

Abstract
Alpha-synuclein is implicated in several neurodegenerative disorders, such as Parkinson’s disease and multiple system atrophy, yet its functions remain obscure. When expressed in yeast, alpha-synuclein associated with the plasma membrane in a highly selective manner, before forming cytoplasmic inclusions through a concentration-dependent, nucleated process. Alpha-synuclein inhibited phospholipase D, induced lipid droplet accumulation, and affected vesicle trafficking. This readily manipulable system provides an opportunity to dissect the molecular pathways underlying normal alpha-synuclein biology and the pathogenic consequences of its misfolding.
Alpha-synuclein (αSyn) is abundant and broadly expressed in the brain, where it interacts with membranes, vesicular structures, and a puzzling variety of other proteins (1). Some cases of Parkinson’s disease (PD) have a genetic basis (2) that implicates protein folding and quality-control (QC) factors, including a ubiquitin ligase (3) and a ubiquitin C-terminal hydrolase (4, 5), in αSyn pathology. In mammalian cells αSyn has been reported in the nucleus, cytosol, associated with membranes and, in diseased brains, in large cytoplasmic inclusions (Lewy bodies) (1). Synucleinopathies are now classified as protein-misfolding disorders (6). Given the strong conservation of protein folding, membrane trafficking, and protein QC mechanisms between yeast and higher eukaryotes, we used Saccharomyces cerevisiae to uncover and establish basic aspects of both normal and abnormal αSyn biology.
To study αSyn dynamics in living cells, we created an αSyn-GFP (green fluorescent protein) fusion that was not subject to proteolysis in yeast cells (Fig. 1A) (7) as related fusions have been in mammalian cells (8). Integrating this construct into the genome under the control of a galactose-inducible promoter allowed routine manipulations in the absence of αSyn expression. Upon induction with galactose, wild-type (WT) αSyn-GFP localized intensely at the plasma membrane; a smaller quantity accumulated in the cytoplasm (Fig. 1B). Compared with other GFP fusion proteins, αSyn did not localize to mitochondrial or nuclear membranes (9). Thus, reminiscent of its selectivity for membranes with particular lipid compositions in vitro (10), αSyn has a high intrinsic selectivity for particular cellular membranes in vivo.
Fig. 1.
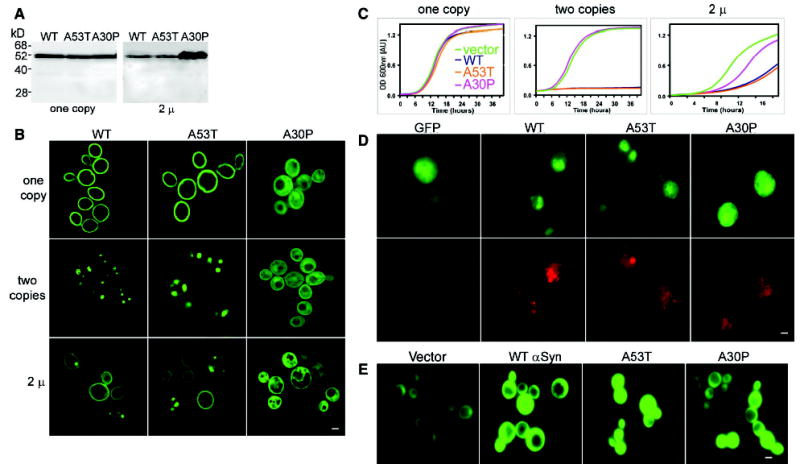
Expression of αSyn in yeast. (A) Immunoblot analysis of cells expressing αSyn-GFP fusions. Single integrated copies (left) of WT, A53T, and A30P αSyn produced similar amounts of protein (supporting online material). 2μ plasmid (variable copy number) expression levels of WT and A53T were similar but lower than those of A30P αSyn (right). (B) Fluorescence microscopy of yeast cells expressing αSyn-GFP. In cells carrying one copy of WT or A53T, the protein concentrated at the plasma membrane, and small amounts concentrated in the cytoplasm. Cells with two copies of WT or A53T showed cytoplasmic inclusions and reduced membrane localization. When expressed from the 2μ plasmid, WT and A53T distributions varied from cell to cell, whereas A30P showed a diffuse cytoplasmic distribution. (C) αSyn inhibits growth. One copy of αSyn WT or A53T had little effect on growth (left), whereas two copies completely inhibited it (middle). When expressed from a 2μ plasmid, A30P inhibited growth but less so than WT or A53T (right). (D) Immunofluorescence of cells expressing αSyn-GFP (2μ) (top) with an antibody to ubiquitin (bottom) showed increased levels of ubiquitin accumulation in cells expressing αSyn ( WT and both mutants). (E) Cells coexpressing αSyn and GFPu showed accumulation of the reporter when compared to cells carrying an empty vector control. Scale bars, 1 μm.
Two αSyn point mutants (A53T and A30P) are associated with rare forms of early-onset familial PD but have distinct physical properties (11, 12). In yeast, each αSyn mutant accumulated at the same level as WT αSyn (Fig. 1A, one copy), but their cellular distributions differed profoundly: Like WT αSyn, A53T concentrated at the plasma membrane; A30P dispersed throughout the cytoplasm (Fig. 1B, one-copy). The effects of the A30P mutation on membrane association in yeast are consistent with its poor membrane-binding capacity in other systems (13, 14).
One hypothesis to explain the late onset of PD (and several other misfolding diseases) is that disease occurs in aging neurons when the capacity of the QC system to cope with accumulating misfolded proteins is exceeded (15, 16). It should be possible to mimic this situation, and exceed the QC system of yeast, by increasing αSyn expression. Indeed, simply doubling αSyn expression, by integrating a second copy in the genome, profoundly changed its fate: The vast majority of αSyn appeared in large cytoplasmic inclusions (Fig. 1B). Notably, inclusions were not formed by excess protein unable to find “docking sites” at the membrane: (i) Much less protein was membrane-localized in two-copy strains than in single-copy strains (Fig. 1B); and (ii) in time courses with two-copy strains, αSyn first accumulated at the membrane and was later recruited away into cytoplasmic inclusions. The distribution of A53T was similar to that of the WT, but an even smaller fraction associated with membranes. A30P maintained its cytoplasmic distribution (Fig. 1B, two-copies). In vitro, purified αSyn undergoes nucleated polymerization, A53T having a greater and A30P a lesser propensity to form large polymers than WT αSyn (17). Our studies demonstrate that the recruitment of αSyn to inclusion bodies is a biologically relevant property in living cells, where total protein concentrations are very high (~300 mg/ml) and αSyn concentrations are low (below detection on stained gels) (9).
Under growth conditions used for microscopy, a single-copy of WT or A53T αSyn had no appreciable effect on growth. Two copies completely inhibited growth (Fig. 1C and fig. S1). A30P inhibited growth only when expressed from a high-copy (2μ) plasmid (Fig. 1C). Curiously, cells were able to grow with 2μ plasmids expressing WT and A53T. Further analysis demonstrated that selective pressures had reduced expression by reducing the average copy number of WT and A53T plasmids, confirming the strong concentration dependence of their toxicity (Fig. 1A) (9). In this and all other assays (9) (supporting online material), similar results were obtained with αSyn constructs not fused to GFP.
At various times after galactose induction, cells were plated on glucose to repress further expression and determine the number of viable cells. The vast majority of cells (~90%) retained colony-forming ability after 12 hours. This lag between αSyn induction and cell death afforded an opportunity to study the protein’s biological effects before toxicity confounded results.
To investigate interactions between αSyn and the QC system, we first stained cells for ubiquitin, a highly conserved polypeptide that is posttranslationally attached to misfolded proteins to mark them for degradation (18). We used the extrachromosomal plasmids to create mixed populations, perfectly matched for growth and culture conditions but expressing αSyn at different levels in different cells. Direct visualization of αSyn-GFP showed the expected diversity of localization patterns from cell to cell (membranes and cytosolic inclusions) (Fig. 1B, 2μ; fig. S2). After fixation and detergent permeabilization (required for immunological detection of ubiquitin), membrane fluorescence was greatly reduced (Fig. 1D; figs. S2 and S3), demonstrating the advantage of direct GFP localization. Nevertheless, the preparations were adequate to unambiguously detect strong accumulation of ubiquitin (Fig. 1D) and establish that it varied with αSyn expression on a cell-by-cell basis. As in mammalian cells and human brains, some but not all αSyn aggregates stained with antibodies to ubiquitin (19, 20). Thus, ubiquitylation was neither required for aggregation nor prevented it (Fig. 1D). We also investigated whether accumulating misfolded αSyn impaired the capacity of the QC system to degrade other proteins. We used an unstable GFP derivative, GFPu, previously established as a reporter for general proteasome activity and αSyn constructs not fused to GFP (21). All three αSyn proteins caused GFPu accumulation, indicating proteasome impairment (Fig. 1E).
Fig. 2.
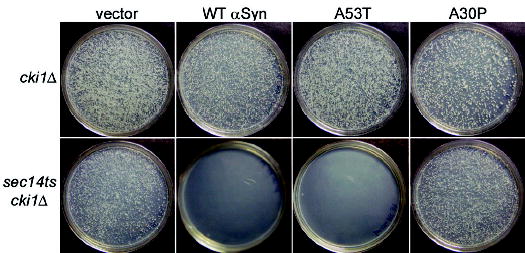
αSyn inhibits PLD. A cki1Δ strain (top) or a sec14ts cki1Δ double-mutant strain (bottom) were transformed with 2μ plasmids expressing αSyn and grown at 37°C (supporting online material). WT and A53TαSyn blocked growth in sec14ts cki1Δ strain, but not in the cki1Δ strain.
Next, we examined properties of αSyn that have been postulated but not directly tested: participation in the regulation of cellular lipid metabolism and vesicle trafficking (22, 23). In a search for inhibitors of phospholipase D (PLD) activity, αSyn was serendipitously found to potently and selectively inhibit mammalian PLD in vitro (24). To determine if αSyn inhibits PLD in vivo, we took advantage of a previously established relation between PLD, Sec14p (a phosphatidylinositol-phosphatidylcholine transfer protein), and Cki1p (a choline kinase) (25). When expressed at a level that had no effect in cki1Δ cells, αSyn completely blocked growth in cki1Δ sec14ts double mutants (Fig. 2). This suggested that αSyn had inhibited endogenous yeast PLD activity. A53T had the same effect; A30P did not. Cells carrying the sec14ts mutation alone can grow at 37°C by acquiring any of several “bypass mutations” (26). PLD mutations block the ability of bypass mutations to rescue growth; expression of αSyn did the same (25, 27) (fig. S4B). Overexpressing PLD (from an extrachromosomal plasmid) restored bypass rescue (fig. S4A), confirming that the effects of αSyn in these assays were due to the inhibition of PLD. A mutant huntingtin exon-1 fragment with 103 glutamines (Q103 Htt) did not inhibit PLD (fig. S4C). Thus, the serendipitously discovered ability of αSyn to inhibit PLD in vitro is a specific, highly conserved, and biologically relevant property of the protein in vivo.
αSyn shares biophysical properties with the broadly distributed family of fatty acid–binding proteins, and its oligomerization is affected by lipids in vitro (28, 29). In mammalian cells cultured with lipid-enriched medium, αSyn accumulates on lipid monolayers surrounding triglyceride-rich lipid droplets and promotes their accumulation by protecting them from hydrolysis (23). We used Nile red, a fluorescent dye that preferentially binds neutral lipids such as triglycerides, to assess the effect of αSyn on lipid accumulation. Cells expressing WT αSyn and A53T stained much more strongly than did cells expressing A30P, even in the absence of extrinsic lipids (Fig. 3A, bottom). Electron microscopy established that WT αSyn and A53T caused the accumulation of discrete lipid droplets; A30P did not (Fig. 3B).
Fig. 3.
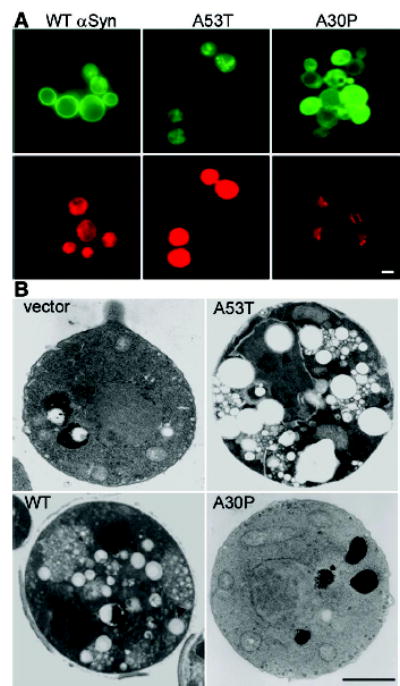
Cells expressing αSyn accumulate lipids. (A) Cells expressing either WT or A53T αSyn-GFP (top) were highly reactive for the lipophylic dye Nile red (bottom); cells expressing A30P were not (supporting online material). (B) Electron microscopy demonstrated the accumulation of lipid droplets (arrowheads) in cells expressing WT or A53T αSyn but not in cells expressing A30P. Scale bars, 1 μm.
Finally, we tested αSyn’s effect on the status of vesicular bodies by monitoring internalization of the membrane-binding fluorescent dye FM4-64. As expected, in cells not expressing αSyn, FM4-64 was rapidly endocytosed and accumulated at the vacuolar membrane (large ringlike structure in Fig. 4, top). In cells expressing any of the three αSyns (WT, A53T, or A30P), FM4-64 distribution was profoundly affected (Fig. 4, bottom). Again, this was not simply the result of protein aggregation or toxicity: The dye accumulated at the vacuolar membrane in cells expressing low, but similarly toxic levels of an aggregation-prone Q103 Htt (Fig. 4) (supporting online material). Notably, the defect in FM4-64 localization was due neither to PLD inhibition nor to lipid droplet accumulation because, in contrast with other assays with the same plasmids, A30P affected this phenomenon as strongly as WT and A53T did.
Fig. 4.
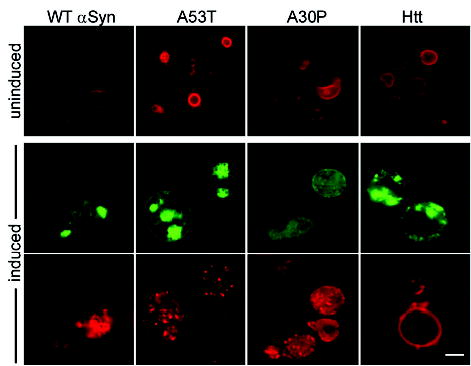
αSyn overexpression perturbs the distribution of vesicular pools. Endocytosis of the fluorescent dye FM4-64 (red) was used to monitor the effects of all three αSyn-GFP variants and of Q103 Htt exon 1 (green) on vesicular trafficking. Cells grown in raffinose (uninduced) show normal ring-like vacuolar staining (top). Expression of αSyn WT, A53T, and A30P markedly altered FM4-64 distributions (punctate structures in red, bottom). Scale bar, 1 μm.
Expressing αSyn and its mutant derivatives in yeast provides a model for studying normal αSyn function and its misfunction in synucleinopathies. We established that the ability of the protein to inhibit PLD, promote the accumulation of lipids, influence the balance of vesicular pools, associate with membranes in a highly selective manner, induce ubiquitin accumulation, and inhibit the proteasome when misfolded are all intrinsic and biologically relevant properties of the protein that can be uncoupled from each other by the effects of αSyn mutations. Constructs expressing mutant Q103 Htt did not produce similar biological effects (supporting online material), and unbiased genetic screens confirmed that distinct pathways are involved in αSyn and Htt toxicities (30). Notably, membrane-bound αSyn is in dynamic equilibrium with cytoplasmic forms. Just a twofold difference in expression was sufficient to cause a catastrophic change in its behavior, inducing nucleated polymerization and recruiting protein previously associated with membranes to cytoplasmic inclusions. This nucleated polymerization process suggests a mechanism by which even small changes in the QC balance of aging neurons could produce a toxic gain of αSyn function concomitantly with a loss of normal function. Thus, two hypotheses [gain of toxic function or loss of normal function (31)] put forward to explain PD can be reconciled by a single molecular mechanism.
Supplementary Material
Footnotes
Supporting Online Material
Note added in proof: Very recently Singleton et al. (32) reported that a triplication of the αSyn locus on one chromosome (presumably doubling the expression of wild-type αSyn) causes premature onset of PD, strongly supporting our model that a small change in the expression of αSyn relative to the cell’s quality-control systems causes disease-related toxicity.
References
- 1.Lucking CB, Brice A. Cell Mol Life Sci. 2000;57:1894. doi: 10.1007/PL00000671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gasser T. J Neurol. 2001;248:833. doi: 10.1007/s004150170066. [DOI] [PubMed] [Google Scholar]
- 3.Kitada T, et al. Nature. 1998;392:605. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 4.Leroy E, et al. Nature. 1998;395:451. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT., Jr Cell. 2002;111:209. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- 6.Muchowski PJ. Neuron. 2002;35:9. doi: 10.1016/s0896-6273(02)00761-4. [DOI] [PubMed] [Google Scholar]
- 7.Materials and methods, additional data, and other supporting materials concerning data not shown are available on Science Online.
- 8.McLean PJ, Kawamata H, Hyman BT. Neuroscience. 2001;104:901. doi: 10.1016/s0306-4522(01)00113-0. [DOI] [PubMed] [Google Scholar]
- 9.T. F. Outeiro, S. Lindquist, data not shown.
- 10.Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE. J Biol Chem. 2000;275:34328. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 11.Polymeropoulos MH, et al. Science. 1997;276:2045. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 12.Kruger R, et al. Nature Genet. 1998;18:106. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 13.Jo E, Fuller N, Rand RP, St George-Hyslop P, Fraser PE. J Mol Biol. 2002;315:799. doi: 10.1006/jmbi.2001.5269. [DOI] [PubMed] [Google Scholar]
- 14.Bussell R, Jr, Eliezer D. J Mol Biol. 2003;329:763. doi: 10.1016/s0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
- 15.Berke SJ, Paulson HL. Curr Opin Genet Dev. 2003;13:253. doi: 10.1016/s0959-437x(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 16.Taylor JP, Hardy J, Fischbeck KH. Science. 2002;296:1991. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 17.Conway KA, et al. Proc Natl Acad Sci USA. 2000;97:571. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hershko A, Ciechanover A, Varshavsky A. Nature Med. 2000;6:1073. doi: 10.1038/80384. [DOI] [PubMed] [Google Scholar]
- 19.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. J Neurosci. 2001;21:9549. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sampathu DM, Giasson BI, Pawlyk AC, Trojanowski JQ, Lee VM. Am J Pathol. 2003;163:91. doi: 10.1016/s0002-9440(10)63633-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bence NF, Sampat RM, Kopito RR. Science. 2001;292:1552. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 22.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. J Neurosci. 2000;20:3214. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole NB, et al. J Biol Chem. 2002;277:6344. doi: 10.1074/jbc.M108414200. [DOI] [PubMed] [Google Scholar]
- 24.Jenco JM, Rawlingson A, Daniels B, Morris AJ. Biochemistry. 1998;37:4901. doi: 10.1021/bi972776r. [DOI] [PubMed] [Google Scholar]
- 25.Bankaitis VA, Aitken JR, Cleves AE, Dowhan W. Nature. 1990;347:561. doi: 10.1038/347561a0. [DOI] [PubMed] [Google Scholar]
- 26.Rudge SA, Pettitt TR, Zhou C, Wakelam MJ, Engebrecht JA. Genetics. 2001;158:1431. doi: 10.1093/genetics/158.4.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sreenivas A, Patton-Vogt JL, Bruno V, Griac P, Henry SA. J Biol Chem. 1998;273:16635. doi: 10.1074/jbc.273.27.16635. [DOI] [PubMed] [Google Scholar]
- 28.Sharon R, et al. Proc Natl Acad Sci USA. 2001;98:9110. doi: 10.1073/pnas.171300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharon R, et al. Neuron. 2003;37:583. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 30.Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Science. 2003;302:1769. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- 31.Dawson TM, Dawson VL. J Clin Invest. 2003;111:145. doi: 10.1172/JCI17575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singleton AB, et al. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 33.This work was funded by NIH/National Institute of Neurological Disorders and Stroke (grant NS044829-01). T.F.O. was partially supported by Programa Prax-is XXI, Fundacao para a Ciencia e Tecnologia, Portugal. We thank P. Lansbury, R. Esposito, J. Engebrecht, H. Chang, and S. Henry for plasmids and strains and members of the Lindquist laboratory for critical reading of this manuscript.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.