

Abstract
In mammals, the suprachiasmatic nuclei (SCN) of the hypothalamus act as a dominant circadian pacemaker, coordinating rhythms throughout the body and regulating daily and seasonal changes in physiology and behavior. This review focuses on the mechanisms that mediate synchronization of circadian rhythms between SCN neurons. Understanding how these neurons communicate as a network of circadian oscillators has begun to shed light on the adaptability and dysfunction of the brain's master clock.
Cell-Autonomous Generation of Circadian Rhythms
Genetic screens and spontaneous mutants in Drosophila, hamsters, mice, and humans have produced a short but growing list of “clock genes,” whose primary function is daily timekeeping (Emery and Reppert, 2004). The SCN and other neural and nonneural tissues show daily rhythms of clock gene expression in vitro that are thought to be coordinated by the SCN in vivo (Abe et al., 2002; Yoo et al., 2004). These findings have led to a standard model where circadian rhythms are generated and sustained intracellularly by a transcription-translation negative-feedback loop (Figure 1). This model captures features of the circadian system, including near-24-hr periodicity, robust oscillation, and resistance to perturbations. For synchronization to local time, the model incorporates light-induced changes in transcription of some clock genes. However, by definition the intracellular model does not include communication among oscillators or, critically, the potential for diverse mechanisms for rhythm generation or coordination between various cell types (Figure 2). Multioscillator organization may explain key features of circadian behavior, including its remarkable daily precision and seasonal plasticity.
Figure 1.

Synchronization of Circadian Timekeeping among SCN Neurons
Pacemaking neurons generate near-24-hr rhythms in gene expression, firing rate, and peptide release through a transcription-translation negative-feedback loop. These neurons are all GABAergic, and a subset in the ventral (core) SCN release VIP. VIP and its receptor VPAC2 are necessary for synchronization of circadian periods among SCN neurons. Daily GABA application can synchronize SCN neurons, and blockade of GABAA receptors interferes with rhythm coordination between the dorsal (shell) and ventral SCN. Gap junctions have also been implicated in spike-for-spike synchrony between neighboring SCN neurons.
Figure 2.
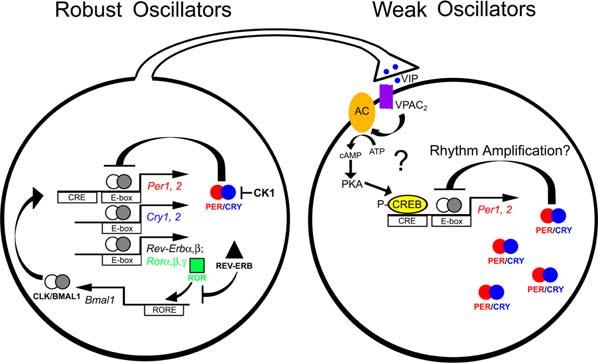
Heterogeneity of Pacemaking Ability among SCN Neurons Robust rhythm generation in SCN neurons is thought to rely on circadian expression of Period (Per1, 2), Cryptochrome (Cry1, 2), Rev-Erb, Ror, and Bmal1 genes, and constitutive expression of Clock. Period and Cryptochrome proteins (PER/CRY) form a negative-feedback loop, repressing transcriptional activation by CLK/BMAL1 through E-Box sequences on Per and Cry promoters. Casein kinase 1 and (CK1Δ) causes a phosphorylation-dependent delay in PER/CRY feedback. REV-ERB and ROR proteins form a second feedback loop, binding to ROR-element (RORE) promoters of the Bmal1 gene to repress and enhance expression, respectively. However, more than half of all SCN neurons require daily VIP-VPAC2 signaling to maintain robust rhythms. This signaling pathway may impinge on the intracellular molecular clockwork through activation of adenylyl cyclase (AC), cAMP, protein kinase A (PKA), and CREB-dependent transcription of the Period genes (Travnickova-Bendova et al., 2002; Itri and Colwell, 2003). This regulation of clock gene transcription may underlie the synchronization and amplification of neuronal rhythms by daily VIP/VPAC2 signaling.
Heterogeneity of Circadian Pacemakers
Although early studies suggested that all 20,000 neurons in the bilateral SCN were cell-autonomous circadian clocks (Welsh et al., 1995), recent studies show that they are not identical in function. SCN neurons differ in their pacemaking ability, neuropeptide expression, their response to environmental timing cues, and the rhythms they control (Antle and Silver, 2005). Some do not fire or express clock genes rhythmically, and some rely on daily signals from other SCN neurons to maintain rhythmicity (Hastings and Herzog, 2004; Aton et al., 2005). Among rhythmic SCN neurons, those in the dorsal “shell” reach their daily peak in firing or gene expression earlier in the day than neurons in the ventral “core” (Yamaguchi et al., 2003; Schaap et al., 2003). Evidence for SCN heterogeneity has led to various models of SCN network function, with each model predicting some features of the organized system (Antle et al., 2003; Gonze et al., 2005). To evaluate the appropriateness of these models, we must learn more about the intrinsic properties of the various cell types (e.g., neurotransmitter and receptor expression) and the specifics of their spatial and temporal connectivity.
Neurotransmission Synchronizes SCN Neurons
In vivo, SCN neurons must synchronize to environmental cycles and each other. Although much is known about mechanisms of entrainment to light cycles, relatively little is known about entrainment of SCN neurons to synchronizing signals within the SCN. “Synchrony” occurs when neurons share the same circadian period, but not necessarily the same phase. In fact, within the “synchronized” SCN, neurons express the same period, but peak at different times during the day (Quintero et al., 2003; Yamaguchi et al., 2003; Schaap et al., 2003; Aton et al., 2005). This is consistent with the prediction that oscillators with differing intrinsic periods synchronize their periods to each other with a distribution of phase relationships (Pittendrigh and Daan, 1976). Multiple studies have highlighted that the SCN is anatomically organized into a dorsal “shell” and ventral “core”—and that these subdivisions may function as independent populations of circadian oscillators. For example, under specific culture conditions, rhythmic release of arginine vasopressin (AVP) from neurons in the dorsal SCN can proceed with different phasing and periodicity from vasoactive intestinal polypeptide (VIP) release rhythms from neighboring neurons in the ventral SCN (Shinohara et al., 1995). In vivo, the dorsal SCN resynchronizes to a shifted light schedule far more slowly than the ventral SCN (Nagano et al., 2003; Albus et al., 2005). As a result, expression of clock genes can be driven nearly 12 hr out of phase between dorsal and ventral SCN when the animal experiences a rapid shift in the light-dark cycle (Nagano et al., 2003), or a 22 hr light cycle (De la Iglesia et al., 2004). Under the extreme conditions of constant light, some animals lose coherent daily rhythms, a phenomenon recently shown to coincide with a loss of circadian synchrony among SCN neurons (Ohta et al., 2005). “Jet lag” seems a likely consequence of this environmentally imposed internal desynchrony within the SCN.
Recent technological developments have greatly facilitated studies of SCN synchrony in vitro. Multielectrode-array recordings allow analyses of weeks of firing from large numbers of individual SCN neurons. High-sensitivity cameras have made it possible to record bioluminescent reporters of clock gene expression from individual cells over many days. Each method has its strengths and weaknesses. Electrical recordings resolve events on a millisecond time scale, but are often from SCN neurons pooled from multiple neonates, are restricted to neurons that coincidentally rest near an electrode, and do not necessarily reveal the state of the intracellular oscillator. Bioluminescence recordings detail intracellular events from cells expressing the reporter, but do not resolve events that transpire in less than an hour. In conjunction with treatments that block or mimic intercellular signals, however, these techniques are being used successfully to address the necessity and sufficiency of specific pathways for circadian synchrony.
Dispersed at low density, individual SCN neurons express firing-rate rhythms with widely varying circadian periods (Welsh et al., 1995; Herzog et al., 1998). However, neurons in SCN explants or high-density dispersals synchronize their periods to one another (Nakamura et al., 2001; Aton et al., 2005). Two clues regarding a mechanism for synchronization within the SCN were revealed by imaging Period1::luciferase expression rhythms in single SCN neurons in vitro (Yamaguchi et al., 2003). First, when the authors separated dorsal and ventral SCN with a knife cut, neuronal rhythms in the dorsal SCN (but not the ventral SCN) lost synchrony. Thus, projections from the ventral SCN synchronize neurons in the dorsal SCN, while the reverse is not true, consistent with dense anatomical projections from ventral to dorsal SCN and sparse reciprocal projections (Abrahamson and Moore, 2001). Second, when the authors applied tetrodotoxin (TTX) to intact SCN slices, Period1::luciferase rhythms throughout the slice lost synchrony. This suggests that sodium-dependent action potentials are required to coordinate daily timing between cells. These data are consistent with the finding that following prolonged TTX administration, correlated firing between pairs of SCN neurons in vitro was prevented (Honma et al., 2000). They are also consistent with in vivo studies in which coherent locomotor rhythms were abolished by TTX infusion into the SCN and recovered several days after the end of infusion (Schwartz et al., 1987). A parsimonious explanation of these results is that a neurotransmitter released by neurons of the ventral SCN is necessary to maintain synchrony throughout the SCN.
A putative neurotransmitter synchronizing signal within the SCN must conform to certain criteria. The transmitter(s) must be expressed by pacemaker neurons and must display a circadian rhythm in activity (e.g., at the level of release). Its receptor must be expressed within the SCN, but not necessarily in a rhythmic fashion. Gap junctions, another candidate synchronizer, could fulfill these criteria through rhythmic electrical coupling between adjacent neurons. Daily activation of the synchronizing pathway should entrain rhythms in SCN neurons and in behavior. Genetic or pharmacologic blockade of this pathway should desynchronize rhythms. Finally, rhythms in signaling activity must entrain to daily environmental cues and to signals that are known to shift rhythms in behavior.
Candidate Synchronizing Factors
GABA
Of the signals that have been studied as potential SCN synchronizers, GABA is unique in that it is expressed by most (if not all) SCN neurons (Figure 1). GABAA and GABAB receptors are found throughout the SCN. There is evidence for a circadian rhythm in GABA release in the dorsal SCN in constant conditions, with higher frequencies of inhibitory postsynaptic potentials during the late day and early subjective night than late subjective night (Itri and Colwell, 2003). Exogenously applied GABA can phase-shift the firing rhythms of individual SCN neurons in vitro. When applied daily, GABA synchronizes firing rhythms of neurons within a culture (Liu and Reppert, 2000). A recent study showed that after a 6 hr shift in the light cycle, dorsal and ventral SCN showed two peaks in daily multiunit firing (Albus et al., 2005). When dorsal and ventral SCN were divided and recorded separately, each region showed a single peak in firing, with the ventral portion shifting rapidly and the dorsal portion lagging behind. These dissociated rhythms corresponded temporally to the two peaks in activity recorded in the intact SCN. When GABAA antagonist bicuculline was applied to intact SCN slices after a phase shift, activity in the dorsal and ventral SCN resembled that of a divided SCN slice. The authors concluded that dorsal and ventral SCN communicate their circadian phases to one another via GABA following shifts in the light cycle. Whether dorsal SCN neurons influence the phase of ventral SCN neurons and whether GABA is required for circadian synchrony remains to be tested.
VIP
Vasoactive intestinal polypeptide (VIP) meets most of the criteria for a synchronizing factor. VIP is synthesized by ventromedial SCN neurons that receive input from retinal ganglion cells (Figure 1). VIPergic neurons comprise around 15% of the 20,000 neurons in the SCN (Abrahamson and Moore, 2001). The VPAC2 receptor for VIP (encoded by the Vipr2 gene) is expressed by about 60% of all SCN neurons, including half of all VIPergic neurons, and almost all arginine-vasopressin (AVP)-expressing neurons of the dorsal SCN. VIP is rhythmically released from the rat SCN in vitro (Shinohara et al., 1995) and shifts both behavioral and SCN firing rhythms (Piggins et al., 1995; Reed et al., 2001). VIP-(Colwell et al., 2003) and VPAC2-deficient (Harmar et al., 2002) mice have strikingly similar behavioral phenotypes. Under a 12 hr light:12 hr dark cycle, their behavior is similar to that of wild-type mice, due to an acute suppression of activity by light. In a skeleton photoperiod (two 11 hr dark periods separated by 1 hr light periods) all VIP−/− and Vipr2−/− mice show equal amounts of activity in both dark phases, while wild-type mice restrict activity to a single dark period each day. In constant darkness, both mutant genotypes show abnormally low-amplitude rhythmicity, with roughly 70% of mice expressing multiple free-running circadian periods simultaneously (Aton et al., 2005). The remaining 30% free-run with shorter periods and lower-amplitude rhythms than wild-type mice. Vipr2−/− SCN have attenuated rhythms in gene expression and multiunit firing (Harmar et al., 2002; Cutler et al., 2003). Long-term recordings of VIP−/− and Vipr2−/− SCN neurons revealed two underlying defects—loss of firing rhythms in the majority and a lack of circadian synchrony among the remaining neurons (Aton et al., 2005). While 70% of wild-type SCN neurons showed firing rhythms with similar circadian periodicity, only about 30% of mutant neurons fired rhythmically, and these showed an abnormally broad range of circadian periods. While rhythmic SCN neurons from wild-type mice were similarly phased within a culture, rhythmic mutant neurons within a culture showed randomly distributed phases. These residual rhythms were also less robust than rhythms from wild-type SCN neurons. Thus, the SCN phenotype of VIP−/− and Vipr2−/− mutants reflects their behavioral phenotype, with respect to both amplitude and circadian synchrony. These data are consistent with findings from Yamaguchi et al., who found that in the presence of TTX, which would prevent the activity-dependent release of VIP (as well as other neurotransmitters), Period1::luciferase rhythms in neurons throughout the SCN lost synchrony and decreased in amplitude (Yamaguchi et al., 2003).
Critically, daily application of a VPAC2 agonist restored rhythmicity to previously arrhythmic VIP−/− neurons and synchronized firing rhythms of neurons within a culture. Restored rhythms showed similar phasing to those of previously rhythmic neurons synchronized by daily agonist treatment and persisted after the daily treatments ended (Aton et al., 2005). This suggests that daily VPAC2 signaling mediates rhythmicity in these neurons by amplifying and entraining weak endogenous rhythms (Figure 2). The data indicate that VIP signaling through VPAC2 is necessary for circadian synchrony of SCN firing rhythms and behavioral rhythms. It is unclear whether the role of VIP in rhythm amplification in SCN neurons is mechanistically related to its role as a synchronizing factor. Disruption of VIP signaling may underlie the behavioral arrhythmicity that can arise in constant, bright light or with aging. Future studies will likely aim to determine whether VIP modulates GABA signaling (or vice versa) to influence interneuronal synchrony (Itri and Colwell, 2003) and identify rhythmic and arrhythmic neuronal populations in mutant SCN. This may be best approached by analysis of Period:luciferase expression rhythms in SCN slice preparations, where SCN anatomy remains largely intact.
Other Neuropeptides
Although VIP meets most criteria for a synchronizing signal within the SCN, it may not be the only one. Other neurotransmitters, such as gastrin-releasing peptide (GRP) and prokineticin 2 (PK2), and their cognate receptors are expressed in a pattern consistent with a role in synchronization. Exogenous GRP shifts behavioral and SCN firing rate rhythms in a manner similar to light and VIP (Piggins et al., 1995; McArthur et al., 2000). Thus, the potential of GRP and PK2 as synchronizing factors in the SCN warrants further investigation.
Gap Junctions
Although TTX disrupts circadian synchrony, mechanisms that modulate membrane potential independent of voltage-gated sodium channels may also participate in coordinated firing in the SCN. Gap junctions have been implicated (Figure 1). The subunits that form gap junctions, connexins, are expressed in the mammalian SCN (Colwell, 2000). Electrical communication between SCN neurons, measured by dye coupling, is high during the daytime peak in firing (Colwell, 2000). Gap junction communication leads to simultaneous firing in approximately 25% of neighboring SCN neurons and is lost in connexin-36 (Cx36) knockout mice (Long et al., 2005). Cx36−/− mice showed slightly lower-amplitude behavioral rhythmicity in constant darkness. Although the behavioral phenotype is not as severe as that seen in VIP−/− or Vipr2−/− mice, it will be important to test whether the observed phenotype results from impaired circadian synchrony between SCN neurons. One possibility is that gap junctions mediate tighter coupling between neurons with similar neuropeptide expression patterns, producing coordinated release from AVPergic or VIPergic neurons. It is also possible that gap junctions, like VIP, are required for rhythmicity in some SCN neurons. In vitro studies of rhythmic gene expression or firing in populations of individual Cx36−/− SCN neurons would likely clarify the role of gap junctions within the SCN.
Postsynaptic Mechanisms of Synchronization
How does receptor activation entrain circadian rhythms in postsynaptic neurons? Some of the candidate synchronizing factors—GABA, VIP, and GRP—change the firing rate of SCN neurons, and electrical coupling causes SCN neurons to fire simultaneously. In light of the recent demonstration that transmembrane calcium conductances regulate circadian oscillation of Period gene expression (Lundkvist et al., 2005), it is likely that changes in firing rate and conductance impinge on core clock events. VIP and GRP induce Period gene transcription in a circadian-phase-dependent manner, perhaps by activating adenylyl cyclase, calcium conductances, and CREB-dependent signaling (Figure 2 [Travnickova-Bendova et al., 2002; Itri and Colwell, 2003]). Importantly, gap junctions may allow passage of cAMP and calcium between neighboring cells. Changes in Period expression within the postsynaptic neuron could directly shift its circadian rhythm of clock-controlled genes and firing rate, bringing it into synchrony with presynaptic neurons.
The Pros and Cons of Multioscillator Organization
A synchronized multioscillator system offers several advantages. Within the SCN, for example, interactions between neurons not only coordinate the population but decrease cycle-to-cycle variability, allowing behavioral rhythms to be more precise than individual neuronal rhythms (Herzog et al., 2004). The result is that free-running SCN rhythms can be accurate to within a few minutes out of the 1440 minutes per day. Critically for the organism, this allows tight temporal scheduling of physiological and behavioral programs. The heterogeneity of the clocks within the SCN has also been postulated to underlie seasonal adaptations. For example, “internal coincidence” models of photoperiodic responses like migration, hibernation, and reproduction posit that when two circadian oscillators assume a critical phase relationship in response to long or short days, a biological response is triggered (Schwartz et al., 2001). The time of day when a SCN neuron peaks may depends upon the neuron's intrinsic period, so that heterogeneity among synchronized neighbors could allow for independent regulation of morning and evening outputs (Schaap et al., 2003). A circadian system composed of multiple circadian oscillators can be, however, disadvantageous for the organism, for example during jet lag, when internal desynchrony among circadian pacemakers results from a rapid change in environmental cycles. More long-term desynchrony may underlie chronic disorders such as depression and sleep disorders.
The study of synchrony among oscillators has attracted much attention and is part of a broader movement toward research on complex systems (Strogatz, 2001). Such systems are inherently difficult to understand because their wiring can be intricate, diverse, and change over time. The in vitro SCN, however, is a model system where current genetic and physiological tools are revealing organizing principles in a network of oscillators.
References
- Abe M, Herzog ED, Yamazaki S, Straume M, Tei H, Sakaki Y, Menaker M, Block GD. J. Neurosci. 2002;22:350–356. doi: 10.1523/JNEUROSCI.22-01-00350.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahamson EE, Moore RY. Brain Res. 2001;916:172–191. doi: 10.1016/s0006-8993(01)02890-6. [DOI] [PubMed] [Google Scholar]
- Albus H, Vansteensel MJ, Michel S, Block GD, Meijer JH. Curr. Biol. 2005;15:886–893. doi: 10.1016/j.cub.2005.03.051. [DOI] [PubMed] [Google Scholar]
- Antle MC, Silver R. Trends Neurosci. 2005;28:145–151. doi: 10.1016/j.tins.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Antle MC, Foley DK, Foley NC, Silver R. J. Biol. Rhythms. 2003;18:339–350. doi: 10.1177/0748730403253840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aton SJ, Colwell CS, Harmar AJ, Waschek J, Herzog ED. Nat. Neurosci. 2005;8:476–483. doi: 10.1038/nn1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS. J. Neurobiol. 2000;43:379–388. doi: 10.1002/1097-4695(20000615)43:4<379::aid-neu6>3.0.co;2-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS, Michel S, Itri J, Rodriguez W, Tam J, Lelievre V, Hu Z, Liu X, Waschek JA. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003;285:R939–R949. doi: 10.1152/ajpregu.00200.2003. [DOI] [PubMed] [Google Scholar]
- Cutler DJ, Haraura M, Reed HE, Shen S, Sheward WJ, Morrison CF, Marston HM, Harmar AJ, Piggins HD. Eur. J. Neurosci. 2003;17:197–204. doi: 10.1046/j.1460-9568.2003.02425.x. [DOI] [PubMed] [Google Scholar]
- De la Iglesia HO, Cambras T, Schwartz WJ, Diez-Noguera A. Curr. Biol. 2004;14:796–800. doi: 10.1016/j.cub.2004.04.034. [DOI] [PubMed] [Google Scholar]
- Emery P, Reppert SM. Neuron. 2004;43:443–446. doi: 10.1016/j.neuron.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Gonze D, Bernard S, Waltermann C, Kramer A, Herzel H. Biophys. J. 2005;89:120–129. doi: 10.1529/biophysj.104.058388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmar AJ, Marston HM, Shen S, Spratt C, West KM, Sheward WJ, Morrison CF, Dorin JR, Piggins HD, Reubi JC, et al. Cell. 2002;109:497–508. doi: 10.1016/s0092-8674(02)00736-5. [DOI] [PubMed] [Google Scholar]
- Hastings MH, Herzog ED. J. Biol. Rhythms. 2004;19:400–413. doi: 10.1177/0748730404268786. [DOI] [PubMed] [Google Scholar]
- Herzog ED, Takahashi JS, Block GD. Nat. Neurosci. 1998;1:708–713. doi: 10.1038/3708. [DOI] [PubMed] [Google Scholar]
- Herzog ED, Aton SJ, Numano R, Sakaki Y, Tei H. J. Biol. Rhythms. 2004;19:35–46. doi: 10.1177/0748730403260776. [DOI] [PubMed] [Google Scholar]
- Honma S, Shirakawa T, Nakamura W, Honma K-I. Neurosci. Lett. 2000;294:113–116. doi: 10.1016/s0304-3940(00)01558-5. [DOI] [PubMed] [Google Scholar]
- Itri J, Colwell CS. J. Neurophysiol. 2003;90:1589–1597. doi: 10.1152/jn.00332.2003. [DOI] [PubMed] [Google Scholar]
- Liu C, Reppert SM. Neuron. 2000;25:123–128. doi: 10.1016/s0896-6273(00)80876-4. [DOI] [PubMed] [Google Scholar]
- Long MA, Jutras MJ, Connors BW, Burwell RD. Nat. Neurosci. 2005;8:61–66. doi: 10.1038/nn1361. [DOI] [PubMed] [Google Scholar]
- Lundkvist GB, Kwak Y, Davis EK, Tei H, Block GD. J. Neurosci. 2005;25:7682–7686. doi: 10.1523/JNEUROSCI.2211-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur AJ, Coogan AN, Ajpru S, Sugden D, Biello SM, Piggins HD. J. Neurosci. 2000;20:5496–5502. doi: 10.1523/JNEUROSCI.20-14-05496.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano M, Adachi A, Nakahama K, Nakamura T, Tamada M, Meyer-Bernstein E, Sehgal A, Shigeyoshi Y. J. Neurosci. 2003;23:6141–6151. doi: 10.1523/JNEUROSCI.23-14-06141.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura W, Honma S, Shirakawa T, Honma K-I. Eur. J. Neurosci. 2001;14:1–10. doi: 10.1046/j.0953-816x.2001.01684.x. [DOI] [PubMed] [Google Scholar]
- Ohta H, Yamazaki S, McMahon DG. Nat. Neurosci. 2005;8:267–269. doi: 10.1038/nn1395. [DOI] [PubMed] [Google Scholar]
- Piggins HD, Antle MC, Rusak B. J. Neurosci. 1995;15:5612–5622. doi: 10.1523/JNEUROSCI.15-08-05612.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittendrigh CS, Daan S. J. Comp. Physiol. [A] 1976;106:291–331. [Google Scholar]
- Quintero JE, Kuhlman SJ, McMahon DG. J. Neurosci. 2003;23:8070–8076. doi: 10.1523/JNEUROSCI.23-22-08070.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed HE, Meyer-Spasche A, Cutler DJ, Coen CW, Piggins HD. Eur. J. Neurosci. 2001;13:839–843. doi: 10.1046/j.0953-816x.2000.01437.x. [DOI] [PubMed] [Google Scholar]
- Schaap J, Albus H, Tjebbe VH, Eilers PH, Detari L, Meijer JH. Proc. Natl. Acad. Sci. USA. 2003;100:15994–15999. doi: 10.1073/pnas.2436298100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz WJ, Gross RA, Morton MT. Proc. Natl. Acad. Sci. USA. 1987;84:1694–1698. doi: 10.1073/pnas.84.6.1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz WJ, De la Iglesia HO, Zlomanczuk P, Illnerova H. J. Biol. Rhythms. 2001;16:302–311. doi: 10.1177/074873001129002024. [DOI] [PubMed] [Google Scholar]
- Shinohara K, Honma S, Katsuno Y, Abe H, Honma K-I. Proc. Natl. Acad. Sci. USA. 1995;92:7396–7400. doi: 10.1073/pnas.92.16.7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strogatz SH. Nature. 2001;410:268–276. doi: 10.1038/35065725. [DOI] [PubMed] [Google Scholar]
- Travnickova-Bendova Z, Cermakian N, Reppert SM, Sassone-Corsi P. Proc. Natl. Acad. Sci. USA. 2002;99:7728–7733. doi: 10.1073/pnas.102075599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DK, Logothetis DE, Meister M, Reppert SM. Neuron. 1995;14:697–706. doi: 10.1016/0896-6273(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Isejima H, Matsuo T, Okura R, Yagita K, Kobayashi M, Okamura H. Science. 2003;302:1408–1412. doi: 10.1126/science.1089287. [DOI] [PubMed] [Google Scholar]
- Yoo S-H, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong H-K, Oh WJ, Yoo OJ, et al. Proc. Natl. Acad. Sci. USA. 2004;102:2608–2613. [Google Scholar]