

Abstract
Objectives
To document the chemical and biological profile of a clinical phase II red clover (Trifolium pratense L.) extract by identifying and measuring the major and minor components visible in the high performance liquid chromatography-ultraviolet (HPLC-UV) chromatogram and evaluating each compound for estrogenic and antioxidant activity.
Design
Individual compounds in the pre-formulated (i.e., no excipients present) extract were identified by either chemical isolation followed by structure elucidation or by matching to retention time and molecular mass of chemical standards via liquid chromatography-mass spectrometry (LC-MS) analysis. Quantitation of the amounts of compounds found in the pre-formulated extract was done using HPLC-UV or LC-MS. Isolated compounds or standards were evaluated for their ability to 1) induce alkaline phosphatase (AP) in an endometrial carcinoma cell line, 2) competitively bind to recombinant human estrogen receptors (ERs) alpha (α) and beta (β), and 3) act as antioxidants by scavenging 2,2-di(4-tert-octylphenyl)-1-picrylhydrazyl (DPPH) free radicals.
Results
The pre-formulated red clover extract had an EC50 of 2.0–2.2 μg/mL in the AP estrogenicity assay, and IC50s of 18.4–32.6 μg/mL and 1.9–3.4 μg/mL in the ERα and ERβ binding assays, respectively. The pre-formulated extract was composed of 35.54% isoflavones, 1.11% flavonoids, 0.06% pterocarpans, ≤ 0.03% coumarins, and ≤ 0.03% tyramine. Daidzein, genistein, formononetin, biochanin A, coumestrol and naringenin were estrogenic in the AP assay, and all of these, except formononetin, bound to one or both ERs.
Conclusions
The major and minor chemical and active estrogenic components of a pre-formulated Phase II red clover clinical extract were identified, quantitatively measured, and the final capsule doses were calculated. The extract is currently under evaluation in a yearlong clinical study for the alleviation of menopausal hot flashes. This is the first report to thoroughly summarize the chemistry and biology of all major peaks observed in the HPLC-UV chromatogram of a clinical red clover dietary supplement.
Keywords: biological activity, chemical profile, clinical extract, estrogenic, HPLC chromatogram, red clover, Trifolium pratense
INTRODUCTION
A recent survey of peri- and postmenopausal female outpatients found that approximately 79% of these women use botanical dietary supplements (Mahady et al., 2003). Red clover is one botanical dietary supplement used by women for menopausal hot flashes. However, significant concerns about the quality of botanical dietary supplements (Cardellina, 2002; Srinivasan, 2002; Glisson et al., 2003; Kroes et al., 2004) complicate current efforts to study the clinical efficacy of botanical supplements.
Commercial extracts of red clover contain high amounts of the mildly estrogenic isoflavones daidzein, genistein, formononetin, and biochanin A. The general premise supporting use of red clover supplements during menopause is that the isoflavones will act as a natural estrogen replacement in older women having low endogenous estradiol levels, and thus alleviate hot flashes and other symptoms. Red clover isoflavone supplements contain several minor components as well, many of which have not been investigated for their estrogenic activity or even quantitatively measured in over-the-counter supplements. Because some of these minor compounds may potentially be converted into active estrogenic metabolites in vivo, identification and measurement of these compounds is useful in the standardization of red clover supplements for clinical trials, allowing researchers to calculate the individual doses of each compound consumed by the subjects. Currently, no published papers thoroughly detail the chemical content of major and minor compounds in red clover botanical supplements used in clinical studies.
Experiments to characterize the chemical content and evaluate the estrogenic activity of 22 major and minor compounds present in a Phase II clinical red clover extract were performed. The coumarin content of this extract was previously reported (Booth et al., 2004). We present a HPLC-UV chromatogram of the extract, and include calculated daily doses of 20 of the compounds, based on the amount of raw extract administered in our phase II clinical trial. Pure compounds or standards identified in the red clover extract were tested in 1) the AP assay, 2) in human recombinant ERα and ERβ competitive binding assays, and 3) in a DPPH free radical scavenging antioxidant assay. We hope that our biological and chemical evaluation of this red clover dietary supplement will help establish a standard approach that clinicians and researchers can use in the study of botanical dietary supplements.
MATERIALS AND METHODS
Materials
Chemical solvents and reagents were purchased from Fisher (Hanover Park, IL) or Sigma (St. Louis, MO), unless otherwise indicated. Isoflavone, flavonoid, and coumarin standards were purchased from Indofine Chemical Co. (Somerville, NJ), Chromadex (Denver, CO), and TimTec (Newark, DE). Irilone was a kind gift from Professor Eckhard Wollenweber (Technische Universit t Darmstadt, Germany). Cell culture media and GlutaMAX™-1 was purchased from Invitrogen (Carlsbad, CA) and fetal bovine serum (FBS) was from Atlanta Biologicals (Norcross, GA).
Plant material
The 30% isoflavone (defined as total content of daidzein + genistein + formononetin + biochanin A, w/w%) red clover Phase II clinical extract was a T. pratense autohydrolyzed extract manufactured by PureWorld Botanicals, Inc. (South Hackensack, NJ) using proprietary methods. The raw unformulated extract, without added excipients, was used in the present investigation. A voucher specimen of the coarsely milled plant source material is being stored at the University of Illinois at Chicago. The plant source material was examined by Dr. D. Doel Soejarto (UIC) and was found to be consistent with authentic T. pratense specimens deposited at the Field Museum of Natural History (Chicago, IL).
General experimental procedures
NMR spectra were recorded on Bruker Avance 300, 360 and 500 MHz spectrometers (Billerica, MA). Exact mass electrospray mass spectra were recorded on a Micromass QTOF2 quadrupole time-of-flight hybrid mass spectrometer (Manchester, UK). Quantitative mass spectra were determined using an Agilent G1946A single quadrupole LC-MS (Palo Alto, CA) equipped with a Model 1100 HPLC system. UV spectra were observed with a Beckman UV/Visible spectrophotometer (Fullerton, CA). CD spectra were recorded using a Jasco J-710 Spectropolarimeter (Great Dunmow, UK). Preparative HPLC was carried out using a Waters Delta 600 controller system and Delta 600 pumps equipped with a Waters 996 UV photodiode array (PDA) detector, Waters 717 plus autosampler, and Millennium™32 Chromatography Manager software (Milford, MA) using a Jones (Genesis™ C18 column, 250 × 4.6 mm, 4 μm particle size), GROM-SIL™ (120 ODS-4 HE Säule 300 × 20 mm, 5 μm particle size), or Phenomenex phenylhexyl (Luna™ 250 × 22.5 mm, 5 μm particle size) column, as noted. Analytical HPLC analyses were done using either a Waters 2695 unit with PDA detector, or an Agilent 1100 analytical HPLC with DAD. Bioassay results were obtained using a Power Wave 200 microplate scanning spectrophotometer (Bio-Tek Instruments, Winooski, VT).
Extraction and isolation
For initial fractionation, 300 g of the unformulated powdered raw red clover extract was mixed with 60 g of dry microcrystalline cellulose and loaded into a vacuum flash column packed with 3 kg of dry microcrystalline cellulose. A total of 25 fractions of 5 L each were collected: 1) 100% petroleum ether (PE) (9.9 g); 2) 95/5 (v/v) PE/chloroform (CHCl3) (5.1 g); 3) 90/10 PE/CHCl3 (2.2 g); 4) 85/15 PE/CHCl3 (3.1 g); 5) 80/20 PE/CHCl3 (2.3 g); 6) 50/50 PE/CHCl3 (6.2 g); 7) 40/60 PE/CHCl3 (5.4 g); 8) 10/90 PE/CHCl3 (10.6 g); 9) 100% CHCl3 (9.6 g); 10) 50/50 CHCl3/toluene (7.4 g); 11) 100% toluene (1.3 g); 12) 50/50 toluene/ethyl acetate (EtOAc) (14.2 g); 13) 100% EtOAc (35.6 g); 14) 10/90 methanol (MeOH)/EtOAc (55.8 g); 15) 20/80 MeOH/EtOAc (32.9 g); 16) 50/50 MeOH/EtOAc (23.0 g); 17) 70/30 MeOH/EtOAc (9.3 g); 18) 80/20 MeOH/EtOAc (7.3 g); 19) 90/10 MeOH/EtOAc (4.8 g); 20) 100% MeOH (3.2 g); 21) 10/90 H2O/MeOH (3.9 g); 22) 20/80 H2O/MeOH (6.0 g); 23) 50/50 H2O/MeOH (5.8 g); 24) 100% H2O (2.7 g); 25) 1% aqueous acetic acid (4.3 g).
Fractions 6 and 7 were combined based on TLC analysis and 10.0 g was dissolved in a small amount of CHCl3 plus PE and mixed with 20 g of C18 silica gel. The evaporated dry sample was loaded into a 50 g C18 gravity column and eluted successively with 1 L 70% aq. MeOH, 325 mL 85% aq. MeOH, 1 L 100% MeOH, 175 mL CHCl3, and 200 mL PE. The first two and last three eluates were combined, separately, to give two fractions. The first fraction (70% aq. MeOH + 85% aq. MeOH) retained some slight green color and comprised 3.5 g of product. The second fraction (100% MeOH + CHCl3 + PE) yielded 6.1 g. The first (decolorized) fraction was dissolved in MeOH and added to 5 g of pre-washed C18 silica gel, dried, and loaded into a pressure flash column containing 200 g of C18. Subfractions A and B were collected using 60/40 (v/v) MeOH/H2O as the eluent, subfractions C, D, and E were collected using 70/30 MeOH/H2O, and subfraction F was collected during a gradient from 70/30 to 80/20 MeOH/H2O.
Pratensein (12) was isolated (9 mg) from subfraction A, formononetin (16) (< 1 mg), maackiain (17) (59 mg), and medicarpin (23) (2 mg) were isolated from subfraction B, and irilone (18) (~1 mg), dihydrobiochanin A (19) (2 mg), and cicerin (20) (2 mg) were isolated from subfraction C by repeated preparative HPLC chromatography on isolated peaks.
LC-MS quantitative analyses
The following compounds were tentatively identified from electrospray LC-MS results by matching m/z values to compounds reported in Trifolium and related species: tyramine (1), fisetin (5), calycosin (7), quercetin (8), naringenin (9), pratensein (12), kaempferol (13), pseudobaptigenin (15), irilone (18), and prunetin (21). These compounds were purchased or isolated and then the mass spectra and retention times of extract peaks were compared with those of the standards (including spiked extract samples), confirming their presence in the red clover Phase II extract. Since 1, 5, and 9 were found to be present in the extract at < 0.05% they were quantitated using LC-MS. Standards were used to determine the weight percent present in the Phase II red clover extract. For tyramine, an isocratic mobile phase of acetonitrile/0.1% formic acid (72:28, v/v) was used at 0.2 mL/min on a TSK Gel-80 2.0 × 250 mm amide column, 5 μm particle size. In this system, tyramine eluted at 7.6 min. For analysis of the two flavonoids, a gradient method of 38% aqueous MeOH → 100% MeOH in 60 min at a flow rate of 0.2 mL/min, was used on an YMC AQ 2.0 × 250 mm column. In this system, 5 eluted at 25.7 min and 9 eluted at 23.6 min. LC-MS quantitation of 2, 3, 4, 11, and 14 was described previously (Booth et al., 2004), and the results are reported in Table 2.
Table 2.
Bioassay activity of compounds detected in the Phase II clinical red clover extract. N.T. = not tested.
| Compound | Estrogenicity in Ishikawa cells [EC50, μM] | Estrogen Receptor α Binding [IC50, μM] | Estrogen Receptor β Binding [IC50, μM] | DPPH Free Radical Scavenging [IC50, μM] |
|---|---|---|---|---|
| Tested 10-2002: | Tested 10-2002: | Tested 10-2002: | ||
| Phase II Red | 2.0 ± 0.1 μg/mL | 18.4 ± 4.9 μg/mL | 1.9 ± 0.8 μg/mL | 143.1 ± 8.2 |
| Clover Extract | Tested 09-2004: | Tested 09-2004: | Tested 09-2004: | μg/mL |
| 2.2 ± 0.2 μg/mL | 32.6 ± 9.0 μg/mL | 3.4 ± 0.7 μg/mL | ||
| Tyramine (1) | > 20 | > 100 | > 100 | > 200 |
| Scopoletin (2) | > 20 | N.T. | N.T. | > 200 |
| Fraxidin (3) | > 20 | N.T. | N.T. | > 200 |
| Xanthotoxol (4) | > 20 | N.T. | N.T. | > 200 |
| Fisetin (5) | > 20 | N.T. | N.T. | 41.6 ± 2.7 |
| Daidzein (6) | 0.5 ± 0.1 | 17.0 ± 3.0 | 1.20 ± 0.01 | > 200 |
| Calycosin (7) | > 20 | N.T. | N.T. | > 200 |
| Quercetin (8) | > 200 | N.T. | N.T. | 49.9 ± 8.3 |
| Naringenin (9) | 4.6–12.2 (n = 2) | > 100 | 7.3 ± 1.8 | > 200 |
| Genistein (10) | 0.3 ± 0.1 | 0.30 ± 0.01 | 0.020 ± 0.002 | > 200 |
| Coumestrol (11) | 0.09–0.1 (n = 2) | 0.06 ± 0.04 | 0.02 ± 0.01 | > 200 |
| Pratensein (12) | > 67 | N.T. | N.T. | > 660 |
| Kaempferol (13) | > 200 | N.T. | N.T. | 64.9 ± 2.8 |
| Daphnoretin (14) | > 20 | N.T. | N.T. | > 200 |
| Pseudobaptigenin (15) | > 20 | N.T. | N.T. | > 200 |
| Formononetin (16) | N.A. | 104 ± 8 | 60.0 ± 7.0 | > 200 |
| Maackiain (17) | > 70 | N.T. | N.T. | > 700 |
| Irilone (18) | > 20 | N.T. | N.T. | > 200 |
| Dihydrobiochanin A (19) | > 20 | N.T. | N.T. | > 200 |
| Cicerin (20) | > 20 | N.T. | N.T. | > 200 |
| Prunetin (21) | > 20 | > 175 | > 175 | > 200 |
| Biochanin A (22) | 4.6 ± 0.8 | 35 ± 1.0 | 4.1 ± 0.8 | > 200 |
N.A. = range of dose-response curve not sufficient to calculate an EC50
HPLC quantitative analyses
The quantitative analyses of 6, 7, 8, 10, 12, 13, 15, 16, 17, 18, 21, and 22 in the red clover Phase II clinical extract were carried out using HPLC-UV instead of LC-MS, since these compounds were present at > 0.05% (w/w). For 12 and 17, the isolated compounds were used as standards. All compounds were analyzed on the Agilent 1100 HPLC instrument, except 6, 10, 16 and 22, which were analyzed on the Waters 2695 HPLC system. Injections of each sample (10 μL) were analyzed in triplicate using a Beckman octadecyl silane (ODS) μBondapak 4.2 × 250 mm column, and the DAD detector was set to record absorbance at 254 nm. A 60-min method with a flow rate of 1.5 mL/min and the following gradient was used: 100% A/0% B to 60% A/40% B from 0–20 min, hold at 60% A/40% B from 20–22 min, 60% A/40% B to 10% A/90% B from 22–60 min, followed by a 10 min column wash of 100% B and then re-equilibration to 100% A for 14 min before the next injection, where A = doubly-deionized water/acetonitrile/glacial acetic acid (85:15:0.1, v/v/v) and B = doubly-deionized water/acetonitrile/glacial acetic acid (50:50:0.1, v/v/v). Areas under the curve from experimental peaks in the chromatogram were determined and the percent weight of each compound present was calculated based on a standard curve and amount (μg) of extract injected onto the column. Data were only used if their relative standard deviations (RSDs, %; [(mean – standard deviation)/mean] ×100) were < 5%. The lowest concentration of compound at which a peak could still be discerned and reproducibly integrated with RSD < 5% is reported as the limit of quantitation (LOQ). LOQs and percents of compounds present in the Phase II red clover extract are reported in Table 1.
Table 1.
Results of HPLC-UV and LC-MS quantitative analyses of compounds in the Phase II clinical red clover extract.
| Compound | LOQ [ng/mL, λ254] | Weight Percent in Red Clover Extract [%] | Daily Clinical Dose of Compound [mg, based on 397 mg extract] |
|---|---|---|---|
| Tyramine (1) | 10 | 0.026 | 0.10 |
| Scopoletin (2) | ≤ 20 ppm | ≤ 0.002 | ≤ 0.008 |
| Fraxidin (3) | ≤ 100 ppm | ≤ 0.01 | ≤ 0.04 |
| Xanthotoxol (4) | ≤ 100 ppm | ≤ 0.01 | ≤ 0.04 |
| Fisetin (5) | 70 | 0.02 | 0.08 |
| Daidzein (6) | 50 | 0.23 | 0.91 |
| Calycosin (7) | 105 | 0.44 | 1.75 |
| Quercetin (8) | 100,000 (poor peak shape) | 1.00 | 3.97 |
| Naringenin (9) | 90 | 0.02 | 0.08 |
| Genistein (10) | 125 | 0.41 | 1.63 |
| Coumestrol (11) | ≤ 100 ppm | ≤ 0.01 | ≤ 0.04 |
| Pratensein (12) | 515 | 1.08 | 4.29 |
| Kaempferol (13) | 51,100 (poor peak shape) | 0.07 | 0.29 |
| Daphnoretin (14) | ≤ 20 ppm | ≤ 0.002 | ≤ 0.008 |
| Pseudobaptigenin (15) | 55 | 0.86 | 3.41 |
| Formononetin (16) | 130 | 14.26 | 56.61 |
| Maackiain (17) | 500 | 0.06 | 0.24 |
| Irilone (18) | 70 | 3.20 | 12.70 |
| Prunetin (21) | 110 | 0.59 | 2.34 |
| Biochanin A (22) | 540 | 14.47 | 57.45 |
| TOTAL COMPOUNDS | 36.77% | 145.89 mg | |
Ishikawa estrogenicity assay
Isolated compounds and standards were tested for estrogenic (agonistic) activity using the Ishikawa endometrial cell AP induction assay described previously (Pisha et al., 1997; Liu et al., 2001), except that the p-nitrophenylphosphate solution used was 1 mg/mL. Results are presented in Table 2.
Estrogen receptor binding assays
The procedure carried out in Liu et al., 2001 was followed except that the ER alpha (α) wash buffer contained only 40mM Tris and 100 mM KCl, pH 7.5, and incubations were performed at room temperature for one hour or at 4 ºC overnight. Results are presented in Table 2.
DPPH antioxidant assay
A published protocol (Burdette et al., 2002) was followed, except that 200 μM DPPH solution in ethanol was added to samples, which were then covered and shaken for 30 sec on a rotary shaker, and incubated at 37 °C for 20 minutes. Absorbance was read at 515 nm. Samples were tested at a final concentration of 200 μg/mL in dimethylsulfoxide. Results are presented in Table 2.
RESULTS AND DISCUSSION
Compound identification
Pratensein (12) was identified based on exact mass determination and comparison of proton and carbon NMR data with that reported by Hanawa et al., 1991 and Jain and Bambah, 1987.
Formononetin (16) was identified based on exact mass determination and comparison of proton and carbon NMR data with that reported by Lin et al., 1998, Balasubramanian and Nair 2000, and Chang et al., 1994.
Maackiain (17) was identified based on exact mass determination and comparison of proton and carbon NMR data with that reported by Banks and Dewick, 1982, Tang et al., 2002, and Pelter et al., 1976.
Irilone (18) was identified based on exact mass determination and comparison of proton and carbon NMR data with that reported by Fraishtat and Popravko, 1979 and Veitch et al., 2003.
(±)-Dihydrobiochanin A (19) was identified based on exact mass determination and comparison of proton and carbon NMR data with that reported by Osawa et al., 1992 and Pauli et al., 2000.
No references containing NMR data for (±)-cicerin (20) could be located, although Kunzru and Sinha apparently first reported cicerin in 1970. Therefore, the assignment of compound 20 was based on its similarity to dihydrobiochanin A. Data are as follows: light tan-orange solid, mp 224–225 °C; UV λmax MeOH nm (log ɛ ) 290 (4.50); δ1H NMR (360 MHz, MeOD-d3) 6.70 (1H, s, H-5′), 6.64 (1H, s, H-2′), 5.90 (2H, s, OCH2O), 5.89 (1H, d overlapping with methylenedioxy resonance, J = 2.2 Hz, H-6), 5.87 (1H, d, J = 2.2 Hz, H-8), 4.48 (1H, t, J = 11.0 Hz, H-2ax), 4.37 (1H, dd, J = 5.5, 10.8 Hz, H-2eq), 4.25 (1H, dd, J = 5.5, 11.3 Hz, H-3ax); 13C NMR (360 MHz, acetone-d6) 198.80 (C=O), 168.24 (C7-OH), 165.89 (C5-OH), 165.16 (C-9), 154.33 (C-6′), 149.46 (C-4′), 142.81 (C-3′), 116.53 (C-1′), 110.99 (CH-2′), 103.74 (C-10), 102.71 (OCH2O), 97.18 (CH-6), 96.47 (CH-5′), 96.03 (CH-8), 71.48 (CH2-2), 57.08 (6′-OCH3), 48.59 (CH-3, buried under MeOD peaks); HRESMS, m/z found: 329.0652 [M-H]−; calc. for C17H13O7: 329.0661.
(-)-Medicarpin (23) was identified based on exact mass determination and comparison of its proton and carbon NMR data with that reported by Yahara et al., 1989, Goda et al., 1992, and Kulesh et al., 2001.
Quantitative analysis of the Phase II clinical red clover extract
Red clover capsules for our Phase II clinical trial were formulated by Pharmavite, LLC (Mission Hills, CA) as follows: 198.5 mg raw red clover extract, 182.5 mg rice flour, 4.0 mg silicon dioxide. Two capsules are administered daily, for a total dose of 397 mg raw extract (containing 120 mg total daidzein + genistein + formononetin + biochanin A) per person per day. Compound doses in Table 1 are based on ingestion of 397 mg of raw extract daily. Chemical structures are provided in Figure 1, and the HPLC-UV chromatogram of the raw red clover extract is presented in Figure 2. Nine measured isoflavones (6, 7, 10, 12, 15, 16, 18, 21, 22) comprise 35.54% of the weight of the raw extract without excipients. Other compound classes were present in the red clover extract in significantly smaller amounts: 1.11% flavonoids (5, 8, 9, 13), 0.06% pterocarpans (17), ≤ 0.03% coumarins (2, 3, 4, 11, 14), and 0.03% tyramine (1). The rationale for coumarin analysis was described previously (Booth et al., 2004). Flavonoids (Wong et al., 1968; Lin et al., 2000; He et al., 1996; Nakatani et al., 1989), pterocarpans (Dewick, 1975; Fraishtat et al., 1981), and a few coumarins (Lyman et al., 1959) are known to occur in Trifolium species. Tyramine was discovered during LC-MS analysis of aqueous fractions of the extract, and it may be formed from decomposition of some amino acids, which were detected using ninhydrin reagent (data not shown). However, the red clover isoflavone supplements currently marketed are aqueous alcoholic extracts that likely contain low protein content, and therefore tyramine is not expected to be a major constituent of these products.
Figure 1.
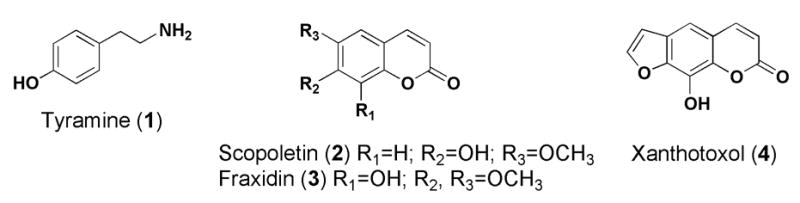
Chemical structures of compounds isolated from or identified in the Phase II clinical red clover extract.
Figure 2.
HPLC-UV chromatogram of the Phase II clinical red clover extract, with individual compounds identified by numbers corresponding to the structures provided in Figure 1. Absorbance at 254 nm was observed.
A total of 36.77% of the raw Phase II clinical red clover extract has been identified and measured. This analysis accounts for the majority of peaks visible in the HPLC-UV chromatogram of this extract.
Bioassay activities of extract components
Results summarizing the estrogenic and antioxidant activity of compounds detected in the Phase II clinical red clover extract are presented in Table 2. Since the raw clinical extract gave similar results in the AP induction and the ER binding assays both initially and after two years of storage, the estrogenic compounds in this preparation of red clover are quite stable. Among the 22 constituents evaluated, six tested positive in the AP assay (6, 9, 10, 11, 16, 22) and of these, five bound to one or both ERs (6, 9, 10, 11, 22). Coumestrol was the most potent estrogenic compound, having an EC50 of 0.09–0.10 μM in the AP assay, and IC50 values of 0.06 ± 0.04 and 0.02 ± 0.01 μM in the ER alpha (α) and ER beta (β) competitive binding assays, respectively. However, coumestrol was only present at a trace amount (≤ 0.01%) in the extract, amounting to a daily human dose of ≤ 0.04 mg. Naringenin was active in the AP induction and ER beta (β)-binding assays, but was present at only 0.02%. Thus, the principal estrogenic components of the extract, based on percent present and relative activity, are the four known isoflavones daidzein, genistein, formononetin and biochanin A. Although formononetin did not bind to the ERs at a physiological concentration, it is metabolized to daidzein in vivo (Piersen et al., 2004; Heinonen et al., 2004) and then to equol (Setchell et al., 2002), both of which are estrogenic in the Ishikawa assay (equol data not shown). Although prunetin was not active in any estrogenic assay, there is evidence that it is metabolically converted into genistein (Hu et al., 2003). Only three compounds were active in the DPPH antioxidant assay (5, 8, 13), and these flavonols all contain 3-hydroxyl and 4′-hydroxyl groups.
CONCLUSIONS
We have identified 22 compounds and quantitatively measured 20 that constitute more than 36% of the weight of the studied phase II clinical red clover extract. This represents a significant improvement in the characterization of red clover supplements, as currently only four of the isoflavone components (daidzein, genistein, formononetin, biochanin A) are measured and reported in clinical studies. Although the majority of compounds identified in the studied red clover extract were not biologically active in vitro, many had never before been tested in the AP induction, ER binding or DPPH antioxidant assays. Since minor constituents that are not estrogenic in vitro may still potentially be converted to active metabolites in vivo, there exists a logical rationale for measuring certain minor compounds (i.e., prunetin as an estrogenic “prodrug”). This work provides a detailed examination of the chemical and in vitro biologically active components of a red clover dietary supplement, and it is hoped that more such studies will be carried out on botanical supplements undergoing clinical evaluation.
Acknowledgments
This work was supported, in part, by grant P50 AT00155 provided jointly by the National Center for Complementary and Alternative Medicine (NCCAM), the Office of Dietary Supplements (ODS), the Office for Research on Women’s Health (ORWH), and the National Institute of General Medicine (NIGMS) of the National Institutes of Health (NIH). NLB is grateful for a National Research Service Award (NRSA) from NCCAM, F31 AT00804, and Drs. John Fitzloff and Jimmy Orjala are acknowledged for their donations of HPLC instrument time for analysis of the red clover extract. CRO acknowledges support from Ruth L. Kirschstein NIH Predoctoral fellowship F31 AT24232. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NCCAM, NCI, ODS, ORWH, NIGMS, or NIH.
References
- Balasubramanian S, Nair MG. An efficient “one pot” synthesis of isoflavones. Synthetic Commun. 2000;30:469–484. [Google Scholar]
- Banks SW, Dewick PM. (-)-Pisatin, an induced pterocarpan metabolite of abnormal configuration from Pisum sativum. Phytochemistry. 1982;21:1605–1608. [Google Scholar]
- Booth NL, Nikolic D, van Breemen RB, Geller SE, Banuvar S, Shulman LP, Farnsworth NR. Confusion regarding anticoagulant coumarins in dietary supplements. Clin Pharmacol Ther. 2004;76:511–516. doi: 10.1016/j.clpt.2004.08.023. [DOI] [PubMed] [Google Scholar]
- Burdette JE, Chen SN, Lu ZZ, Xu H, White BE, Fabricant DS, Liu J, Fong HH, Farnsworth NR, Constantinou AI, van Breemen RB, Pezzuto JM, Bolton JL. Black cohosh (Cimicifuga racemosa L.) protects against menadione-induced DNA damage through scavenging of reactive oxygen species: bioassay-directed isolation and characterization of active principles. J Agric Food Chem. 2002;50:7022–7028. doi: 10.1021/jf020725h. [DOI] [PubMed] [Google Scholar]
- Cardellina JH. 2nd. Challenges and opportunities confronting the botanical dietary supplement industry. J Nat Prod. 2002;65:1073–1084. doi: 10.1021/np0200515. [DOI] [PubMed] [Google Scholar]
- Chang YC, Nair MG, Santell RC, Helferich WG. Microwave-mediated synthesis of anticarcinogenic isoflavones from soybeans. J Agric Food Chem. 1994;42:1869–1871. [Google Scholar]
- Dewick PM. Pterocarpan biosynthesis: 2′-hydroxy-isoflavone and -isoflavanone precursors of demethylhomopterocarpin in red clover. Chem Commun (Camb) 1975;656 [Google Scholar]
- Fraishtat PD, Popravko SA. Secondary metabolites of clover. II. Isolation of 4′,5-dihydroxy-6,7-methylenedioxyisoflavone and its 4′-O-beta-D-glucoside from the roots of red clover (Trifolium pratense. Sov J Bioorg Chem. 1979;5:162–166. [Google Scholar]
- Fraishtat PD, Popravko SA, Vul’fson NS. Secondary metabolites of clover. 8. Isolation and identification of pterocarpans from the roots of cultured clover species. Bioorg Khim. 1981;7:927–936. [Google Scholar]
- Glisson JK, Rogers HE, Chambliss WG. Dietary supplements: important concerns for the clinician. J Miss State Med Assoc. 2003;44:35–38. [PubMed] [Google Scholar]
- Goda Y, Kiuchi F, Shibuya M, Sankawa U. Inhibitors of prostaglandin biosynthesis from Dalbergia odorifera. Chem Pharm Bull. 1989;37:979–987. doi: 10.1248/cpb.40.2452. [DOI] [PubMed] [Google Scholar]
- Hanawa F, Tahara S, Mizutani J. Isoflavonoids produced by Iris pseudacorus leaves treated with cupric chloride. Phytochemistry. 1991;30:157–163. [Google Scholar]
- He XG, Lin LZ, Lian LZ. Analysis of flavonoids from red clover by liquid chromatography-electrospray mass spectrometry. J Chromatogr A. 1996;755:127–132. [Google Scholar]
- Heinonen SM, Wahala K, Adlercreutz H. Identification of urinary metabolites of the red clover isoflavones formononetin and biochanin a in human subjects. J Agric Food Chem. 2004;52:6802–6809. doi: 10.1021/jf0492767. [DOI] [PubMed] [Google Scholar]
- Hu M, Krausz K, Chen J, Ge X, Li J, Gelboin HL, Gonzalez FJ. Identification of CYP1A2 as the main isoform for the phase I hydroxylated metabolism of genistein and a prodrug converting enzyme of methylated isoflavones. Drug Metab Dispos. 2003;31:924–931. doi: 10.1124/dmd.31.7.924. [DOI] [PubMed] [Google Scholar]
- Jain AC, Bambah PK. Synthesis of naturally occurring pratensein and 6,8-di-C-prenylpratensein. Indian J Chem. 1987;26B:488–490. [Google Scholar]
- Kroes R, Walker R. Safety issues of botanicals and botanical preparations in functional foods. Toxicology. 2004;198:213–220. doi: 10.1016/j.tox.2004.01.028. [DOI] [PubMed] [Google Scholar]
- Kulesh NI, Maksimov OB, Denisenko VA, Glazunov VP. Isoflavonoids from heartwood of Maackia amurensis. Rupr et Maxim Chem Nat Compd. 2001;37:29–31. [Google Scholar]
- Kunzru R, Sinha S. ‘Cicerin’ a new phytoalexin associated with blight of gram. In: Raychaudhuri SP, editor. Plant Dis Probl, Proc Int Symp; 1st 1970; Meeting Date 1966–1967; pp. 724–732. [Google Scholar]
- Lin LZ, He XG, Lindenmaier M, Yang J, Cleary M, Qiu SX, Cordell GA. LC-ESI-MS study of the flavonoid glycoside malonates of red clover (Trifolium pratense) J Agric Food Chem. 2000;48:354–365. doi: 10.1021/jf991002+. [DOI] [PubMed] [Google Scholar]
- Lin YL, Tsai WJ, Chen IS, Kuo YH. Chemical constitutents from Mucuna membranacea. J Chin Chem Soc (Taip) 1998;45:213–217. [Google Scholar]
- Liu J, Burdette JE, Xu H, Gu C, van Breemen RB, Bhat KP, Booth N, Constantinou AI, Pezzuto JM, Fong HH, Farnsworth NR, Bolton JL. Evaluation of estrogenic activity of plant extracts for the potential treatment of menopausal symptoms. J Agric Food Chem. 2001;49:2472–2479. doi: 10.1021/jf0014157. [DOI] [PubMed] [Google Scholar]
- Lyman RL, Bickoff EM, Booth AN, Livingston AL. Detection of coumestrol in leguminous plants. Arch Biochem Biophys. 1959;80:61. [Google Scholar]
- Mahady GB, Parrot J, Lee C, Yun GS, Dan A. Botanical dietary supplement use in peri- and postmenopausal women. Menopause. 2003;10:65–72. doi: 10.1097/00042192-200310010-00011. [DOI] [PubMed] [Google Scholar]
- Nakatani M, Tajiri M, Kaga T, Fukami R, Takada Y, Hase T. A dihydroxycyclopentadienone and other constituents from the seeds of Trifolium repens. Phytochemistry. 1989;289:2499–2501. [Google Scholar]
- Osawa K, Yasuda H, Maruyama T, Morita H, Takeya K, Itokawa H. Isoflavanones from the heartwood of Swartzia polyphylla and their antibacterial activity against cariogenic bacteria. Chem Pharm Bull. 1992;40:2970–2974. doi: 10.1248/cpb.40.2970. [DOI] [PubMed] [Google Scholar]
- Pauli GF. Higher order and substituent chemical shift effects in the proton NMR of glycosides. J Nat Prod. 2000;63:834–838. doi: 10.1021/np990527t. [DOI] [PubMed] [Google Scholar]
- Pelter A, Ward RS, Gray TI. The carbon-13 nuclear magnetic resonance spectra of flavonoids and related compounds. J Chem Soc [Perkin 1] 1976;22:2475–2483. [PubMed] [Google Scholar]
- Piersen CE, Booth NL, Sun Y, Liang W, Burdette JE, van Breemen RB, Geller SE, Gu C, Banuvar S, Shulman LP, Bolton JL, Farnsworth NR. Chemical and biological characterization and clinical evaluation of botanical dietary supplements: a Phase I red clover extract as a model. Curr Med Chem. 2004;11:1361–1373. doi: 10.2174/0929867043365134. [DOI] [PubMed] [Google Scholar]
- Pisha E, Pezzuto JM. Cell-based assay for the determination of estrogenic and anti-estrogenic activities. Methods Cell Sci. 1997;19:37–43. [Google Scholar]
- Setchell KD, Brown NM, Lydeking-Olsen E. The clinical importance of the metabolite equol-a clue to the effectiveness of soy and its isoflavones. J Nutr. 2002;132:3577–3584. doi: 10.1093/jn/132.12.3577. [DOI] [PubMed] [Google Scholar]
- Srinivasan VS. Considerations in the development of public standards for botanicals and their dosage forms. Adv Phytomedicine. 2002;1:259–265. [Google Scholar]
- Tang YP, Hu J, Wang JH, Lou FC. A new coumaronochromone from Sophora japonica. J Asian Nat Prod Res. 2002;4:1–5. doi: 10.1080/10286020290019622. [DOI] [PubMed] [Google Scholar]
- Veitch NC, Sutton PSE, Kite GC, Ireland HE. Six new isoflavones and a 5-deoxyflavonol glycoside from the leaves of Atelia herbert-smithii. J Nat Prod. 2003;66:210–216. doi: 10.1021/np020425u. [DOI] [PubMed] [Google Scholar]
- Wong EE, Francis CM. Flavonoids in genotypes of Trifolium subterraneum. III. Varietal differences Phytochemistry. 1968;7:2139–2142. [Google Scholar]
- Yahara S, Ogata T, Saijo R, Konishi R, Yamahara J, Miyahara K, Nohara T. Isoflavan and related compounds from Dalbergia odorifera. Chem Pharm Bull. 1989;37:979–987. [Google Scholar]