations, we vary δP1, k21, and kPTEN and determine the remaining parameters using the steady-state PI concentrations from above: IP1 = P1bδP1, kPI5K = P2bk21/P1b, kPI3K = P3bkPTEN/P2b. We found that the parameter values shown in Table 2 give the appropriate time course for the PIs. It has been determined experimentally that PIP3 diffuses at the rate DP = 0.5 − 5 μm2/s, depending on the source (99,100). The diffusion rate of active (membrane-bound) Rho proteins is ∼Dm = 0.1 μm2/s.
Robustness to parameter variations
To explore parameter sensitivity, we varied many parameters by 10% or more. PI parameter values could be changed up or down by a factor of 10 without significantly changing the simulation results. Other than the Hill coefficient, n, Rho protein parameters can also be varied without much qualitative change. Increasing the following actin parameters leads to a delay in initiation of motility and slower speed: P2half (PIP2 threshold for Arp2/3 activation), Km (threshold for nucleation), and kmax (maximum capping rate). Increasing μP (rate of Arp activation) has no apparent effect for strong stimuli but decreases the time to initiation of motion for weak stimuli. Steady-state profiles are quantitatively (but not qualitatively) modified. Increasing η (magnitude of side-branching rate) decreases time to initiate motility and increases the cell speed, with no qualitative difference in spatial profiles.
APPENDIX C: SIMULATIONS AND SIMULATION PROTOCOLS
Equations 6, 8, 9, and 13 (the full-default model) were simulated on a one-dimensional domain, representing a thin strip of lamellipod, oriented in the direction of motion, as shown in Fig. 2. All equations were transformed to a coordinate system moving with the protruding cell edge, (τ = t, z = x – x′edget). (The edge is then at x = 0 for convenience of visualization.) Second derivatives are discretized using centered differencing while first derivatives are discretized using forward Euler. This (explicit) system of discretized equations was coded using the C programming language, and simulated on a 10-μm grid with a step size of 0.01 μm. The time step was chosen according to the fastest diffusion rate to ensure numerical stability. At the beginning of a simulation, all variables were set to their steady-state levels.
Graded stimuli
An internal gradient of PI3K and PTEN is observed as the most upstream graded cellular response. To simulate the translocation of PI3K and PTEN observed in motile cells exposed to a gradient of chemoattractant, we imposed a gradient in kPTEN and kPI3K across the cell. A typical linear gradient would be a graded decrease (from some value at one boundary) by some percentage (at the other boundary). For example, we refer to a stimulus that decreases from baseline to zero as a 100% gradient. To represent a weak stimulus, we applied a 1–10% graded stimulus across the cell. To mimic the accumulation of PI3K in the front (respectively, PTEN in the back) we generally decreased kPTEN, at the front of the cell and decreased kPI3K, at the back. This gradient was applied for 10 s, unless otherwise noted. We tested stimuli with and without an (additive) constant background level.
Competing (random) stimuli
A random stimulus is applied in either the Cdc42 activation rate, Ic, or the PTEN/PI3K activity levels (as explained above). At each grid space, the given parameter value (Ic or kPI3K, and kPTEN) is randomly assigned a value from zero to twice its baseline value (given in Table 2). This random stimulus is held constant during the stimulus application to mimic the effect of (static) competing random influences. To simulate a near-uniform stimulus with low noise level, we followed a similar protocol, but with small amplitude (e.g., ∼1%) randomly assigned parameter values, which were either static or temporally fluctuating. As above, we also considered random stimuli superimposed on a constant background.
Dissecting the pathways
To model the effects of abolishing one or more arrows in the signaling pathway of Fig. 1 a, we selectively set to zero the corresponding expression in the appropriate model equation. For example, to delete the effect of PIP3 on the activation of Cdc42 or Rac, we set P3 = 0 in the activation term QC or QR for Cdc42 or Rac, respectively, in Eq. 10. To remove the effect of Rac on PI5K and PI3K, R was set to zero in Eq. 12. The activity of PTEN is represented by the level of Rho based on its form in Eq. 11. To relieve Cdc42-mediated downregulation of Rho, we set C = 0 in Qρ in Eq. 10
Feedback from actin polymerization
In Polymerization-Dependent Rac Activation of PI5K and PI3K Causes Loss of PI Asymmetry, but Not Rho Protein Asymmetry, we modified the PI equations so that the activity of PI5K and PI3K is enhanced by Rac only when active polymerization is taking place. To do so, we modified the QSS terms for PTEN and PI3K in Eqs. 11–12 so that these depended jointly on Rac and on newly polymerizing actin. The polymerizing actin was represented by the product Bv of barbed end density and polymerization velocity. Accordingly,
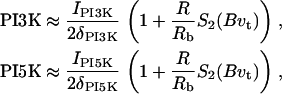 |
(15) |
where B is the current number of barbed ends and vt is the rate of polymerization in a treated cell. The conversion rates kPI5K and kPI3K are similarly modified. We choose S2(Bvt) to have switchlike properties so that in the absence of barbed ends or free actin monomers (which prevents polymerization), Rac cannot enhance the activity of PI5K or PI3K: 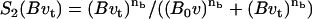 . Using B0 = 0.1 μm−1, we ensured that even very little polymerization activity allows Rac to enhance PI5K and PI3K activity. Note that S2(0) = 0, so Rac has no influence in Eq. 15 if there are no actively polymerizing filament tips or no available monomers. The choice of a switching function here is arbitrary; we achieve similar model results when we use a Michaelis-Menten or linear term.
. Using B0 = 0.1 μm−1, we ensured that even very little polymerization activity allows Rac to enhance PI5K and PI3K activity. Note that S2(0) = 0, so Rac has no influence in Eq. 15 if there are no actively polymerizing filament tips or no available monomers. The choice of a switching function here is arbitrary; we achieve similar model results when we use a Michaelis-Menten or linear term.
References
- 1.Ridley, A. J., M. A. Schwartz, K. Burridge, R. A. Firtel, M. H. Ginsberg, G. Borisy, J. T. Parsons, and A. R. Horwitz. 2003. Cell migration: integrating signals from front to back. Science. 302:1704–1709. [DOI] [PubMed] [Google Scholar]
- 2.Servant, G., O. D. Weiner, P. Herzmark, T. Balla, J. W. Sedat, and H. R. Bourne. 2000. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 287:1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiner, O. D., G. Servant, M. D. Welch, T. J. Mitchison, J. W. Sedat, and H. R. Bourne. 1999. Spatial control of actin polymerization during neutrophil chemotaxis. Nat. Cell Biol. 1:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang, F., P. Herzmark, O. D. Weiner, S. Srinivasan, G. Servant, and H. R. Bourne. 2002. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat. Cell Biol. 4:513–518. [DOI] [PubMed] [Google Scholar]
- 5.Fisher, P. R., R. Merkl, and G. Gerisch. 1989. Quantitative analysis of cell motility and chemotaxis in Dictyostelium discoideum by using an image-processing system and a novel chemotaxis chamber providing stationary chemical gradients. J. Cell Biol. 108:973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iijima, M., Y. E. Huang, and P. Devreotes. 2002. Temporal and spatial regulation of chemotaxis. Dev. Cell. 3:469–478. [DOI] [PubMed] [Google Scholar]
- 7.Merlot, S., and R. A. Firtel. 2003. Leading the way: directional sensing through phosphatidylinositol 3-kinase and other signaling pathways. J. Cell Sci. 116:3471–3478. [DOI] [PubMed] [Google Scholar]
- 8.Weiner, O. D., P. O. Neilsen, G. D. Prestwich, M. W. Kirschner, L. C. Cantley, and H. R. Bourne. 2002. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat. Cell Biol. 4:509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levchenko, A., and P. A. Iglesias. 2002. Models of eukaryotic gradient sensing: application to chemotaxis of amoebae and neutrophils. Biophys. J. 82:50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Haastert, P. J., and P. N. Devreotes. 2004. Chemotaxis: signaling the way forward. Nat. Rev. Mol. Cell Biol. 6:626–634. [DOI] [PubMed] [Google Scholar]
- 11.Rickert, P., O. Weiner, F. Wang, H. Bourne, and G. Servant. 2000. Leukocytes navigate by compass: roles of PI3Kγ and its lipid products. Trends Cell Biol. 10:466–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parent, C. A., and P. N. Devreotes. 1999. A cell's sense of direction. Science. 284:765–770. [DOI] [PubMed] [Google Scholar]
- 13.Xu, J. S., F. Wang, A. Van Keymeulen, P. Herzmark, A. Straight, K. Kelly, Y. Takuwa, N. Sugimoto, T. Mitchison, and H. R. Bourne. 2003. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 114:201–214. [DOI] [PubMed] [Google Scholar]
- 14.Wedlich-Soldner, R., and R. Li. 2003. Spontaneous cell polarization: undermining determinism. Nat. Cell Biol. 5:267–270. [DOI] [PubMed] [Google Scholar]
- 15.Meinhardt, H. 1999. Orientation of chemotactic cells and growth cones: models and mechanisms. J. Cell Sci. 112:2867–2874. [DOI] [PubMed] [Google Scholar]
- 16.Narang, A., K. K. Subramanian, and D. A. Lauffenburger. 2001. A mathematical model for chemoattractant gradient sensing based on receptor-regulated membrane phospholipid signaling dynamics. Ann. Biomed. Eng. 29:677–691. [DOI] [PubMed] [Google Scholar]
- 17.Subramanian, K. K., and A. Narang. 2004. A mechanistic model for eukaryotic gradient sensing: spontaneous and induced phosphoinositide polarization. J. Theor. Biol. 231:49–67. [DOI] [PubMed] [Google Scholar]
- 18.Narang, A. 2006. Spontaneous polarization in eukaryotic gradient sensing: a mathematical model based on mutual inhibition of frontness and backness pathways. J. Theor. Biol. 240:538–553. [DOI] [PubMed] [Google Scholar]
- 19.Ma, L., C. Janetopoulos, L. Yang, P. N. Devreotes, and P. A. Iglesias. 2004. Two complementary, local excitation, global inhibition mechanisms acting in parallel can explain the chemoattractant-induced regulation of PI(3,4,5)P-3 response in Dictyostelium cells. Biophys. J. 87:3764–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishnan, J., and P. A. Iglesias. 2004. A modeling framework describing the enzyme regulation of membrane lipids underlying gradient perception in Dictyostelium cells. J. Theor. Biol. 229:85–99. [DOI] [PubMed] [Google Scholar]
- 21.Haugh, J. M., and I. C. Schneider. 2004. Spatial analysis of 3′ phosphoinositide signaling in living fibroblasts: I. Uniform stimulation model and bounds on dimensionless groups. Biophys. J. 86:589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maly, I. V., H. S. Wiley, and D. A. Lauffenburger. 2004. Self-organization of polarized cell signaling via autocrine circuits: computational model analysis. Biophys. J. 86:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakumura, Y., Y. Tsukada, N. Yamamoto, and S. Ishii. 2005. A molecular model for axon guidance based on cross talk between Rho GTPases. Biophys. J. 89:812–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gamba, A., A. de Candia, S. Di Talia, A. Coniglio, F. Bussolino, and G. Serini. 2005. Diffusion-limited phase separation in eukaryotic chemotaxis. Proc. Natl. Acad. Sci. USA. 102:16927–16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skupsky, R., W. Losert, and R. J. Nossal. 2005. Distinguishing modes of eukaryotic gradient sensing. Biophys. J. 89:2806–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krishnan, J., and P. A. Iglesias. 2004. Uncovering directional sensing: where are we headed? Syst. Biol. 1:54–61. [DOI] [PubMed] [Google Scholar]
- 27.Devreotes, P., and C. Janetopoulos. 2003. Eukaryotic chemotaxis: distinctions between directional sensing and polarization. J. Biol. Chem. 278:20445–20448. [DOI] [PubMed] [Google Scholar]
- 28.Marée, A. F. M., A. Jilkine, A. T. Dawes, V. A. Grieneisen, and L. Edelstein-Keshet. 2006. Polarization and movement of keratocytes: a multiscale modeling approach. Bull. Math. Biol. 68:1169–1211. [DOI] [PubMed] [Google Scholar]
- 29.Svitkina, T. M., A. B. Verkhovsky, K. M. McQuade, and G. G. Borisy. 1997. Analysis of the actin-myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J. Cell Biol. 139:397–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Small, J. V., M. Herzog, and K. Anderson. 1995. Actin filament organization in the fish keratocyte lamellipodium. J. Cell Biol. 129:1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verkhovsky, A. B., O. Y. Chaga, S. Schaub, T. M. Svitkina, J. J. Meister, and G. G. Borisy. 2003. Orientational order of the lamellipodial actin network as demonstrated in living motile cells. Mol. Biol. Cell. 14:4667–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Redmond, T., and S. H. Zigmond. 1993. Distribution of F-actin elongation sites in lysed polymorphonuclear leukocytes parallels the distribution of endogenous F-actin. Cell Mot. Cytoskel. 26:7–18. [DOI] [PubMed] [Google Scholar]
- 33.Ponti, A., M. Machacek, S. L. Gupton, C. M. Waterman-Storer, and G. Danuser. 2004. Two distinct actin networks drive the protrusion of migrating cells. Science. 305:1782–1786. [DOI] [PubMed] [Google Scholar]
- 34.Bailly, M., F. Macaluso, M. Cammer, A. Chan, J. Segall, and J. Condeelis. 1999. Relationship between Arp2/3 complex and the barbed ends of actin filaments at the leading edge of carcinoma cells after epidermal growth factor stimulation. J. Cell Biol. 145:331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wood, W., and P. Martin. 2002. Structures in focus—filopodia. Int. J. Biochem. Cell Biol. 34:726–730. [DOI] [PubMed] [Google Scholar]
- 36.Small, J. V., T. Stradal, E. Vignal, and K. Rottner. 2002. The lamellipodium: where motility begins. Trends Cell Biol. 12:112–120. [DOI] [PubMed] [Google Scholar]
- 37.Bailly, M., I. Ichetovkin, W. Grant, N. Zebda, L. M. Machesky, J. E. Segall, and J. Condeelis. 2001. The F-actin side binding activity of the Arp2/3 complex is essential for actin nucleation and lamellipod extension. Curr. Biol. 11:620–625. [DOI] [PubMed] [Google Scholar]
- 38.Falet, H., K. M. Hoffmeister, R. Neujahr, J. E. Italiano, T. P. Stossel, F. S. Southwick, and J. H. Hartwig. 2002. Importance of free actin filament barbed ends for Arp2/3 complex function in platelets and fibroblasts. Proc. Natl. Acad. Sci. USA. 99:16782–16787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Svitkina, T. M., and G. G. Borisy. 1999. Arp2/3 complex and actin depolymerizing factor cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145:1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amann, K. J., and T. D. Pollard. 2001. Direct real-time observation of actin filament branching mediated by Arp2/3 complex using total internal reflection fluorescence microscopy. Proc. Natl. Acad. Sci. USA. 98:15009–15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujiwara, I., S. Suetsugu, S. Uemura, T. Takenawa, and S. Ishiwata. 2002. Visualization and force measurement of branching by Arp2/3 complex and N-WASP in actin filament. Biochem. Biophys. Res. Commun. 293:1550–1555. [DOI] [PubMed] [Google Scholar]
- 42.Mogilner, A., and G. Oster. 1996. Cell motility driven by actin polymerization. Biophys. J. 71:3030–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mogilner, A., and G. Oster. 2003. Force generation by actin polymerization. II: The elastic ratchet and tethered filaments. Biophys. J. 84:1591–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carlsson, A. E. 2001. Growth of branched actin networks against obstacles. Biophys. J. 81:1907–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mogilner, A., and L. Edelstein-Keshet. 2002. Regulation of actin dynamics in rapidly moving cells: a quantitative analysis. Biophys. J. 83:1237–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carlsson, A., M. Wear, and J. Cooper. 2004. End versus side branching by Arp2/3 complex. Biophys. J. 86:1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rubinstein, B., K. Jacobson, and A. Mogilner. 2005. Multiscale two-dimensional modeling of a motile simple-shaped cell. Multiscale Model. Sim. 3:413–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chung, C. Y., S. Lee, C. Briscoe, C. Ellsworth, and R. A. Firtel. 2000. Role of Rac in controlling the actin cytoskeleton and chemotaxis in motile cells. Proc. Natl. Acad. Sci. USA. 97:5225–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wedlich-Soldner, R., S. J. Altschuter, L. F. Wu, and R. Li. 2003. Spontaneous cell polarization through actomyosin-based delivery of the Cdc42 GTPase. Science. 299:1231–1235. [DOI] [PubMed] [Google Scholar]
- 50.Ridley, A. J. 2001. Rho GTPases and cell migration. J. Cell Sci. 114:2713–2722. [DOI] [PubMed] [Google Scholar]
- 51.Mackay, D. J. G., and A. Hall. 1998. Rho GTPases. J. Biol. Chem. 273:20685–20688. [DOI] [PubMed] [Google Scholar]
- 52.Ridley, A. J. 2001. Rho family proteins: coordinating cell responses. Trends Cell Biol. 11:471–477. [DOI] [PubMed] [Google Scholar]
- 53.Srinivasan, S., F. Wang, S. Glavas, A. Ott, F. Hofmann, K. Aktories, D. Kalman, and H. R. Bourne. 2003. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P-3 and polarity during neutrophil chemotaxis. J. Cell Biol. 160:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kraynov, V. S., C. Chamberlain, G. M. Bokoch, M. A. Schwartz, S. Slabaugh, and K. M. Hahn. 2000. Localized Rac activation dynamics visualized in living cells. Science. 290:333–337. [DOI] [PubMed] [Google Scholar]
- 55.Nalbant, P., L. Hodgson, V. Kraynov, A. Toutchkine, and K. M. Hahn. 2004. Activation of endogenous Cdc42 visualized in living cells. Science. 305:1615–1619. [DOI] [PubMed] [Google Scholar]
- 56.Wong, K., O. Pertz, K. Hahn, and H. Bourne. 2006. Neutrophil polarization: spatiotemporal dynamics of RhoA activity support a self-organizing mechanism. Proc. Natl. Acad. Sci. USA. 103:3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmidt, A., and A. Hall. 2002. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 16:1587–1609. [DOI] [PubMed] [Google Scholar]
- 58.Zheng, Y. 2001. DBl family guanine nucleotide exchange factors. Trends Biochem. Sci. 26:724–732. [DOI] [PubMed] [Google Scholar]
- 59.Moon, S. Y., and Y. Zheng. 2003. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 13:13–22. [DOI] [PubMed] [Google Scholar]
- 60.Miura, Y., A. Kikuchi, T. Musha, S. Kuroda, H. Yaku, T. Sasaki, and Y. Takai. 1993. Regulation of morphology by Rho P21 and its inhibitory GDP/GTP exchange protein (Rho GDI) in Swiss 3T3 cells. J. Biol. Chem. 268:510–515. [PubMed] [Google Scholar]
- 61.Nobes, C. D., and A. Hall. 1995. Rho, Rac and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 81:53–62. [DOI] [PubMed] [Google Scholar]
- 62.Burridge, K. 1999. Signal transduction—crosstalk between Rac and Rho. Science. 283:2028–2029. [DOI] [PubMed] [Google Scholar]
- 63.Li, Z., C. D. Aizenman, and H. T. Cline. 2002. Regulation of Rho GTPases by crosstalk and neuronal activity in vivo. Neuron. 33:741–750. [DOI] [PubMed] [Google Scholar]
- 64.Yamaguchi, Y., H. Katoh, and H. Yasui. 2001. Rho-A inhibits the nerve growth factor-induced Rac1 activation through Rho-associated kinase-dependent pathway. J. Biol. Chem. 276:18977–18983. [DOI] [PubMed] [Google Scholar]
- 65.Ohta, Y., J. H. Hartwig, and T. P. Stossel. 2006. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodeling. Nat. Cell Biol. 8:803–814. [DOI] [PubMed] [Google Scholar]
- 66.Giniger, E. 2002. How do Rho family GTPases direct axon growth and guidance? A proposal relating signaling pathways to growth cone mechanics. Differentiation. 70:385–396. [DOI] [PubMed] [Google Scholar]
- 67.Jilkine, A. 2005. Mathematical study of Rho GTPases in motile cells. Master's thesis, University of British Columbia.
- 68.Jilkine, A., A. F. M. Marée, and L. Edelstein-Keshet. 2006. Mathematical model for spatial segregation of the Rho-family GTPases based on inhibitory crosstalk. [DOI] [PubMed]
- 69.Cicchetti, G., P. G. Allen, and M. Glogauer. 2002. Chemotactic signaling pathways in neutrophils: From receptor to actin assembly. Crit. Rev. Oral Biol. Med. 13:220–228. [DOI] [PubMed] [Google Scholar]
- 70.Schmitz, A. A. P., E. E. Govek, B. Bottner, and L. van Aelst. 2000. Rho GTPases: signaling, migration, and invasion. Exp. Cell Res. 261:1–12. [DOI] [PubMed] [Google Scholar]
- 71.Insall, R. H., and O. D. Weiner. 2001. PIP3, PIP2, and cell movement—similar messages, different meanings? Dev. Cell. 1:743–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rameh, L. E., and L. C. Cantley. 1999. The role of phosphoinositide 3-kinase lipid products in cell function. J. Biol. Chem. 274:8347–8350. [DOI] [PubMed] [Google Scholar]
- 73.Toker, A. 2002. Phosphoinositides and signal transduction. Cell. Mol. Life Sci. 59:761–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yin, H. L., and P. A. Janmey. 2003. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Phys. 65:761–789. [DOI] [PubMed] [Google Scholar]
- 75.Funamoto, S., R. Meili, S. Lee, L. Parry, and R. A. Firtel. 2002. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell. 109:611–623. [DOI] [PubMed] [Google Scholar]
- 76.Iijima, M., and P. Devreotes. 2002. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 5:599–610. [DOI] [PubMed] [Google Scholar]
- 77.Huang, Y. E., M. Iijima, C. A. Parent, S. Funamoto, R. A. Firtel, and P. Devreotes. 2003. Receptor-mediated regulation of PI3Ks confines PI(3,4,5)P-3 to the leading edge of chemotaxing cells. Mol. Biol. Cell. 14:1913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Higgs, H. N., and T. D. Pollard. 2000. Activation by Cdc42 and PIP2 of Wiskott-Aldrich Syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J. Cell Biol. 150:1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rohatgi, R., P. Nollau, H. Y. H. Ho, M. W. Kirschner, and B. J. Mayer. 2001. NCK and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J. Biol. Chem. 276:26448–26452. [DOI] [PubMed] [Google Scholar]
- 80.Rohatgi, R., L. Ma, H. Miki, M. Lopez, T. Kirchhausen, T. Takenawa, and M. W. Kirschner. 1999. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 97:221–231. [DOI] [PubMed] [Google Scholar]
- 81.Hawkins, P. T., A. Eguinoa, R. G. Qiu, D. Stokoe, F. T. Cooke, R. Walters, S. Wennstrom, L. Claessonwelsh, T. Evans, M. Symons, and L. Stephens. 1995. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr. Biol. 5:393–403. [DOI] [PubMed] [Google Scholar]
- 82.Li, Z., M. Hannigan, Z. C. Mo, B. Liu, W. Lu, Y. Wu, A. V. Smrcka, G. Q. Wu, L. Li, M. Y. Liu, C. K. Huang, and D. Q. Wu. 2003. Directional sensing requires Gβγ-mediated PAK1 and PIX α-dependent activation of Cdc42. Cell. 114:215–227. [DOI] [PubMed] [Google Scholar]
- 83.Welch, H. C. E., W. J. Coadwell, C. D. Ellson, G. J. Ferguson, S. R. Andrews, H. Erdjument-Bromage, P. Tempst, P. T. Hawkins, and L. R. Stephens. 2002. P-Rex1, a PtdIns(3,4,5)P-3- and Gβγ-regulated guanine-nucleotide exchange factor for Rac. Cell. 108:809–821. [DOI] [PubMed] [Google Scholar]
- 84.Aoki, K., T. Nakamura, K. Fujikawa, and M. Matsuda. 2005. Local phosphatidylinositol 3,4,5-trisphosphate accumulation recruits Vav2 and Vav3 to activate Rac1/Cdc42 and initiate neurite outgrowth in nerve growth factor-stimulated PC12 cells. Mol. Biol. Cell. 16:2207–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.van Hennik, P. B., J. P. ten Klooster, J. R. Halstead, C. Voermans, E. C. Anthony, N. Divecha, and P. L. Hordijk. 2003. The C-terminal domain of Rac1 contains two motifs that control targeting and signaling specificity. J. Biol. Chem. 278:39166–39175. [DOI] [PubMed] [Google Scholar]
- 86.Tolias, K. F., J. H. Hartwig, H. Ishihara, Y. Shibasaki, L. C. Cantley, and C. L. Carpenter. 2000. Type I α-phosphatidylinositol-4-phosphate 5-kinase mediates Rac-dependent actin assembly. Curr. Biol. 10:153–156. [DOI] [PubMed] [Google Scholar]
- 87.Tolias, K. F., L. C. Cantley, and C. L. Carpenter. 1995. Rho-family GTPases bind to phosphoinositide kinases. J. Biol. Chem. 270:17656–17659. [DOI] [PubMed] [Google Scholar]
- 88.Bokoch, G. M., C. J. Vlahos, Y. Wang, U. G. Knaus, and A. E. Traynor-Kaplan. 1996. Rac GTPase interacts specifically with phosphatidylinositol 3-kinase. Biochem. J. 315:775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zheng, Y., S. Bagrodia, and R. A. Cerione. 1994. Activation of phosphoinositide 3-kinase activity by Cdc42HS binding to P85. J. Biol. Chem. 269:18727–18730. [PubMed] [Google Scholar]
- 90.Li, Z., X. M. Dong, Z. L. Wang, W. Z. Liu, N. Deng, Y. Ding, L. Y. Tang, T. Hla, R. Zeng, L. Li, and D. Q. Wu. 2005. Regulation of PTEN by Rho small GTPases. Nat. Cell Biol. 7:399–442. [DOI] [PubMed] [Google Scholar]
- 91.Niggli, V. 2003. Signaling to migration in neutrophils: importance of localized pathways. Cell Biol. 35:1619–1638. [DOI] [PubMed] [Google Scholar]
- 92.Meinhardt, H. 2003. Complex pattern formation by a self-destabilization of established patterns: chemotactic orientation and phyllotaxis as examples. Comput. Rend. Biol. 326:223–237. [DOI] [PubMed] [Google Scholar]
- 93.Sohrmann, M., and M. Peter. 2003. Polarizing without a c(l)ue. Trends Cell Biol. 13:526–533. [DOI] [PubMed] [Google Scholar]
- 94.Jones, G. E., W. E. Allen, and A. J. Ridley. 1998. The Rho GTPases in macrophage motility and chemotaxis. Cell Adhes. Commun. 6:237–245. [DOI] [PubMed] [Google Scholar]
- 95.Welch, H. C. E., W. J. Coadwell, L. R. Stephens, and P. T. Hawkins. 2003. Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett. 546:93–97. [DOI] [PubMed] [Google Scholar]
- 96.Gracheva, M. E., and H. G. Othmer. 2004. A continuum model of motility in amoeboid cells. Bull. Math. Biol. 66:167–193. [DOI] [PubMed] [Google Scholar]
- 97.Mogilner, A., and G. Oster. 2003. Polymer motors: pushing out the front and pulling up the back. Curr. Biol. 13:R721–R733. [DOI] [PubMed] [Google Scholar]
- 98.Dawes, A. T., G. B. Ermentrout, E. N. Cytrynbaum, and L. Edelstein-Keshet. 2006. Actin filament branching and protrusion velocity in a simple one-dimensional model of a motile cell. J. Theor. Biol. 242:265–279. [DOI] [PubMed] [Google Scholar]
- 99.Postma, M., L. Bosgraaf, H. M. Loovers, and P. J. M. Van Haastert. 2004. Chemotaxis: signaling modules join hands at front and tail. EMBO Rep. 5:35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schneider, I. C., and J. M. Haugh. 2004. Spatial analysis of 3′ phosphoinositide signaling in living fibroblasts: II. Parameter estimates for individual cells from experiments. Biophys. J. 86:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Michaelson, D., J. Silletti, G. Murphy, P. Eustachio, M. Rush, and M. R. Philips. 2001. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 152:111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dawes, A. T. 2006. Phosphoinositides and Rho proteins conspire to spatially regulate actin polymerization in motile cells. Ph.D. thesis, University of British Columbia.
- 103.Akasaki, T., H. Koga, and H. Sumimoto. 1999. Phosphoinositide 3-kinase-dependent and -independent activation of the small GTPase Rac2 in human neutrophils. J. Biol. Chem. 274:18055–18059. [DOI] [PubMed] [Google Scholar]
- 104.Lacalle, R. A., C. Gomez-Mouton, D. F. Barber, S. Jimenez-Baranda, E. Mira, C. Martinez, A. C. Carrera, and S. Manes. 2004. PTEN regulates motility but not directionality during leukocyte chemotaxis. J. Cell Sci. 117:6207–6215. [DOI] [PubMed] [Google Scholar]
- 105.Kimmel, A. R., and C. A. Parent. 2003. The signal to move: D. discoideum go orienteering. Science. 300:1525–1527. [DOI] [PubMed] [Google Scholar]
- 106.Gunst, S. J. 2004. Actions by actin: reciprocal regulation of cortactin activity by tyrosine kinases and F-actin. Biochem. J. 380:e7–e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Woodring, P. J., T. Hunter, and J. Y. J. Wang. 2001. Inhibition of c-Abl tyrosine kinase activity by filamentous actin. J. Biol. Chem. 276:27104–27110. [DOI] [PubMed] [Google Scholar]
- 108.Fan, L. Z., C. Di Ciano-Oliveira, S. A. Weed, A. W. B. Craig, P. A. Greer, O. D. Rotstein, and A. S. Kapus. 2004. Actin depolymerization-induced tyrosine phosphorylation of cortactin: the role of Fer kinase. Biochem. J. 380:581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Postma, M., and P. J. M. Van Haastert. 2001. A diffusion-translocation model for gradient sensing by chemotactic cells. Biophys. J. 81:1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meili, R., and R. A. Firtel. 2003. Two poles and a compass. Cell. 114:153–156. [DOI] [PubMed] [Google Scholar]
- 111.Weiner, O. D. 2002. Regulation of cell polarity during eukaryotic chemotaxis: the chemotactic compass. Curr. Opin. Cell Biol. 14:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Turing, A. M. 1952. The chemical basis of morphogenesis. Philos. Trans. R. Soc. (B). 237:37–72. [Google Scholar]
- 113.Rivero, F., H. Dislich, G. Glockner, and A. A. Noegel. 2001. The Dictyostelium discoideum family of Rho-related proteins. Nucleic Acids Res. 29:1068–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rivero, F., and B. P. Somesh. 2002. Signal transduction pathways regulated by Rho GTPases in Dictyostelium. J. Muscle Res. Cell Mot. 23:737–749. [DOI] [PubMed] [Google Scholar]
- 115.Kucik, D. F., E. L. Elson, and M. P. Sheetz. 1990. Cell migration does not produce membrane flow. J. Cell Biol. 111:1617–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bonder, E., D. Fishkind, and M. Mooseker. 1983. Direct measurement of critical concentrations and assembly rate constants at the two ends of an actin filament. Cell. 34:491–501. [DOI] [PubMed] [Google Scholar]
- 117.Pollard, T. D. 1986. Rate constants for the reactions of ATP-actin and ADP-actin with the ends of actin-filaments. J. Cell Biol. 103:2747–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pollard, T. D., L. Blanchoin, and R. D. Mullins. 2000. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29:545–576. [DOI] [PubMed] [Google Scholar]
- 119.Schafer, D. A., P. B. Jennings, and J. A. Cooper. 1996. Dynamics of capping protein and actin assembly in vitro: uncapping barbed ends by polyphosphoinositides. J. Cell Biol. 135:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Watanabe, N., and T. J. Mitchison. 2002. Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science. 295:1083–1086. [DOI] [PubMed] [Google Scholar]
- 121.Sako, Y., K. Hibino, T. Miyauchi, Y. Miyamoto, M. Ueda, and T. Yanagida. 2000. Single-molecule imaging of signaling molecules in living cells. Single Mol. 2:159–163. [Google Scholar]
- 122.Zhang, B. L., and Y. Zheng. 1998. Regulation of RhoA GTP hydrolysis by the GTPase-activating proteins p190, p50RhoGAP, Bcr, and 3BP-1. Biochemistry. 37:5249–5257. [DOI] [PubMed] [Google Scholar]
- 123.Niv, H., O. Gutman, Y. I. Henis, and Y. Kloog. 1999. Membrane interactions of a constitutively active GFP-Ki-Ras 4B and their role in signaling—evidence from lateral mobility studies. J. Biol. Chem. 274:1606–1613. [DOI] [PubMed] [Google Scholar]
- 124.Sanger, J. M., R. Chang, and F. Ashton. 1996. Novel form of actin-based motility transports bacteria on the surfaces of infected cells. Cell Mot. Cytoskel. 34:279–287. [DOI] [PubMed] [Google Scholar]
- 125.Pollard, T. D., and G. G. Borisy. 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 112:453–465. [DOI] [PubMed] [Google Scholar]
- 126.Sternmark, H. 2000. Phosphatidylinositol 3-Kinase and Membrane Trafficking. Oxford University Press, Oxford, UK, 32–108.
- 127.Schneider, I. C., and J. M. Haugh. 2005. Quantitative elucidation of a distinct spatial gradient-sensing mechanism in fibroblasts. J. Cell Biol. 171:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Herrmann, K. H., S. V. M. Satyanarayana, V. Sridhar, and K. P. N. Murthy. 2003. Monte Carlo simulation of actin filament based cell motility. Int. J. Mod. Phys. B. 17:5597–5611. [Google Scholar]
- 129.Grimm, H. P., A. B. Verkhovsky, A. Mogilner, and J. J. Meister. 2003. Analysis of actin dynamics at the leading edge of crawling cells: implications for the shape of keratocyte lamellipodia. Eur. Biophys. J. Biophys. Lett. 32:563–577. [DOI] [PubMed] [Google Scholar]
- 130.Civelekoglu-Scholey, G., A. W. Orr, I. Novak, J. J. Meister, M. A. Schwartz, and A. Mogilner. 2005. Model of coupled transient changes of Rac, Rho, adhesions and stress fibers alignment in endothelial cells responding to shear stress. J. Theor. Biol. 232:569–585. [DOI] [PubMed] [Google Scholar]
- 131.Ma, L., L. C. Cantley, P. A. Janmey, and M. W. Kirschner. 1998. Corequirement of specific phosphoinositides and small GTP binding protein Cdc42 in inducing actin assembly in Xenopus egg extracts. J. Cell Biol. 140:1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.DiNubile, M. J., and S. Huang. 1997. High concentrations of phosphatidylinositol-4,5-bisphosphate may promote actin filament growth by three potential mechanisms: inhibiting capping by neutrophil lysates, severing actin filaments and removing capping protein-β(2) from barbed ends. Biochim. Biophys. Acta Mol. Cell Res. 1358:261–278. [DOI] [PubMed] [Google Scholar]