

Abstract
Exposure to stressors that elicit fear and feelings of hopelessness can cause severe vagal activation leading to bradycardia, syncope, and sudden death. These phenomena though documented, are difficult to diagnose, treat clinically, and prevent. Therefore, an animal model incorporating these cardiovascular conditions could be useful. The present study examined ‘sinking’ during a 2-h swim stress, a phenomenon that occurs in 50% of rats during 25°C water exposure. Concurrent measurements of body temperature, immobility, heart rate (HR), and PR interval (a measure of vagal activity) were made. Neither decreases in immobility nor variations in hypothermia during swim were correlated with sinking. Bradycardia was more severe in sinking rats (average minimum HR±SEM; 143±13 vs 247±14; p<0.01), and PR interval was elevated (p<0.0001). To examine potential modulation of vagal activity during stress, corticotropin-relasing factor (CRF) receptor antagonists (antalarmin, R121919 and astressin B), a glucocorticoid receptor antagonist (RU486), and a peripherally acting cholinergic antagonist (methylatropine nitrate) were administered. The centrally acting CRF antagonist, antalarmin (32 mg/kg), produced elongation of the PR interval (p<0.0001), robust bradycardia (135±18; p<0.001), and increased sinking (92%; p<0.05), and methylatropine nitrate (3.2 mg/kg) blocked these effects. Corroborating these data, two different CRF antagonists, R121919 (30 mg/kg) and astressin B (intracerebroventricular (i.c.v.), 0.03 μg/rat) increased sinking to 100%. RU486 (20 mg/kg) blocked HPA axis negative feedback and decreased percent sinking to 25%. From these studies, we concluded that sinking during a 2-h water exposure was a result of extreme vagal hyperactivity. Furthermore, stress-induced CRF release may serve to protect against elevated cardiac vagal activity.
Keywords: CRF antagonists, antalarmin, astressin B, R121919, cardiac vagal activity, swim stress
INTRODUCTION
Emotional factors are well documented to affect autonomic control of the heart. Emotions such as anger and anxiety are thought to increase sympathetic output, thereby increasing heart rate (HR) and risk of coronary heart disease or sudden cardiac death (Agelink et al, 2002; Johnsen et al, 2003; Kawachi et al, 1994; Nahshoni et al, 2004; Watkins et al, 1998; Wittstein et al, 2005; Yeragani et al, 2001, 2002). Alternatively, fear and feelings of hopelessness cause severe vagal activation resulting in bradycardia, which can lead to vasovagal syncope and asystole (Angrilli et al, 1997; Friedman et al, 1993; Hartel, 1987). Psychologically influenced syncope and sudden death in humans is often supposed, but difficult to diagnose and document clinically.
In vivo analysis of vagally stimulated syncope and sudden death has also proven to be daunting in experimental animal models. Different forms of stress and fear predominantly elicit increases in sympathetic activity, leaving vagal activity largely unaffected. Sgoifo et al 1999 evaluated autonomic activation in response to commonly used experimental stress paradigms (social defeat, restraint, shock, and forced swim); all studies elicited sympathetic activation, however, forced swim was the only model to result in robust vagal activation. In fact, increased cardiac vagal activity in response to water immersion has been documented in humans (Schipke and Pelzer, 2001; Miwa et al, 1997) and has been found to elicit an impact on the cardiovascular system said to be equivalent to that of a radical blood transfusion (Hood et al, 1968). Forced swim has previously been used as a model to evaluate the phenomenon of sudden cardiac death in rats (Richter, 1957). These studies revealed that water temperatures deviating above or below body temperature decreased swim duration markedly, however, great variation existed at all water temperatures; for example, rats exhibited swim durations from 15 min to 81 h in 35°C water. Further examination of sinking rats in cold water revealed an initial tachycardia; however, with prolongation of the stressor the rats exhibited a gradual yet, steady decrease in heart rate until the heart stopped. The effect of atropine, a cholinergic antagonist, was also studied during forced swim and although no direct vagal measurements were reported, atropine increased swim duration in rats. Individual differences in swim duration prior to sinking were, therefore, attributed to differences in vagal activation.
Over three decades later, the phenomenon of individual differences among rats during forced swim was re-evaluated (Nishimura et al, 1988). In 25°C water, 50% of rats sank prior to the end of a 2-h swim. Sinking rats exhibited decreased immobility during swim compared with nonsinking rats, and sinkers defecated more. Some prior experience was found to protect against sinking; for example, repeatedly holding rats briefly and then freeing them, in addition to immersing them in water for a few minutes, resulted in significantly lengthened swim duration (Richter, 1957). From these data and studies indicating that wild rats and domestic rats without whiskers exhibit bradycardia and sink very rapidly upon forced swim, Richter (1957) concluded that sinking was a sign of extreme emotion.
Although it is suggested that sinking was related to the severity of emotional reaction, Nishimura and co-workers did not speculate as to what may facilitate elevated vagal activity and sinking during a 2-h swim test. A reasonable physiologic factor that may modulate vagal activation during forced swim is the hypothalamic–pituitary–adrenal (HPA) axis hormone, corticotropin-releasing factor (CRF). Two CRF receptor subtypes, CRF-R1 and CRF-R2, have been identified in mammalian rat brain (Chalmers et al, 1996). The central distribution of subtype R1 and R2 differ markedly, suggesting each receptor exhibits a distinct functional role (Chalmers et al, 1995). Additionally, one CRF-R2 subtype, CRF-R2b, is highly localized in the heart (Lovenberg et al, 1995).
Both CRF-R1 and -R2 activation have been found to elicit cardiovascular changes. Peripherally administered CRF is reported to increase HR and decrease mean arterial pressure (MAP) (Briscoe et al, 2000; Gardiner et al, 2005; Mackay et al, 2003). This cardiovascular response was unaffected by antalarmin, a selective CRF-R1 antagonist that readily crosses the blood–brain barrier, but was inhibited by peripheral administration of α-helical CRF9–41, which does not cross the blood–brain barrier. (Briscoe et al, 2000; Overton and Fisher, 1991). Centrally administered CRF, on the other hand, increases both HR and MAP (Briscoe et al, 2000; Overton and Fisher, 1991; Richter and Mulvany, 1995). Additionally, CRF infusion in the central amygdala also increases resting HR without changing plasma norepinephrine or epinephrine (Wiersma et al, 1993). These central hemodynamic effects were not blocked by peripherally acting CRF antagonists (Overton and Fisher, 1991), but rather, by either central administration of α-helical CRF9–41 (Richter and Mulvany, 1995) or antalarmin (Briscoe et al, 2000). These data suggest that the cardiovascular response to peripheral CRF is mediated via CRF-R2 and the cardiovascular changes associated with centrally administered CRF are mediated primarily through CRF-R1.
More specifically, many studies have also established a relationship between CRF and vagal activity; for example, intracerebroventricular (i.c.v.) administration of CRF was found to inhibit Fos expression in the dorsal motor nucleus of the vagus nerve in rats (Wang et al, 1996). Also, intracisternal injections of CRF or sauvagine inhibit basal gastric vagal efferent discharge (Kosoyan et al, 1999) and i.c.v. CRF alters cardiac baroreflex function predominantly through inhibition of cardiac vagal outflow (Fisher, 1989). Few studies have utilized selective CRF receptor antagonists; however, nonselective antagonists are documented to elevate vagal activity, leaving epinephrine and norepinephrine unaffected; Nijsen et al 2000 found that during conditioned fear, vagal activity was significantly increased in rats that had received CRF antagonists. This effect was blocked by methylatropine nitrate, further substantiating one source of the observed CRF antagonist-induced increase in cardiac vagal activity. These data indicate a clear relationship between CRF and altered vagal activity during conditioned fear.
In addition to findings that water immersion increases vagal activity, forced swim has also been well documented to increase the release of HPA axis hormones; adreno-corticotropic hormone (ACTH) and corticosterone. Taken together, we suggest that forced swim may be a stress model well designed for the analysis of emotionally induced increases in vagal activity and the regulation of cardiac vagal activity by CRF. In the present study, we quantified changes in vagal activity by measuring the PR interval of the electrocardiogram (ECG). Vagal stimulation increases atrioventricular conduction time and thus directly increases the duration of the PR interval (Kinoshita and Konishi, 1989; Levy and Zieske, 1969; Smith et al, 1976). Elongation of the PR interval can be obtained by either an increase in vagal activity and/or a decrease in sympathetic activity. It is recognized that swim stress increases sympathetic activity (Sgoifo et al, 1999), therefore, a swim stress-induced increase in PR interval can be attributed predominately to elevated vagal activity. Therefore, by evaluating changes in HR and PR interval, one aim of this study was to characterize elevated vagal activation as a major catalyst of sinking during forced swim. Furthermore, we hypothesized that CRF antagonists would exaggerate vagal hyperactivity during forced swim and ultimately lead to sinking.
MATERIALS AND METHODS
Animals and Surgical Procedures
Male, outbred, Sprague–Dawley rats, weighing 250–350 g, were used in these studies (Harlan Sprague Dawley Inc., Indianapolis, IN, USA). Food and water were available ad libitum and rats were housed in groups of three in clear acrylic cages in a 12-h light/dark cycle, climate-controlled room. Rats were allowed to acclimate to the room for at least 3 days upon which they were randomly assigned to either a vehicle or treatment group. Rats committed to a surgery group were also allowed to acclimate to the room at least 2 days before surgery.
Rats undergoing surgery were anesthetized using intramuscular injections of ketamine (100 mg/kg) and xylazine (10 mg/kg). For ECG implantation, a small longitudinal incision was made in the skin and the linea alba of the abdominal muscle. Telemetric ECG transmitters (Data Sciences, Transoma Medical Inc., St Paul, MN, USA) were implanted into the abdominal cavity and sutured to the abdominal wall. Two electrodes, from the bottom of the transmitter were sutured subcutaneously. One electrode was located on muscle above the zyphoid process and the other electrode was placed to the right of the clavicle. For i.c.v. surgery, permanent stainless-steel brain cannulae were aimed at the right lateral ventricle according to the coordinates: 0.8 mm posterior and 1.5 mm lateral to bregma (Paxinos and Watson, 1982). Following each experiment, methylene blue was injected prior to euthanization with an overdose of pentobarbital sodium. Placement of the cannula was confirmed by inspecting the distribution of dye throughout the ventricular space; only rats with wide distribution of dye in the ventricles were included in the study. Following surgery, rats were singly housed and recovered for 6–8 days. The University of Michigan’s University Committee on the Care and Use of Laboratory Animals approved the protocols.
2-h Swim Stress
Rats swam in clear acrylic containers (46 cm tall ×20 cm in diameter) filled with 30 cm of 25°C (±0.5) water. One swim session was preceded by a treatment with either drug or vehicle. Rats were exposed to 25°C water for up to 2 h. Rats were monitored continuously during swim. In the event that they sank below the water and failed at their attempt to return to the water’s surface, the rats were removed from the water tank immediately, towel dried, and placed under a heat lamp for 30 min. Percent sinking was averaged across naïve, nonsurgery rats.
A time-sampling technique first described by Detke et al 1995 proved to be both reliable and valid for detecting levels of immobility during the first 15 min of exposure to the water tank. Immobility is defined as floating in the water with minimal movements to keep the nose above water. Behavior classifications were made at the end of each 5-s period during test sessions. The observers were blind to the treatments. The respective behaviors were averaged within each treatment group across rats.
Blood Sampling
Blood was drawn just prior to and at the end of the swim test, either at the end of 2 h or at the time of sinking, whichever occurred first. Blood samples (240 μl) were collected by nicking the tip of the tail of unrestrained rats. Heparinized microhematocrit capillary tubes were used to collect the blood (Fisher Scientific, Chicago, IL, USA). The samples were stored on ice (<20 min) prior to being centrifuged (4100 r.p.m.). Plasma was separated and stored at −80°C for later analysis. All pre- and post-swim blood samples were collected between 1200 and 1600.
ACTH Measurements
Plasma samples collected in these experiments were assayed for adrenocorticotropic hormone (ACTH; Nichols Institute Diagnostics, San Juan Capistrano, CA, USA). The ACTH assay kit measures levels of intact ACTH molecules and has a sensitivity of 1.0 pg/ml.
Drug Treatments
All treatments were administered 1 h prior to homecage test or swim exposure with the exception of methylatropine nitrate given 15 min before testing. Naïve, nonsurgery rats were administered either antalarmin (3.2, 32, 56 mg/kg, intraperitoneally (i.p.)), methylatropine nitrate (3.2 mg/kg, subcutaneously (s.c.)), antalarmin plus methylatropine nitrate (32 and 3.2 mg/kg, respectively), R121919 (1, 10, 30 mg/kg, i.p.), astressin B (0.032 and 0.1 mg/kg, s.c.; 0.003 and 0.03 μg/rat, i.c.v.), or RU486 (2, 20 mg/kg, s.c.). Antalarmin and RU486 (mifepristone, TOCRIS) were dissolved in a 1 : 1 : 9 solution of ethanol, emulphor, and sterile water, respectively. Methylatropine nitrate (Inland Alkaloid Inc.) was dissolved in sterile water. R121919 was dissolved in ~10% of a 0.1 M tartaric acid solution and 90% sterile water. Astressin B 1 mg/ml was dissolved in ~10% DMSO and 90% sterile water and diluted with either sterile water for s.c. injections or with aCSF for i.c.v. injections. i.c.v. injections were administered in 10 μl volumes. Vehicle controls were conducted using appropriate solutions. Vehicle pH was comparable to each drug.
The dose ranges for CRF antagonists were selected based on previous studies demonstrating antalarmin’s (Deak et al, 1999; Webster et al, 1996), R121919’s (Heinrichs et al, 2002; Jutkiewicz et al, 2005), and astressin B’s (Rivier et al, 2003) ability to inhibit stress-induced release of ACTH. Previous studies have reported active doses of RU486 ranging from 3 (Cook, 2002) to 100 mg/kg (Dal-Zotto et al, 2003). The methylatropine nitrate dosage was carefully chosen as low doses of muscarinic receptor antagonists are known to produce a parasympathomimetic effects via elevated vagal efferent activity (Das et al, 1975; Perlstein et al, 2002); previous studies have shown that 1.5 (Castro et al, 1999) and 3 mg/kg (Negrao et al, 1992) methylatropine nitrate are within the compound’s antagonistic range.
No significant differences were found among vehicle treatments in naïve, non-ECG rats, therefore, only sterile water was used as a vehicle in ECG experiments. ECG rats were administered vehicle (n = 12), 3.2 mg/kg methylatropine nitrate (n = 9), 32 mg/kg antalarmin (n = 9), 32 mg/kg antalarmin + 3.2 mg/kg methylatropine nitrate (n = 9), 0.1 mg/kg astressin B (n = 9), or 20 mg/kg RU486 (n = 9). The effects of each treatment on HR and PR interval were evaluated in the presence or absence of experimental stress, swim exposure or homecage, respectively.
Telemetry System
The telemetry system consisted of battery operated transmitters (Model TA11CTA-F40, Data Sciences, Transoma Medical Inc., St Paul, MN, USA) and receivers (Data Sciences, TMI, St Paul, MN, USA). Each rat, either in its home cage or swim tank, was placed on the receiver where temperature and ECG output were recorded. An ECG analysis program (4.02; Data Sciences, St Paul, MN, USA) was used to analyze PR interval and HR during swim. Core body temperature was also recorded from the ECG transmitters and collected using Dataquest A.R.T Gold analysis 3.01.
Experimental Design
Behavior and hormone measures
Rats were randomly assigned to a vehicle or drug group. Treatments were administered 15 or 60 min prior to water exposure. Swim durations were recorded for each rat and percent sinking was calculated for each dose of drug. Pre- and postswim blood samples were collected from six rats in each group and assayed for ACTH levels. From these results, we concluded which doses of drugs significantly altered HPA axis activity during the 2-h swim exposure. Doses found to alter percent sinking and/or postswim ACTH levels in naïve rats were administered to ECG implanted rats.
Cardiovascular and temperature measures
The effects of each treatment on temperature, HR, and vagal activity at rest and during swim exposure were evaluated. At 30 min of resting measurements were collected from each rat in its homecage; rats were then treated with either drug or water and assigned to either a stress or nonstress group. Rats assigned to the stress group (n = 12/vehicle, n = 9/drug) were placed in the water tank following the designated pretreatment time to evaluate stress-induced changes in temperature, HR, and PR interval in the presence of each drug compared with the vehicle-treated group. Rats in the nonstress group (n = 5/treatment) were returned to their homecage following injection for the pretreatment duration (15 or 60 min), where they remained for an additional 2 h. This study permitted evaluation of thermal and cardiovascular changes produced by drug treatment in the absence of water exposure. The 15 or 60 min pretreatment time was analyzed separately from the following 2 h to identify drug-induced cardiovascular changes that occur in the absence of water exposure.
Data Analysis
Postswim ACTH levels were analyzed using a one-way analysis of variance (ANOVA) for each separate drug group (GraphPad Prism, GraphPad Software Inc., San Diego, CA, USA). Additionally, minimum core body temperature, percent immobility and minimum HR were averaged (±SEM) within each treatment group and compared with vehicle-treated rats using a one-way ANOVA. Tukey’s post hoc tests were used to identify significant changes among vehicle and treatment groups.
The percentage of rats sinking prior to completion of 2 h of water exposure was calculated for each treatment. We utilized a two-tailed Fisher’s Exact Test (GraphPad Prism 3.03) to evaluate whether the proportion of rats sinking following each separate drug treatment was significantly different from the appropriate vehicle.
The ECG analysis program provided an average (±SEM) of both PR intervals and heart rate for each ten second epoch. Data was collected during baseline before and after vehicle or drug treatment. Baseline temperature, HR, and PR intervals collected prior to treatment were analyzed using a one-way ANOVA and Tukey’s post hoc test to identify pre-existing variations between the randomly selected groups. A paired t-test was then used to establish significant changes in these parameters after drug administration. Thermal and cardiovascular changes were also evaluated during swim or homecage exposure. The first 10 min of swim or homecage exposure were averaged over 1-min bins of time followed by 5 min bins for the remaining duration of swim or homecage exposure. Separate two-way repeated measures ANOVA (treatment vs time) were performed for HR and PR values during the 2-h homecage or water exposure. All statistical results with a p<0.05 were considered significant.
RESULTS
Swim Exposure: ACTH
Average baseline measurements of ACTH in all treatment groups prior to swim exposure ranged from 18 to 47 pg/ml. In 15 min of forced swim has been documented to increase ACTH levels to 200–340 pg/ml, four- to sevenfold above baseline (Jutkiewicz et al, 2005). In the current studies, the 2-h water exposure ACTH had recovered to levels of 143 (±11) pg/ml in control rats. No significant difference was found in postswim ACTH levels between sinking and nonsinking control rats (data not shown); therefore, postswim ACTH levels were averaged among all controls.
Pretreatment with the CRF antagonists antalarmin and R121919 significantly attenuated swim-stress induced increases in ACTH; antalarmin (32 and 56 mg/kg; p<0.001) produced a robust decrease (F(3, 28) = 34.15; p<0.001) in postswim ACTH levels that was unaffected by addition of methylatropine nitrate (3.2 mg/kg) (Figure 1a and b; F(3, 16) = 26.39; p<0.0001). R121919 (30 mg/kg, p<0.05) also produced, although to a lesser extent, a decrease in postswim ACTH (Figure 1c; F(3, 24) = 3.97; p = 0.02). RU486 (20 mg/kg) produced a significant (p<0.05) increase in ACTH following forced swim (Figure 1d; F(2, 12) = 5.79; p = 0.02).
Figure 1.
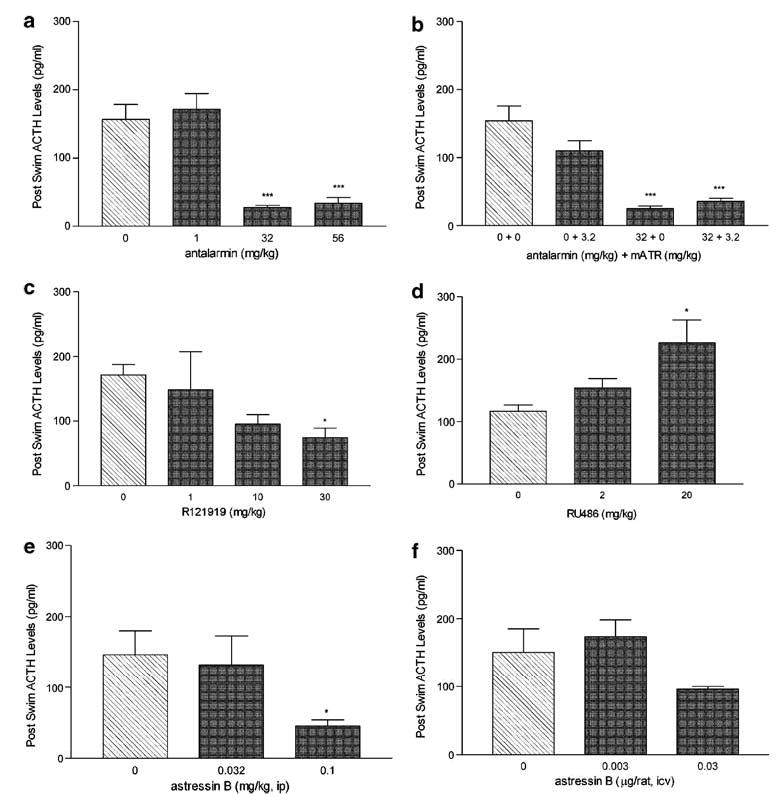
The effects of CRF antagonists, GR antagonists and methylatropine challenge on postswim ACTH levels. Each panel represents the dose-dependent effects of each drug (dark bars) vs vehicle (hatched bar) treatment on ACTH at the time of sinking or at the end of the 2 h swim (n = 6 rats per dose of drug) expressed as mean pg/mL (±SEM); (a), antalarmin; (b) antalarmin + methylatropine nitrate (mATR); (c), R121919; (d), RU486; (e), astressin B—ip; and (f) astressin B—icv. Blood was collected at time of sinking or at the end of the two hour swim. *p<0.05, ***p<0.001 vs 0 mg/kg treatment.
Administration of astressin B was effective in lowering ACTH during forced swim (Figure 1e and f); peripheral administration of astressin B (0.1 mg/kg) markedly decreased ACTH (p<0.05) (F(2, 21) = 5.89; p = 0.01) whereas i.c.v. administration of astressin B (0.03 μg/rat) produced a trend toward decreasing postswim ACTH (F(2, 15) = 2.78; p = 0.09).
Swim Exposure: Percent Sinking
Each treatment group consisted of 8–12 rats; the percentage of rats sinking were compared with the appropriate controls. Systemically administered vehicle treatment resulted in 42–58% sinking, whereas i.c.v. administered vehicle resulted in 63% sinking. Doses of centrally acting CRF receptor antagonists that decreased postswim ACTH levels, also affected percent sinking; antalarmin, for example, produced a robust, dose dependent increase in percent sinking following 32 mg/kg (p<0.05) and 56 mg/kg (p<0.01) (Figure 2a). This effect was blocked by the additional administration of 3.2 mg/kg methylatropine nitrate prior to swim (p<0.01, Figure 2b). The highest dose of R121919 also increased percent sinking to 88%, however, this effect was not found to be significant (p = 0.1, Figure 2c). Peripheral administration of astressin B failed to affect percent sinking (p = 1.3, Figure 2e) despite the decrease in stress-induced release of ACTH elicited by 0.1 mg/kg; i.c.v. administration of astressin B (0.03 μg/rat), however, produced a trend toward increased sinking (p = 0.2, Figure 2f).
Figure 2.
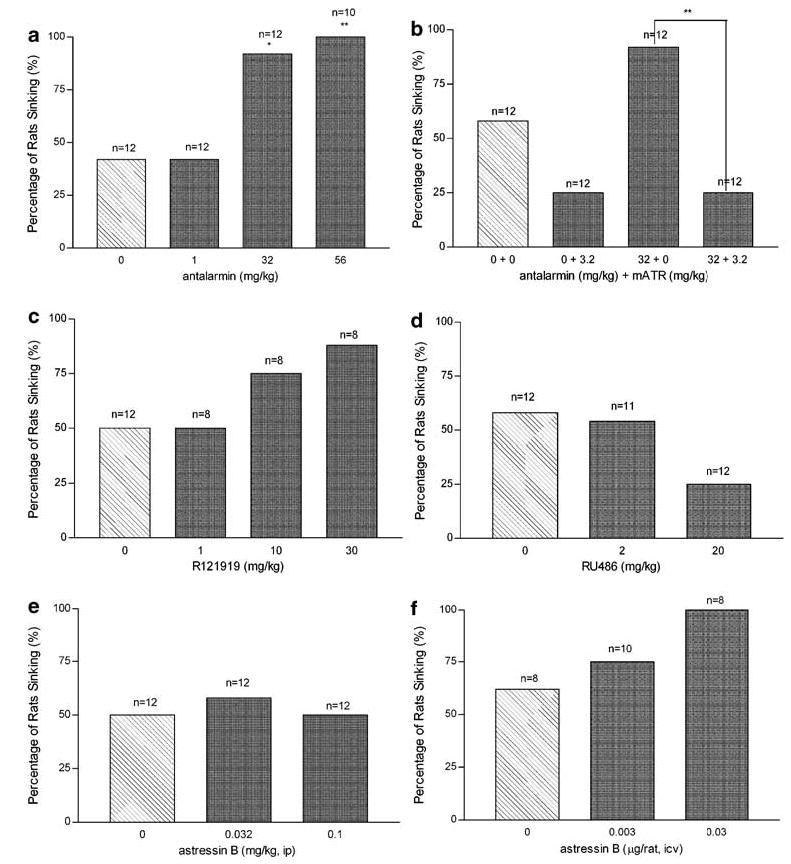
The effects of CRF antagonists, GR antagonists and methylatropine challenge on the percentage of rats sinking during a 2 h swim test. Each panel represents the dose-dependent effects of each drug (dark bars) vs vehicle (hatched bar) treatment on the percentage of rats sinking in each group (total number of rats in each group indicated within each bar). Data expressed as the percentage of rats sinking within the total number of rats treated. See Figure 1 for panel descriptions. *p<0.05, **p<0.01 vs 0 mg/kg treatment.
Methylatropine nitrate (3.2 mg/kg) decreased percent sinking to 25%, however, this effect was not significant (p = 0.2, Figure 2b). RU486 (20 mg/kg) also decreased the percentage of sinking rats to 25%, however, this effect was also not quite significant (p = 0.2, Figure 2d).
Swim Exposure: Changes in Immobility, HR, and Hypothermia
Significant overall differences were found in immobility levels (F(8, 135) = 12.65; p<0.0001), minimum HR (F(7, 40) = 15.01; p<0.0001), and minimum core body temperature (F(7, 64) = 4.17; p = 0.0008) among treatment groups (Table 1). Evaluation of vehicle-treated rats revealed a modest decrease in immobility (p<0.05) in sinking control rats as compared with nonsinking; sinking controls also exhibited a significantly lower minimum HR (p<0.001) than nonsinking rats.
Table 1.
Mean (±SEM) Thermal (Minimum Core Body Temp, °C), Behavioral (% Time Spent Immobile), and Cardiovascular (Minimum Heart Rate = HR, b.p.m.) Changes During Swim Stress
| Treatment | Min. core temp | % Immobile | Min. HR |
|---|---|---|---|
| Vehicle (nonsink) | 24.9 (0.3) | 74 (2) | 247 (14) |
| Vehicle (sink) | 24.8 (0.4) | 67 (1.7)# | 142 (13)*** |
| Average vehicle | 24.8 (0.2) | 70.8 (1.8) | 193 (26) |
| 3.2 mg/kg methylatropine | 24.8 (0.2) | 82 (6.2)* | 235 (11) |
| 32 mg/kg antalarmin | 25.6 (0.3) | 76 (2.3) | 135 (18)* |
| 3.2 methylatropine+32 antalarmin | 23.8 (0.3)* | 85 (1.9)*** | 240 (3) |
| 30 mg/kg R121919 | ND | 80 (2.6)* | ND |
| 0.1 mg/kg astressin B | 24.8 (0.5) | 77 (2) | 229 (11) |
| 20 mg/kg RU486 | 24.4 (0.2) | 75 (3.2) | 243 (6) |
p<0.05,
p<0.001 (vs average vehicle) N = 6–9 rats/treatment.
p<0.05 (vs vehicle nonsink), ND = not determined.
Bold values represent measurements significantly changed versus vehicle-treated rats.
Methylatropine nitrate alone (3.2 mg/kg; p<0.05) and in combination with antalarmin (32 mg/kg; p<0.001) produced an increase in immobility compared with vehicle rats. R121919 (30 mg/kg) elicited a similar increase in immobility (P<0.01).
Antalarmin (32 mg/kg) was the only treatment evaluated in ECG implanted rats that significantly reduced minimum HR (p<0.05) as compared with vehicle. Addition of methylatropine nitrate (3.2 mg/kg) blocked the effect of antalarmin on HR.
Body temperature in the rat under the normal conditions of the laboratory was 37.4°C (±0.18); water exposure produced a profound hypothermia which ranged in different groups from 23.8°C (±0.3) to 25.6°C (±0.3). Antalarmin (32 mg/kg) + methylatropine nitrate (3.2 mg/kg) treated rats maintained a significantly (p<0.05) lower minimum core body temperature as compared with vehicle (F(7,64) = 4.17, p = 0.0001).
Drug-Induced Changes in HR, PR Interval, and Temperature in the Homecage
There were no variations in resting HR (F(7, 88) = 1.2; p = 0.29, Table 2) or temperature (F(5, 24) = 1.6; p = 0.2) among groups prior to vehicle or drug administration. Baseline PR interval was found to differ (F(7, 88) = 2.9; p = 0.01), however, PR narrowly ranged from 50 (±1) to 53 (±1) and Tukey’s post hoc analysis found no significant difference among individual groups (Table 2). Significant thermal variations were not detected; however, cardiovascular changes were observed in response to treatment (Table 2). The average resting HR of all vehicle rats was found to decrease following a 1 h pretreatment of sterile water (t(9) = 2.6; p = 0.05). Methylatropine nitrate (3.2 mg/kg) significantly increased resting HR (t(4) = 3.2: p = 0.05) and decreased PR interval (t(4) = 7.5; p = 0.01) following the 15 min pretreatment. Subsequent to a 1 h pretreatment of antalarmin (32 mg/kg), resting PR interval increased by 6 (±1) msec (t(4) = 3.8; p = 0.05). This effect was blocked by addition of methylatropine nitrate (3.2 mg/ kg; t(4) = 0.8; p = 0.5).
Table 2.
Mean (±SEM) Resting HR (b.p.m.) and PR Interval (msec) Prior to Drug Treatment and Resting HR and PR Interval After Drug Treatment (Post-Treatment HR and PR Collected 5 min Prior to Swim Exposure)
|
Before treatment |
After treatment |
|||
|---|---|---|---|---|
| Treatment | HR | PR interval | HR | PR interval |
| Vehicle | 393 (7) | 53 (1) | 365 (7)* | 53 (1) |
| 3.2 mg/kg methylatropine | 388 (11) | 52 (0.5) | 436 (10)* | 48 (1)** |
| 32 mg/kg antalarmin | 388 (13) | 50 (1) | 399 (7) | 56 (1)* |
| 3.2 methylatropine+32 antalarmin | 399 (10) | 50 (1) | 428 (14) | 52 (1) |
| 0.1 mg/kg astressin B | 379 (8) | 52 (1) | 364 (8) | 53 (1) |
| 20 mg/kg RU486 | 399 (10) | 51 (1) | 397 (3) | 52 (1) |
p<0.05,
p<0.01 vs ‘before treatment’ value (n = 5 rats/treatment).
Bold values represent measurements significantly changed versus before-treatment values.
Drug-induced changes were also observed in resting PR interval and HR at the same time interval that rats in the ‘stressed’ groups were in the water tanks (data not shown). Antalarmin elicited a consistent increase in PR interval of 2.7 (±0.6) ms throughout the 2 h following the end of the drug treatment (F(1, 136) = 111.8; p<0.0001), however, no changes were observed in HR (F(1, 144) = 0.69: p = 0.85). Addition of methylatropine nitrate inhibited antalarmin-induced elevations in PR interval (F(1, 136) = 2.8; p = 0.1) and increased HR by 26 (±2.3) b.p.m. over the first hour and by 17(±1.6) b.p.m. during the second hour (F(1, 142) = 2.25; p<0.0001). Methylatropine nitrate alone decreased PR interval by 2 (±0.7) msec for the first 30 min following the end of the treatment duration (F(1, 147) = 9.23; p = 0.005) and increased HR by 21 (±2.8) b.p.m. during the first hour and 19 (±1) b.p.m. during the second hour (F(1, 146) = 39.9; p<0.0001).
Forced Swim Exposure: ECG Analysis
There was no difference in resting heart rate or PR interval among vehicle rats (Table 2). Although all vehicle control rats exhibited increased PR interval in response to water immersion (F(25, 442) = 86, p<0.0001), rats that sank maintained a significantly elevated PR interval over that of nonsinking rats (F(1, 234) = 84, p<0.0001). An increase in HR immediately upon being placed in the water tank was also observed in vehicle rats. This initial tachycardia is followed by a state of bradycardia that is maintained for the duration of the swim exposure. Sinking vehicle rats, however, exhibit more severe bradycardia within 30 min of swim exposure as compared with nonsinking rats (Figure 3; F(1, 257) = 134; p<0.0001).
Figure 3.
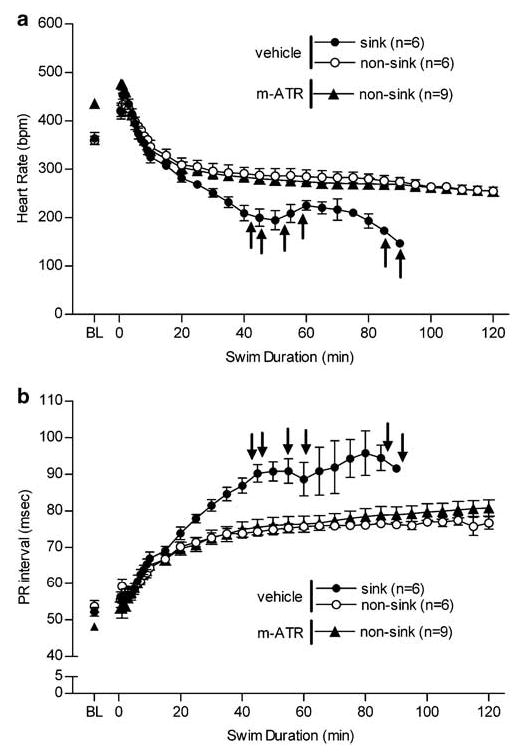
Changes in heart rate (a) and PR interval (b) among vehicle-treated (n = 12) and 3.2 mg/kg methylatropine nitrate (mATR; n = 9) treated rats during swim exposure. Of vehicle treated rats, 6/12 sank whereas 0/9 mATR-treated rats sank. Baseline (BL) HR and PR interval were collected following vehicle or drug treatment while each rat was in its homecage, BL data reported was collected just prior to the swim stress. Rats were placed in the swim tank at time zero and recorded until sinking or 2 h. ↑ indicate when each vehicle-treated rat sank.
Six out of 12 vehicle-treated rats exhibited severely elevated PR interval corresponding with extreme bradycardia; whereas methylatropine nitrate treated rats did not display this radical cardiovascular effect (n = 9, Figure 3a and b). In fact, during swim stress, these rats displayed alterations in HR (F(1, 419) = 0.16; p = 0.7) and PR interval (F(1, 424) = 2.9; p = 0.1) similar to that of nonsinking rats (Figure 3a and b).
Antalarmin elicited an increase in PR interval (F(1, 324) = 90; p<0.0001) and a decrease in HR (F(1, 297) = 156; p<0.0001) during swim in all rats treated with 32 mg/kg; this dose produced 100% sinking in ECG rats (n = 9, Figure 4a and b). This effect was inhibited by administration of methylatropine nitrate (n = 9).
Figure 4.
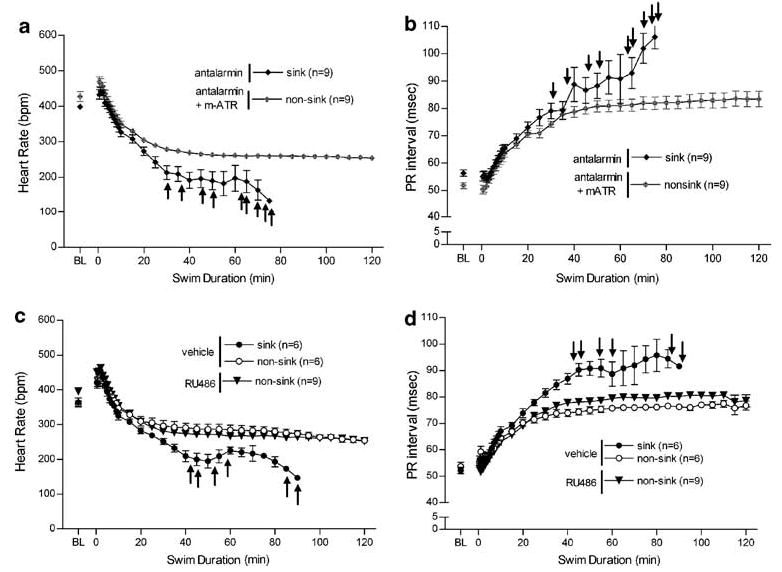
The effects of 32 mg/kg antalarmin (n = 9) on HR (a) and PR interval (b) and the blockade of these effects after administration of 3.2 mg/kg methylatropine nitrate (mATR; n = 9). 100% of ECG implanted rats treated with 32 mg/kg antalarmin sank during the 2-hour swim test, whereas the addition of mATR blocked the behavioral effect of sinking in all antalarmin-treated rats. The cardiovascular effects of 20 mg/kg RU486 (n = 9) on HR (c) and PR interval (d) compared with vehicle-treated rats (n = 12) during swim exposure are also depicted. As previously mentioned, half of vehicle-treated rats sank, whereas no RU486-treated ECG rats sank. Baseline (BL) HR and PR interval were collected subsequent to drug or vehicle treatment while each rat was in its homecage, just prior to swim stress. Swim began at time zero, and data collection continued until sinking or the end of the 2 h swim. ↑ indicates when each antalarmin-treated rat sank.
RU486 treatment (20 mg/kg), similar to methylatropine nitrate alone, also inhibited the severe cardiovascular changes observed in half of vehicle controls such that the modest changes in HR during swim was comparable to that of only nonsinking vehicle rats (F(1, 375) = 0.3; p = 1, Figure 4c). On the other hand, RU486 treated rats (n = 9) did exhibit slight elevations in PR interval during swim stress (Figure 4d) over that of nonsinking vehicle-treated rats (F(1, 378) = 13; p = 0.001) however, they were also significantly below that of sinking vehicle rats (F(1, 273) = 280; p<0.0001).
DISCUSSION
All control rats exhibited bradycardia during swim in 25°C water; however, rats that sank exhibited a significantly lower minimum heart rate than those that did not sink. It was previously reported that decreased core body temperature directly correlates with decreased HR and cardiac output in rats, independent of exercise (Baker and Horvath, 1964; Dawson et al, 1968). Therefore, variations in core body temperature during cold swim were evaluated as a source of the disparity observed in HR among sinking and nonsinking rats. Baseline core temperature and rate of development of hypothermia following water immersion was similar among treatment groups (rate data not shown); additionally, minimum core body temperature was maintained within a narrow range (23.8–25.6°C). Nevertheless, minimum core body temperature was comparable between sinking and nonsinking vehicle rats. Therefore, a more severe state of hypothermia was not responsible for the bradycardia exhibited by sinking rats. These results support Richter’s (1957) original finding that sinking was associated with bradycardia.
Richter (1957) also noted that upon sinking, rats exhibited large, distended hearts postmortem, a sign of elevated cardiac vagal activity. Increased rates of defecation were also observed in sinking rats (Nishimura et al, 1988), suggesting a higher level of parasympathetic activity in sinking as compared with nonsinking rats. In the current 2-h swim study, control rats exhibited increased PR interval (Figure 3b), as was expected following water immersion; however, half of control rats responded with severely elevated PR interval. Decreased HR (Figure 3a) directly correlated with increased in PR interval suggesting that bradycardia and sinking may, in part, be a reflection of elevated vagal activity.
To evaluate whether the increased PR interval and decreased HR observed in sinking rats were a result of cardiac vagal activity, we administered methylatropine nitrate (3.2 mg/kg), a peripherally acting cholinergic antagonist. At the conclusion of the 15 min pretreatment, resting HR was elevated and PR interval was reduced (Table 2). Following the pretreatment time, in the absence of swim stress, the chronotropic effect persisted for at least the next 2 h, whereas the PR interval was only reduced for an additional 30 min. This dose of methylatropine nitrate was found to be the lowest dose capable of decreasing percent sinking from 58 to 25% in naïve, non-ECG rats and resulted in 0/9 ECG rats sinking. Cardiovascular observations revealed that although methylatropine nitrate did not block the development of bradycardia or increased PR interval completely, it did inhibit the development of severely elevated PR interval and extreme bradycardia that was found in 50% of control rats. These data lend further support to the notion that rats sank due to elevated cardiac vagal activity.
One behavior during swim exposure that may affect the expression of vagal activity is the level of immobility exhibited by vehicle rats. Exercise has been shown to decrease vagal activity both in normoxia and hypoxia (Buchheit et al, 2004). Therefore, rats adopting a more immobile posture throughout swim stress, as opposed to swimming and climbing, would be more likely to exhibit increased vagal activity. Our studies showed that sinking and nonsinking rats only exhibited differing levels of immobility during the first 15 min of swim; however, sinking rats remained less immobile during the first 15 min of swim (Table 1), despite exhibiting elevated vagal activity shortly thereafter (Figure 3b). From these data, we concluded that variations in immobility did not contribute to the differences in HR and PR interval among vehicle sinking and nonsinking rats.
Previous literature has also proposed a relationship between CRF and vagal activity; i.c.v. administration of CRF antagonists resulted in elevated PQ interval (also a measure of vagal activity) during conditioned fear compared with control (Nijsen et al, 2000). CRF antagonist-induced increases in PQ interval during conditioned fear were blocked by pretreating rats with methylatropine nitrate. These data also support the role of the vagus nerve in elevating PQ or PR interval in response to CRF antagonists. Therefore, in our studies we evaluated CRF antagonists’ effects on percent sinking. Antalarmin (Hsin et al, 2002; Webster et al, 1996) and R121919 (Heinrichs et al, 2002), centrally acting pyrrolopyrimidine CRF-R1 selective antagonists, both blocked swim stress-induced release of ACTH (Figure 1a and c). Additionally, their administration increased the percentage of rats sinking during swim stress (Figure 2a and c). Evaluation of the cardiovascular effects of antalarmin during water immersion revealed a shift in the distribution of cardiovascular changes compared with vehicle; all rats exhibited robust elevations in PR interval during swim exposure (Figure 4b). In fact, slight, yet significant elevations in PR interval were also observed in the absence of swim stress; subsequent to the one hour pretreatment, PR interval was elevated from 50±1 to 56±1 ms. Although these major elevations in vagal activity were present only during the pretreatment hour, throughout the next 2 h, PR interval remained elevated by 2.7±0.6 ms. Antalarmin treated rats also exhibited severe bradycardia during swim stress (Figure 4a). The behavioral and cardiovascular effects elicited by antalarmin at rest and during swim stress were also antagonized by methylatropine nitrate as was reported in previous conditioned fear studies (Nijsen et al, 2000). These data suggest strongly that CRF antagonists increase swim stress-induced vagal activity and, as a result, facilitate sinking.
Although antalarmin (Webster et al, 1996) and R121919 (Heinrichs et al, 2002) both cross the blood–brain barrier to alter central CRF-1 receptors, we conducted additional studies to confirm that the cardiovascular effects of CRF antagonists are mediated via central receptors. Peripheral administration of astressin B, a peripherally acting CRF 1, 2 receptor antagonist with a long duration of action (Rivier et al, 1999), significantly decreased ACTH (at the level of the pituitary) but had no effect on percent sinking (Figures 1e and 2e). Our ECG data also confirmed that peripherally administered astressin B had no significant effect on PR interval (data not shown) or heart rate (Table 1) compared to vehicle. Astressin B administered centrally (i.c.v.) also produced a trend to decrease ACTH (Figure 1f), indicating blockade of central CRF receptors, and elicited behavioral changes as the highest dose of astressin B produced 100% sinking (Figure 2f). These data suggest that the CRF antagonist-induced cardiovascular effects are mediated via central CRF receptors.
The CRF antagonist results described herein lend support to the hypothesis that central CRF-1 receptor blockade leads to increased vagal activation during swim stress. To challenge the notion that endogenous, central CRF receptor activation may protect against severe elevations in cardiac vagal activity, thus preventing sinking, we administered RU486, a glucocorticoid receptor (GR) antagonist. Active doses of RU486 are suggested to block the HPA axis’ negative feedback associated with the GR, allowing CRF to elevate over that of normal stress-induced release. In these studies, doses that significantly blocked negative feedback were measured by elevated postswim ACTH levels (Figure 1d). RU486 (20 mg/kg) decreased the percentage of rats sinking to 25% in naïve rats (Figure 2d) and to 0/9 ECG rats. Cardiovascular measurements revealed that, similar to methylatropine nitrate treatment, RU486 did not inhibit the modest increase in PR interval or bradycardia that develop as a result of water immersion but rather, it blocked the progression of severe bradycardia and extreme vagal hyperactivity (Figure 4c and d). RU486 maintained these profound effects despite the fact that, unlike methylatropine nitrate, it did not alter PR interval or HR under resting conditions. These data suggest that CRF receptors may modulate vagal activity during stress; moreover, increased CRF-1 receptor activation may inhibit the development of severely elevated cardiac vagal activity.
The present data demonstrate that sinking during forced swim in 25°C water is facilitated by bradycardia stimulated by severely elevated vagal activity and not by differences in immobility or variations in core body temperature. Although further cardiovascular analysis is necessary, this model may prove a valuable tool to better investigate the mechanism and possible treatments for stress-induced alterations in vagal activity. This model would benefit from additional measurements of vagal activity and blood pressure analysis during sinking. Evaluation of central CRF-R1 antagonists’ cardiovascular effects confirms their role in elevating cardiac vagal activity and generating severe bradycardia. Furthermore, endogenous CRF may also play a role in protecting against robust stress-induced vagal elevations.
Acknowledgments
We would like to thank Dr Jean Rivier and his laboratory (Salk Institute; La Jolla, CA) for the generous donation of astressin B used in these studies. This research was supported by USPHS Grants DA14349 and GM07767.
References
- Agelink MW, Boz C, Ullrich H, Andrich J. Relationship between major depression and heart rate variability. Clinical consequences and implication for antidepressive treatment. Psych Res. 2002;113:139–149. doi: 10.1016/s0165-1781(02)00225-1. [DOI] [PubMed] [Google Scholar]
- Angrilli A, Sarlo M, Palomba D, Schincaglia M, Stegagno L. Respiratory sinus arrhythmia in blood phobic subjects. Percept Mot Skills. 1997;84:505–506. doi: 10.2466/pms.1997.84.2.505. [DOI] [PubMed] [Google Scholar]
- Baker MA, Horvath SM. Influence of water temperature on heart rate and rectal temperature of swimming rats. Am J Physiol. 1964;207:1073–1076. doi: 10.1152/ajplegacy.1964.207.5.1073. [DOI] [PubMed] [Google Scholar]
- Briscoe RJ, Cabrera CL, Baird TJ, Rice KC, Woods JH. Antalarmin blockade of corticotropin releasing hormone-induced hypertension in rats. Brain Res. 2000;881:204–207. doi: 10.1016/s0006-8993(00)02742-6. [DOI] [PubMed] [Google Scholar]
- Buchheit M, Richard R, Doutreleau S, Lonsdorfer-Wolf E, Brandenberger G, Simon C. Effect of acute hypoxia on heart rate variability at rest and during exercise. Int J Sports Med. 2004;25:264–269. doi: 10.1055/s-2004-819938. [DOI] [PubMed] [Google Scholar]
- Castro JL, Ricci D, Taira CA, Ramirez A. Central benzodiazepine involvement in clonidine cardiovascular actions. Can J Physiol Pharmacol. 1999;77:844–851. [PubMed] [Google Scholar]
- Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: Comparison with CRF1 receptor mRNA expression. J Neurosci. 1995;15:6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers DT, Lovenberg TW, Grigoriadis DE, Behan DP, De Souza EB. Corticotropin-releasing factor receptors: from molecular biology to drug design. Trends Pharmacol Sci. 1996;17:166–172. doi: 10.1016/0165-6147(96)81594-x. [DOI] [PubMed] [Google Scholar]
- Cook CJ. Glucocorticoid feedback increases the sensitivity of the limbic system to stress. Phys Behav. 2002;75:755–764. doi: 10.1016/s0031-9384(02)00650-9. [DOI] [PubMed] [Google Scholar]
- Dal-Zotto S, Marti O, Armario A. Glucocorticoids are involved in the long-term effects of a single immobilization stress on the hypothalamic-pituitary-adrenal axis. Psychoneuro-endocrinology. 2003;28:992–1009. doi: 10.1016/s0306-4530(02)00120-8. [DOI] [PubMed] [Google Scholar]
- Das G, Talmers FN, Weissler AM. New observations of the effects of atropine on the sinoatrial and atrioventricular nodes in man. Am J Cardiol. 1975;36:281–285. doi: 10.1016/0002-9149(75)90476-2. [DOI] [PubMed] [Google Scholar]
- Dawson CA, Nadel ER, Horvath SM. Cardiac output in the cold-stressed swimming rat. Am J Physiol. 1968;214:320–325. doi: 10.1152/ajplegacy.1968.214.2.320. [DOI] [PubMed] [Google Scholar]
- Deak T, Nguyen KT, Ehrlich AL, Watkins LR, Spencer RL, Maier SF, et al. The impact of the nonpeptide corticotropin-releasing hormone antagonist antalarmin on behavioral and endocrine responses to stress. J Endocrinol. 1999;140:79–86. doi: 10.1210/endo.140.1.6415. [DOI] [PubMed] [Google Scholar]
- Detke MJ, Rickels M, Lucki I. Active behaviors in the rat forced swimming test differentially produced by serotonergic and noradrenergic antidepressants. J Psychopharmacol. 1995;121:66–72. doi: 10.1007/BF02245592. [DOI] [PubMed] [Google Scholar]
- Fisher LA. Central autonomic modulation of cardiac baroreflex by corticotropin-releasing factor. Am J Physiol. 1989;256:H949–H955. doi: 10.1152/ajpheart.1989.256.4.H949. [DOI] [PubMed] [Google Scholar]
- Friedman BH, Thayer JF, Borkovec TD, Tyrrell RA, Johnson BH, Columbo R. Autonomic characteristics of nonclinical panic and blood phobia. Biol Psychiatry. 1993;34:293–310. doi: 10.1016/0006-3223(93)90087-t. [DOI] [PubMed] [Google Scholar]
- Gardiner SM, March JE, Kemp PA, Davenport AP, Wiley KI, Bennett T. Regional hemodynamic actions of selective corticotropin-releasing factor type 2 receptor ligands in conscious rats. J Pharmacol Exp Ther. 2005;312:53–60. doi: 10.1124/jpet.104.075259. [DOI] [PubMed] [Google Scholar]
- Hartel G. Psychological factors in cardiac arrhythmias. Ann Clin Res. 1987;19:104–109. [PubMed] [Google Scholar]
- Heinrichs SC, De Souza EB, Schulteis G, Lapsansky JL, Grigoriadis DE. Brain penetrance, receptor occupancy and antistress in vivo efficacy of a small molecule corticotropin releasing factor type I receptor selective antagonist. Neuropsychopharmacology. 2002;27:194–202. doi: 10.1016/S0893-133X(02)00299-3. [DOI] [PubMed] [Google Scholar]
- Hood WB, Murray RH, Urschel CW, Bowers JA, Goldman J. Circulatory effects of water immersion upon human subjects. Aerosp Med. 1968;39:579–584. [PubMed] [Google Scholar]
- Hsin LW, Tian X, Webster EL, Coop A, Caldwell TM, Jacobson AE, et al. CRHR1 receptor binding and lipophilicity of pyrrolopyrimidines, potential nonpeptide corticotropin-releasing hormone type 1 receptor antagonists. Bioorg Med Chem. 2002;10:175–183. doi: 10.1016/s0968-0896(01)00261-9. [DOI] [PubMed] [Google Scholar]
- Johnsen BH, Thayer JF, Laberg JC, Wormnes B, Raadal M, Skaret E, et al. Attentional and physiological characteristics of patients with dental anxiety. Anxiety Disord. 2003;17:75–87. doi: 10.1016/s0887-6185(02)00178-0. [DOI] [PubMed] [Google Scholar]
- Jutkiewicz EM, Wood SK, Houshyar H, Hsin LW, Rice KC, Woods JH. The effects of CRF antagonists, antalarmin, CP154,526, LWH234, and R121919 in the forced swim test and on swim-induced increases in adrenocorticotropin in rats. J Psychopharmacol. 2005;180:215–223. doi: 10.1007/s00213-005-2164-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawachi I, Colditz GA, Ascherio A, Rimm EB, Giovannucci E, Stampher MJ, et al. Prospective study of phobic anxiety and risk of coronary heart disease in men. Circulation. 1994;89:1992–1997. doi: 10.1161/01.cir.89.5.1992. [DOI] [PubMed] [Google Scholar]
- Kinoshita S, Konishi G. Mechanisms of atypical atrioventricular Wenckelbach periodicity. A theroretical model derived from the concepts of inhomogeneous excitability and electrotonically mediated conduction. J Electrocardiol. 1989;22:227–233. doi: 10.1016/0022-0736(89)90033-2. [DOI] [PubMed] [Google Scholar]
- Kosoyan HP, Wei JY, Tache Y. Intracisternal sauvagine is more potent than corticotropin-releasing factor to decrease gastric vagal efferent activity in rats. Peptides. 1999;20:851–858. doi: 10.1016/s0196-9781(99)00072-8. [DOI] [PubMed] [Google Scholar]
- Levy MN, Zieske H. Autonomic control of cardiac pacemaker activity and atrioventricular transmission. J App Physiol. 1969;27:465–470. doi: 10.1152/jappl.1969.27.4.465. [DOI] [PubMed] [Google Scholar]
- Lovenberg TW, Chalmers DT, Liu C, De Souza EB. CRF2 alpha and CRF2 beta receptor mRNAs are differentially distributed between the rat central nervous system and peripheral tissues. J Endocrinol. 1995;136:4139–4142. doi: 10.1210/endo.136.9.7544278. [DOI] [PubMed] [Google Scholar]
- Mackay KB, Stiefel TH, Ling N, Foster AC. Effects of a selective agonist and antagonist of CRF2 receptors on cardiovascular function in the rat. Eur J Pharmacol. 2003;469:111–115. doi: 10.1016/s0014-2999(03)01725-4. [DOI] [PubMed] [Google Scholar]
- Miwa C, Sugiyama Y, Mano T, Iwase S, Matsukawa T. Sympathovagal responses in humans to thermoneutral head-out water immersion. Aviat Space Enivron Med. 1997;68:1109–1114. [PubMed] [Google Scholar]
- Nahshoni E, Gur S, Marom S, Levin JB, Weizman A, Hermesh H. QT dispersion in patients with social phobia. J Affect Disord. 2004;78:21–26. doi: 10.1016/s0165-0327(02)00052-6. [DOI] [PubMed] [Google Scholar]
- Negrao CE, Moreira ED, Brum PC, Denadai ML, Krieger EM. Vagal and sympathetic control of heart rate during exercise by sedentary and exercise trained rats. Braz J Med Biol Res. 1992;25:1045–1052. [PubMed] [Google Scholar]
- Nijsen MJMA, Croiset G, Diamant M, Stam R, Kamphuis PJGH, Bruijnzeel A, et al. Endogenous corticotropin-releasing hormone inhibits conditioned fear-induced vagal activation in the rat. Eur J Pharmacol. 2000;389:89–98. doi: 10.1016/s0014-2999(99)00870-5. [DOI] [PubMed] [Google Scholar]
- Nishimura H, Tsuda A, Oguchi M, Ida Y, Tanaka M. Is immobility in the forced swim test ‘behavioral despair?’. Physiol Behav. 1988;42:93–95. doi: 10.1016/0031-9384(88)90266-1. [DOI] [PubMed] [Google Scholar]
- Overton J, Fisher L. Differentiated hemodynamic responses to central versus peripheral administration of corticotropin-releasing factor in conscious rats. J Autonom Nerv Syst. 1991;35:43–52. doi: 10.1016/0165-1838(91)90037-4. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; New York: 1982. [Google Scholar]
- Perlstein I, Stepensky D, Krzyzanski W, Hoffman A. A signal transduction pharmacodynamic model of the kinetics of the parasympathomimetic activity of low-dose scopolamine and atropine in rats. J Pharm Sci. 2002;92:2500–2510. doi: 10.1002/jps.10243. [DOI] [PubMed] [Google Scholar]
- Richter CP. On the phenomenon of sudden death in animals and man. J Psychosom Med. 1957;19:191–197. doi: 10.1097/00006842-195705000-00004. [DOI] [PubMed] [Google Scholar]
- Richter RM, Mulvany MJ. Comparison of hCRF and oCRF effects on cardiovascular responses after central, peripheral, and in vitro application. Peptides. 1995;16:843–849. doi: 10.1016/0196-9781(95)00035-i. [DOI] [PubMed] [Google Scholar]
- Rivier CL, Grigoriadis DE, Rivier JE. Role of corticotropin-releasing factor receptors type 1 and 2 in modulating the rat adrenocorticotropin response to stressors. J Endocrinol. 2003;144:2396–2403. doi: 10.1210/en.2002-0117. [DOI] [PubMed] [Google Scholar]
- Rivier JE, Kirby DA, Lahrichi SL, Corrigan A, Vale WW. Constrained corticotrophin releasing factor (CRF) antagonists (astressin analogues) with long duration of action in the rat. J Med Chem. 1999;42:3175–3182. doi: 10.1021/jm9902133. [DOI] [PubMed] [Google Scholar]
- Schipke JD, Pelzer M. Effect of immersion, submersion, and scuba diving on heart rate variability. Br J Sports Med. 2001;35:174–180. doi: 10.1136/bjsm.35.3.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgoifo A, Koolhaas JM, Musso E, De Boer SF. Different sympathovagal modulation of heart rate during social and nonsocial stress episodes in wild-type rats. Phys Behav. 1999;67:733–738. doi: 10.1016/s0031-9384(99)00134-1. [DOI] [PubMed] [Google Scholar]
- Smith RF, Stanton K, Stoop D, Janusz W, King PH. Quantitative electrocardiography during extended space flight: the second manned skylab mission. Aviat Space Environ Med. 1976;47:353–359. [PubMed] [Google Scholar]
- Wang L, Cardin S, Martinez V, Tache Y. Intracerebroventricular CRF inhibits cold restraint-induced c-fos expression in the dorsal motor nucleus of the vagus and gastric erosions in rats. Brain Res. 1996;736:44–53. doi: 10.1016/0006-8993(96)00726-3. [DOI] [PubMed] [Google Scholar]
- Watkins LL, Grossman P, Krishnan R, Sherwood A. Anxiety and vagal control of heart rate. J Psychosom Med. 1998;60:498–502. doi: 10.1097/00006842-199807000-00018. [DOI] [PubMed] [Google Scholar]
- Webster EL, Lewis DB, Torpy DJ, Zachman EK, Rice KC, Chrousos GP. In vivo and in vitro characterization of antalarmin, a nonpeptide corticotropin-releasing hormone (CRH) receptor antagonist: suppression of pituitary ACTH release and peripheral inflammation. J Endocrinol. 1996;137:5747–5750. doi: 10.1210/endo.137.12.8940412. [DOI] [PubMed] [Google Scholar]
- Wiersma A, Bohus B, Koolhaas JM. Corticotropin-releasing hormone microinfusion in the central amygdala diminishes a cardiac parasympathetic outflow under stress-free conditions. Brain Res. 1993;625:219–227. doi: 10.1016/0006-8993(93)91062-w. [DOI] [PubMed] [Google Scholar]
- Wittstein IS, Thiemann DR, Lima JAC, Baughman KL, Schulman SP, Gerstenblith G, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. 2005;352:539–548. doi: 10.1056/NEJMoa043046. [DOI] [PubMed] [Google Scholar]
- Yeragani VK, Rao KA, Pohl R, Jampala VC, Balon R. Heart rate and QT variability in children with anxiety disorders: A preliminary report. Depress Anxiety. 2001;13:72–77. doi: 10.1002/da.1019. [DOI] [PubMed] [Google Scholar]
- Yeragani VK, Rao KA, Smitha M, Pohl RB, Balon R, Srinivasan K. Diminished chaos of heart rate time series in patients with major depression. Biol Psychiatry. 2002;51:733–744. doi: 10.1016/s0006-3223(01)01347-6. [DOI] [PubMed] [Google Scholar]