

Abstract
In this issue of Immunity, Hatton et al. (2006) identify evolutionarily conserved non-coding sequences (CNSs) upstream of the interferon-γ gene, then show using a BAC transgene reporter that CNS-22 is a key regulator of interferon-γ expression.
CD4 T cells adopt a diverse set of functional phenotypes each of which makes a unique contribution to proper immunity. Th1 and Th2 CD4 T cells were defined some time ago and shown to thwart infection by intracellular bacteria and viruses, and extracellular parasites, respectively. More recently regulatory T cells (Treg), which moderate adaptive immune responses, and Th17 CD4 T cells, which are thought to protect against extracellular bacteria, have been defined. The choice between these lineages is made in response to the environmental conditions present when CD4 T cells first encounter antigen. These choices are entrained by lineage-specific transcription factors, which for Th1, Th2, Treg and Th17 cells are T-bet, GATA3, FoxP3 and, based on recent data, ROR γt (Ivanov et al., 2006), respectively. How these ‘master regulators’ of CD4 lineage choice enforce commitment is incompletely understood, due in part to our limited knowledge of the genes and transcriptional regulatory elements to which they bind. Th1, Th2 and Th17 cells are defined by the signature cytokines they express, and these ‘master regulators’ appear to act in part by binding to positive regulatory elements of the genes encoding these cytokines and, perhaps also to negative regulatory elements of the cytokines whose expression is forbidden in that lineage.
In vertebrates, transcription is governed by the promoter and additional regulatory elements located in introns and at distances of 50kb or more upstream or downstream of the target gene. A clear example of this paradigm is the evolutionarily conserved Th2 locus containing the Il4, Il13 and Il5 genes. This locus, like the β-globin locus, has been fertile ground for the discovery of distal transcriptional regulatory elements (Lee et al., 2006), and over the past decade, multiple groups using a variety of computational and experimental approaches have identified enhancers, silencers and locus control regions that collaborate to assure proper expression of the Th2 cytokines.
By contrast, only recently have regulatory elements other than the promoter and introns been identified for Ifng, the gene encoding the signature Th1 cytokine interferon-γ. There is considerable evidence to indicate that distal regulatory are required for proper regulation of Ifng. The core Ifng promoter is T-bet responsive in vitro (Cho et al., 2003) and directs Th1-specific but low level expression of transgenic reporters in mice (Soutto et al., 2002). However, addition of 3.4 kb of 5’ flank to the core promoter leads to non-specific expression of the transgene (Zhu et al., 2001), and addition of introns 1 and 3 or the use of an 8.6 kb genomic human IFNG clone markedly augments expression but abolishes Th1-specificity (Soutto et al., 2002). By contrast, high-level, Th-1 specific expression of human IFNG is observed in mice transgenic for a bacterial artificial chromosome (BAC) containing IFNG and ∼95 kb of upstream and downstream flank (Soutto et al., 2002), indicating that key regulatory elements not contained in the 8.6 kb transgene are present in this BAC. Subsequently, using evolutionary conservation as a guide and Th1-specific DNase hypersensitivity and transcriptionally favorable histone modifications as landmarks, two conserved non-coding sequences (CNSs) that bind T-bet in vivo and enhance Ifng expression in vitro were identified 5.5 kb upstream (CNS1) and 17-19 kb downstream (CNS2) of murine Ifng, respectively (Lee et al., 2004; Shnyreva et al., 2004). Additional CNSs have been recently identified by cross-species comparison of sequences upstream or downstream of Ifng. Some of the newly identified CNSs were shown to lie in regions marked by transcriptionally permissive histone modifications, but their function was not evaluated (Chang and Aune, 2005).
In this issue of Immunity, by comparing sequences up to 60 kb upstream of Ifng in diverse vertebrate species, Hatton et al (2006) also identified CNS1 (which they refer to as CNS-5) and the three additional upstream CNSs first described by Chang and Aune (2005). Using luciferase reporters in vitro, they demonstrated that CNS-22 and CNS-34 are T-bet dependent enhancers when linked to the proximal Ifng promoter, with CNS-22 the stronger of the two (Figure 1a). T-bet bound to both of these CNSs in vivo, as demonstrated by chromatin immunoprecipitation, and also bound weakly to CNS-55 and CNS-5, though they did not detect enhancer activity with these latter two CNSs. The failure to demonstrate enhancer activity for CNS-5 is surprising, since this CNS was previously shown to enhance Ifng expression by two other groups (Lee et al., 2004; Shnyreva et al., 2004); this difference will need to be resolved by further study. A number of features led Hatton et al to focus their attention more closely on CNS-22: 1) T-bet-dependent enhancer activity was lost when a predicted T-bet binding site was mutated; 2) In addition to T-bet, putative binding sites for a number of transcription factors involved in T cell differentiation and cytokine production were found in CNS-22 and conserved across species; 3) The histones at CNS-22 had multiple marks of permissive chromatin not only in Th1 cells but also in naïve and Th2 cells, suggesting that CNS-22 might have multiple context-dependent functions. Then, taking advantage of recent advances that permit the facile manipulation of BACs, they introduced a Thy1.1 reporter into exon 1 of Ifng and placed this reporter into a BAC containing ∼60 kb of upstream and ∼100kb of downstream flanking sequences (Figure 1b). The CNS-22 region of this BAC was then flanked with loxP sites. Two lines of transgenic mice were generated with this BAC, one with a single copy and another with greater than 20 copies. In both lines, the Thy1.1 reporter was expressed only in cells that also expressed endogenous interferon-γ, including Th1 and CD8 T cells and NK cells, following activation via the TCR, ionomycin plus PMA, or IL-12 plus IL-18. Strikingly, when CNS-22 was deleted from the single copy transgene by Cre recombinase, expression of the Thy1.1 reporter was reduced >90% in each of these contexts. The Thy1.1 reporter was not expressed by Th2 cells, regardless of whether CNS-22 was deleted. These results show convincingly that CNS-22 is essential to assure proper, high-level expression of the Thy1.1 Ifng reporter, but not to silence reporter expression in Th2 cells.
Figure 1.
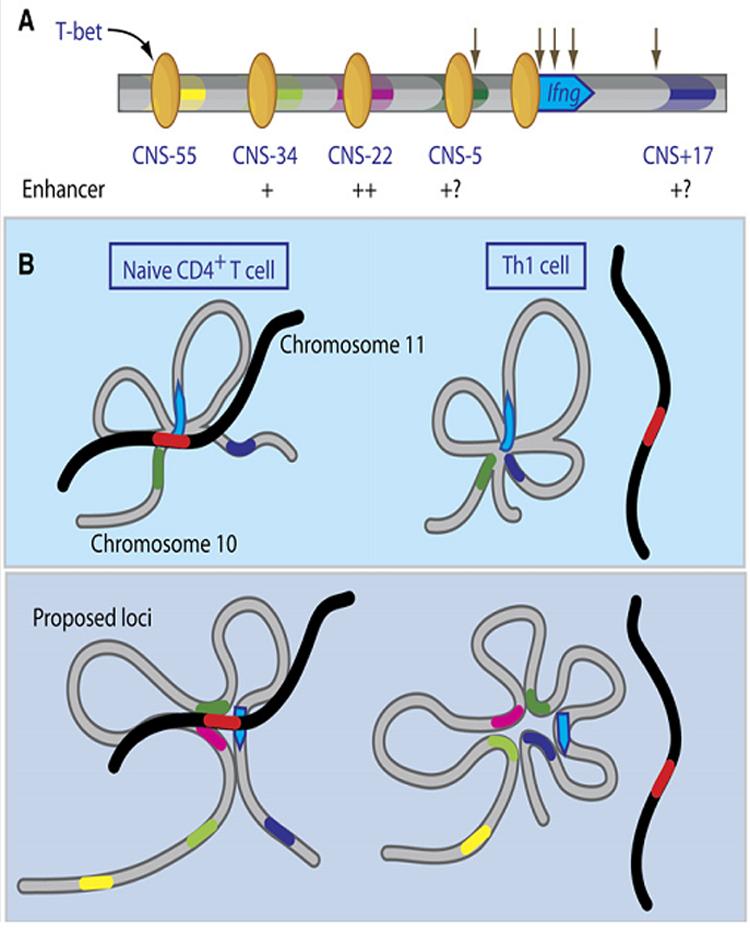
Function of upstream conserved non-coding sequences (CNSs) and hypothetical architecture of the murine Ifng locus. (a) As shown by Hatton et al (2006), T-bet (yellow ovals) binds to each of the upstream CNSs, and CNS-34 and CNS-22 enhance Ifng-driven luciferase expression in the presence of T-bet (degree of enhancement indicated by + marks). CNS-5 and CNS+17 are shown as +?, since they did not enhance expression in this study, but in two previous studies enhanced Ifng expression in vitro and bound T-bet in vivo (Lee et al 2005, Shnyreva et al 2005). Previously defined DNase hypersensitive sites are denoted by downward arrows. (b) Top, constitutive approximation of CNS-5 (dark green region on gray chromosome 10) to the Ifng promoter (blue arrow) by intrachrosomal looping and interchromosomal interactions between the Ifng promoter and the Th2 locus (red region on black chromosome 11) in naïve and Th1 CD4 T cells, as shown by Spilianakis et al (2005). Bottom, hypothetical interactions based on Hatton et al (2006). Like CNS-5, CNS-22 (pink region) is hypothesized to be approximated to the Ifng promoter and with the Th2 cytokine locus in naïve CD4 T cells, whereas CNS-34 (light green region) and CNS+17 (dark blue region) are only recruited into the chromatin hub in IFN-γ producing Th1 cells.
These findings provide new insights regarding Ifng regulation and open up avenues for future investigation. A logical next step will be to define more fully the mechanism(s) by which CNS-22 facilitates proper expression of Ifng. The most parsimonious interpretation of the findings by Hatton et al (2006) is that CNS-22 functions as an enhancer in vivo as it does in vitro. However, as the authors note, this conclusion may be too simplistic, in part because this was the only function assayed in vitro and the failure to identify additional functions for CNS-22 in vivo (e.g., in silencing of Ifng expression in Th2 cells) might have been masked by the loss of positive regulatory functions when this region was deleted in its entirety. Given the presence of CNS-22 in open chromatin in naïve T cells, it is also possible that CNS-22 facilitates expression of Ifng in part by helping to create a locus architecture that is permissive to expression. Such a function has been suggested for the two Ifng CNSs previously identified. In naïve CD4 T cells, the Ifng promoter is approximated by intrachromosomal looping to CNS-5 (CNS1) and by interchromosomal interactions to the Th2 cytokine locus on murine chromosome 11 (Figure 1c). Differentiation of naïve CD4 T cells into Th1 cells, markedly reduces the interaction of the Ifng promoter with the Th2 cytokine locus and results in the de novo approximation of CNS+17 (CNS-2) to the Ifng promoter (Spilianakis et al., 2005). Might CNS-22 participate in these context-dependent intragenic and intergenic interactions?
It will also be important to determine whether deletion of CNS-22 from the endogenous Ifng locus abolishes expression as it did expression of the BAC-encoded Thy1.1 reporter. There are reasons to suspect that CNS-22 may be important but not absolutely essential in its native context. While the Thy1.1 reporter was expressed only in cells that also expressed interferon-γ, only 13-50% of interferon-γ expressing cells also expressed Thy1.1. Moreover, expression from the >20 copy and single copy BAC transgenes was similar. Together these two findings indicate that this BAC lacks a locus control region, which, if present, would have resulted in copy number-dependent expression of the transgene by insulating it from local environment in which the transgene integrated (Lee et al., 2006). Thus, assuming that there is an Ifng locus control region, it does not appear to lie within ∼60kb upstream or ∼100 kb downstream of the murine Ifng gene. Future studies should also address the possibility that the CNSs now identified have functions not yet addressed and determine if any non-CNS sequences also contribute to proper Ifng expression. While, evolutionarily conserved regions are more likely to contain regulatory elements, a considerable fraction of regulatory elements that lack the requisite degree of sequence conservation to be considered CNSs have been identified by non-targeted approaches, such as comprehensive DNase hypersensitivity site or chromatin immunoprecipitation assays. Thus, an unbiased search of the Ifng locus may identify important regulatory elements in addition to those that have now been identified.
References
- Chang S, Aune TM. Proc Natl Acad Sci U S A. 2005;102:17095–17100. doi: 10.1073/pnas.0502129102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JY, Grigura V, Murphy TL, Murphy K. Int Immunol. 2003;15:1149–1160. doi: 10.1093/intimm/dxg113. [DOI] [PubMed] [Google Scholar]
- Hatton RD, Harrington LE, Luther RJ, Wakefield T, Janowski KM, Oliver JR, Lallone RL, Murphy KM, Weaver CT. Immunity. 2006 doi: 10.1016/j.immuni.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Ivanov, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Lee DU, Avni O, Chen L, Rao A. J Biol Chem. 2004;279:4802–4810. doi: 10.1074/jbc.M307904200. [DOI] [PubMed] [Google Scholar]
- Lee GR, Kim ST, Spilianakis CG, Fields PE, Flavell RA. Immunity. 2006;24:369–379. doi: 10.1016/j.immuni.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Shnyreva M, Weaver WM, Blanchette M, Taylor SL, Tompa M, Fitzpatrick DR, Wilson CB. Proc Natl Acad Sci U S A. 2004;101:12622–12627. doi: 10.1073/pnas.0400849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutto M, Zhou W, Aune TM. J Immunol. 2002;169:6664–6667. doi: 10.4049/jimmunol.169.12.6664. [DOI] [PubMed] [Google Scholar]
- Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Nature. 2005;435:637–645. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- Zhu H, Yang J, Murphy TL, Ouyang W, Wagner F, Saparov A, Weaver CT, Murphy KM. J Immunol. 2001;167:855–865. doi: 10.4049/jimmunol.167.2.855. [DOI] [PubMed] [Google Scholar]