

Abstract
Dendritic cells (DC) carry antigen from peripheral tissues via lymphatics to lymph nodes (LN). We report that differentiated DC can also travel from the periphery into the blood. Circulating DC migrated to the spleen, liver and lung, but not LN. They also homed to the bone marrow (BM) where they were better retained than in most other tissues. DC homing to the BM depended on constitutively expressed VCAM-1 and endothelial selectins in BM microvessels. Two-photon intravital microscopy in BM cavities revealed that DC formed stable antigen-dependent contacts with BM-resident central memory T cells. Moreover, using this novel migratory pathway, antigen-pulsed DC could trigger central memory T cell-mediated recall responses in the BM.
Dendritic cells (DC) are key players in innate and adaptive immune responses1. The prevalent model of DC migration is an unidirectional pathway whereby precursor DC arise from progenitors in the bone marrow (BM), enter the blood and traffic into secondary lymphoid organs (SLO) and peripheral tissues, such as skin or gut2, where they contribute to the front-line of defense against pathogens. When DC encounter inflammatory stimuli, they undergo a switch in chemokine receptor expression enabling their egress into lymphatic vessels and transport to draining lymph nodes (LN)3. Maturing DC also become fully immuno-stimulatory by up-regulating major histocompatibility complex (MHC) and costimulatory molecules to prime naïve T cells.
Normal peripheral blood contains some immature DC, which capture blood-borne bacteria and transport them to the spleen4. However, whether these DC originate directly in the BM or reenter the blood after migrating within other tissues is unclear. There is ample experimental evidence that draining LN are the terminal targets for most DC that leave peripheral tissues. However, a few observations suggest that tissue-resident DC can somehow return to the blood and carry antigen (Ag) to organs other than LN. For example, DC carry fluorescent beads, dyes or Ag to the spleen after either i.c. injection or instillation into the lung5-7. However, whether and to what extent blood-borne DC carry Ag to other tissues is unclear. This question has clinical relevance, since cancer vaccine trials have infused ex vivo modified autologous DC into patients8. The migration of DC to non-lymphoid organs might be advantageous to boost memory responses to previously encountered pathogens, because Ag-experienced T cells disseminate to many non-lymphoid tissues9. Importantly, different organs harbor T cells with distinct functional properties. The BM is particularly conspicuous here; BM-resident CD8+ memory cells undergo more vigorous homeostatic proliferation and respond faster to Ag-stimulation than in other tissues10,11. The BM actively accumulates central memory CD8+ T cells (TCM), which are more frequent among T cells in the BM than elsewhere12. In cancer patients, memory T cells in the BM possess enhanced anti-tumor reactivity compared to peripheral blood memory cells13. However, it is still unclear whether and how recall Ag from tumors or pathogens gain access to the BM.
Here, we report that small numbers of DC traffic constitutively from peripheral tissues to blood. Moreover, circulating DC possess considerable BM tropism. Using intravital microscopy (IVM) in murine skull BM, we show that DC homing to the BM depends on microvascular P- and E-selectin as well as VCAM-1. Once in the BM, DC induced rapid proliferation of Ag-specific TCM. Using two-photon IVM to visualize T cell receptor (TCR)-transgenic TCM and homed DC in BM cavities we found that both cell types interacted rarely in the absence of Ag, while Ag-presenting DC rapidly formed stable conjugates with TCM. These data suggest a pathway for DC migration that allows DC to collect Ag in peripheral sites and, subsequently, to traffic to the BM to elicit recall responses by resident TCM.
Results
In vivo migration pathways of DC
The first objective of this study was to elucidate the trafficking routes of differentiated DC upon adoptive transfer to the circulation of normal mice. To obtain a sufficiently large number of DC to perform these experiments, DC were expanded in vivo by implanting donor mice with tumors secreting Flt3-ligand14,15. ∼2 weeks after tumor implantation, donor splenocytes yielded large numbers of CD11c+ cells representing every conventional DC subset15. Donor DC uniformly exhibited an immature phenotype (i.e. MHC class I-IIhi-lo, CD80lo, CD86lo and CCR7neg). Maturation was induced by 24-48h culture in LPS, as evidenced by upregulation of MHC and costimulatory molecules (Table 1).
Table 1.
Adhesion molecule expression in immature and mature DC
| Adhesion molecule |
CD11c+CD8α- |
CD11c+CD8α+ |
||
|---|---|---|---|---|
| Immature MFI±SEMa (% positive) | Mature MFI±SEM (% positive) | Immature MFI±SEM (% positive) | Mature MFI±SEM (% positive) | |
| CD11a (αL)b | 1154±214 (99.3) | 562±3 (89.7) | 2056±387 (99.9) | 1387±334 (98.5) |
| CD11b (αM) | 1002±209 (78.7) | 453±56 (62.3) | 245±50 (42.3) | 942±302 (37.3) |
| CD49d (α4)c | 284±63 (94.6) | 306±16 (78.0) | 293±57 (97.9) | 348±28 (70.7) |
| CD49e (α5) | 139± 39 (57.8) | 175±9 (56.3) | 176±47 (81.9) | 492±55 (41.9) |
| CD18 (β2) | 1618±403 (100) | 634±18 (97.1) | 2453±489 (100) | 1216±263 (97.6) |
| CD61 (β3) | 96±19 (57.1) | 60±6 (25.1) | 45±9 (15.7) | 70±4 (8.1) |
| β7 | 358±87 (80.4) | 107±9 (40.5) | 250±29 (95.8) | 155±7 (34.8) |
| α4β7 | 148±41 (50.8) | 41±5 (21.1) | 52±8 (22.9) | 48±3 (12.3) |
| CD44 | 2839±534 (99.2) | 1776±266 (99.0) | 4638±698 (99.7) | 4120±535 (99.9) |
| CD54 | 1109±270 (99.2) | 2347±227 (99.2) | 1482±204 (99.9) | 3863±283 (97.9) |
| CD103 (αIEL) | 41±11 (15.6) | 25±2 (6.2) | 127±32 (48.1) | 40±7 (9.3) |
| CD62L (L-selectin) | 834±325 (50.1) | 128±51 (28.7) | 1456±647 (36.3) | 222±84 (41.4) |
| CD162 (PSGL-1) | 1563±104 (98.8) | 2123±103 (99.2) | 2384±62 (99.7) | 2503±9 (100) |
| P-selectin ligand | 1722±414 (69.3) | 888 (52.9) | 4363±672 (73.9) | 4556 (90.4) |
| CCL19 ligand | 655±292 (31.4) | 905±451 (87.9) | 1983±746 (56.7) | 2219±1269 (63.2) |
| CXCR4 | 186 (99) | 554 (99) | 252 (99) | 550 (99) |
| Activation marker | ||||
| MHC II | 1101±270 (76.0) | 1645±5 (94.9) | 1500±307 (95.7) | 1705±90 (80.1) |
| CD80 | 80±20 (45.3) | 1816±384 (91.8) | 133±32 (78.4) | 2326±711 (77.6) |
| CD86 | 93±30 (37.9) | 2138±398 (95.0) | 153±42 (70.9) | 3575±897 (83.8) |
| CD40 | 66±18 (35.2) | 654±20 (87.9) | 112± 31 (60.9) | 693±11 (73.5) |
Expression levels are displayed as mean fluorescence intensity (MFI) as well as percentage of cells above background; n = 2-7/group
The αL integrin chain forms an obligate heterodimer (called LFA-1) exclusively with the β2 integrin chain; hence, αL levels must reflect αLβ2/LFA-1 expression
The α4 integrin chain forms obligate heterodimer with either β1 (VLA-4) or β7; therefore, α4 surface levels represent the sum of α4β1/VLA-4 and α4β7 heterodimers
After tail vein injection, the distribution of donor DC in recipient blood and tissues was analyzed by flow cytometry (Fig. 1a). At 2h after injection of both immature and mature DC (Fig. 1b,d), the largest numbers were recovered from the liver followed by the lung and the spleen. At 24h after injection (Fig. 1c,e), immature DC were retained to a greater extent than mature DC in the liver, whereas the number of immature and mature DC in the lung had dropped by 90% and 95%, respectively. Unexpectedly, there was also a substantial accumulation of preferentially immature DC in the recipients’ BM. At 2h after injection, the number of immature and mature DC in the BM corresponded to ∼50% and ∼20% of the respective splenic subset. Compared to this early time-point, most mature DC (88%) disappeared from the BM at 24h after injection, while the number of immature DC decreased by only 39%. By contrast, immature DC were rapidly cleared from the spleen (64% reduction).
Figure 1.

In vivo migration pathways of immature and mature DC. (a) DC-enriched CD45.2+ splenocytes were injected i.v. into CD45.1+ mice and allowed to circulate for 2 h. Homed donor DC in recipient organs were identified by coexpression of CD45.2 and CD11c (gated subset). (b-e) Organ distribution of freshly-prepared, CFSE-labeled immature (empty bars) or LPS-matured (solid bars) DC after i.v. injection. Recipient mice were injected with allogeneic or syngeneic DC, and organs harvested after 2 (b,e) or 24 h (c,e). The absolute number of homed DC in each tissue was calculated by multiplying the frequency of CD11c+ transferred cells by the total number of resident leukocytes. Data are presented as number of recovered DC per 106 injected. (f) Homing of CD8α+CD11c+ DC, compared with total CD11c+ DC, to various organs after i.v. injection. Dashed line indicates the percentage of CD8α+CD11c+ DC in the input (* p<0.01, n = 4-8/group).
Circulating DC were virtually excluded from all SLO other than the spleen. This is consistent with the finding that neither immature nor mature DC co-express sufficient levels of L-selectin and CCR7 (Table 1), the prerequisite traffic molecules for lymphocyte homing to LN and Peyer’s patches16. Equivalent patterns of DC distribution were seen when non-fluorescent CD45.2+ DC were injected i.v. into congenic CD45.1+ recipients (Fig. 1a) or when DC were obtained from donors without Flt3ligand-secreting tumor (data not shown), indicating that DC trafficking was not affected by the labeling procedure or cytokine-induced expansion. Adoptive transfer of DC (C57BL/6) into allogeneic (Balb/c or FVB) recipients yielded similar results (data not shown), indicating that DC recruitment is independent of the recipients’ genetic background.
Subset-specific recruitment of circulating DC
Differential expression of CD8α identifies discrete DC subsets with distinct migratory and functional properties as well as tissue distribution17. Thus, we asked whether there are subset-specific differences in trafficking after i.v. injection. At 2h after transfer of input cells containing ∼25% CD8α+ DC, the frequency of this subset among donor DC was lower in peripheral blood and in all non-pulmonary recipient organs analyzed (Fig. 1f). Conversely, CD8α+ DC represented 38% of donor-derived DC in the lung, suggesting that these relatively large cells are preferentially trapped in pulmonary capillaries. However, the ratio of CD8α+ to CD8α- DC in non-pulmonary sites, including the BM, was similar to their ratio in blood, suggesting that CD8α+ and CD8α- DC subsets migrate equivalently once they have passed the pulmonary sieve.
These experiments suggest that DC recruitment to non-inflamed peripheral organs other than the lung is governed by tissue-specific factors, regardless of the DC’s maturation state or subset membership. However, the retention and/or survival of DC depends on their maturation state and the tissue environment, which seems especially favorable for immature DC in the BM.
DC reach the BM from peripheral tissues
Having determined that circulating DC possess BM tropism, we asked whether DC can reach the BM from peripheral sites. Thus, we deposited 2-8×106 CFSE-labeled immature DC in a footpad and monitored their appearance in the draining popliteal LN, blood, spleen and BM. When DC-maturation was induced by simultaneous injection of 10ng LPS, large numbers of DC migrated to the popliteal LN, but even without LPS a few DC appeared within 24h in the popliteal LN and this population increased considerably at 48h (Fig. 2a,b). The transferred immature DC were almost undetectable in blood, spleen and BM after 24h. However, labeled DC were readily recoverable from all three tissues at 48h. Maturation accelerated DC dissemination from the footpad, since significant numbers were recovered from blood, BM and spleen as early as 24h after injection.
Figure 2.



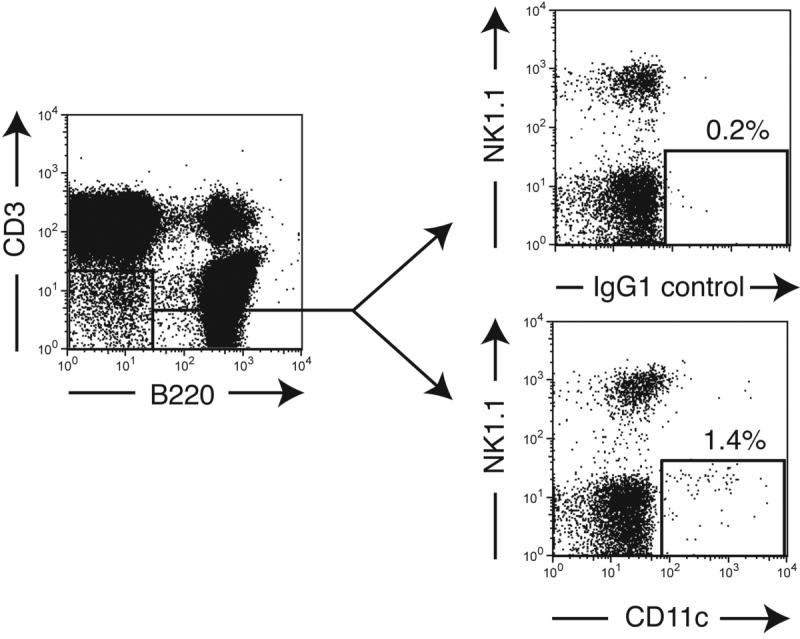
DC traffic constitutively to BM. Partially purified immature (empty bars) or LPS-matured (solid bars) DC were labeled with CFSE and injected in the footpad of recipient mice. After 24h (a) or 48h (b), blood, BM and lymphoid organs were harvested and the number of CFSE+ DC determined (* p<0.05, ** p<0.01, n =4-6/group). (c) CD45.1+ and CD45.2+ mice were parabiosed, and BM harvested at various times after surgery. Left panel shows the time course of appearance of partner-derived leukocytes in the BM of CD45.2+ (closed symbols), or CD45.1+ parabiosed partners (open symbols). Right panel shows the time course of appearance of partner-derived DC in BM (n = 2 mice/group at 3 and 5 d, and 1 mouse/group at other times). (d) Representative FACS dot plots showing small numbers of NK1.1- CD11c+ DC contained within the CD3-B220- leukocyte fraction in thoracic duct lymph.
Constitutive trafficking via blood to BM
Since immature DC are present in normal peripheral blood18, we asked whether these endogenous DC migrate to the BM. CD45.1+ and CD45.2+ congenic mice were surgically joined at the flanks, which results within 3 days in a shared circulation. The BM of parabiotic animals was then screened for partner-derived leukocytes19.
BM was harvested 3 to 15 days after surgery, and in one experiment, mice were separated 28 days after parabiosis and BM was analyzed 28 days later. As soon as a shared circulation was established, partner-derived leukocytes appeared in the BM of parabiosed mice (Fig. 2c). At 5 days post surgery, 1% and 1.9% of DC in the BM were partner-derived in CD45.2 and CD45.1 hosts, respectively (Fig. 2c). By day 15, the average frequency of BM-resident partner-derived DC was 7%. Consistent with the short life-span of DC20, partner-derived DC persisted poorly after separation. Nonetheless, it is likely that at later time-points some of the partner-derived CD11c+ arose from homed precursor cells19. However, the rapid kinetics with which DC became detectable after parabiosis indicate that full-fledged DC migrate constitutively to normal BM.
Thoracic duct lymph contains DC
Having determined that a stream of circulating endogenous DC enters the BM, we asked whether this might include erstwhile tissue-resident DC. Such peripheral DC might enter draining lymphatics and bypass or traverse downstream LN to reach the circulation via the thoracic duct. Indeed, thoracic duct lymph fluid from normal mice contained a small population of NK1.1-CD11c+ cells that constituted 0.013±0.004% of viable cells (Fig. 2d), suggesting that a subtle, but continuous stream of DC migrates from peripheral tissues via the lymph to the blood and from there to select targets, including the BM.
Molecular mechanisms of DC recruitment to BM
Using IVM in murine skull BM21 we characterized the molecules that enable blood-borne DC to adhere to BM microvessels. Both immature and mature DC engaged in rolling in BM venules and sinusoids (Fig. 3a), but immature DC rolled more frequently than mature DC (25.2±1.4% vs. 15.3±1.3%, respectively; p<0.05). There was no difference in the frequency at which rolling cells arrested (Fig. 3b). Injection of anti-P-selectin reduced DC rolling by >50%. Even more pronounced inhibition was achieved with combined anti-P- and anti-E-selectin, although anti-E-selectin alone was ineffective (Fig. 3c).
Figure 3.

Molecular mechanisms of DC homing to BM. CD11c+ immature or mature DC were labeled with calcein and injected into the right carotid artery of an anesthetized mouse. Interactions of injected cells with skull BM microvessels were analyzed using epi-fluorescence IVM21. (a) Rolling fraction (percentage of rolling cells in the total flux of cells passing through a vessel) of immature or LPS-matured DC. (b) Sticking fraction (percentage of rolling cells that arrested for ≥30 s) of immature and mature DC. (c,d) IVM experiments were performed in VCAM-1-deficient mice and in WT animals before and after mAb treatment to characterize the adhesion pathways involved in (c) rolling and (d) sticking of DC in BM microvessels. Results after mAb treatment were normalized to those obtained from the same vessel prior to mAb injection. Rolling and sticking fractions in VCAM-1-deficient mice were normalized to those in WT littermates (*p<0.05, **p<0.01).
VCAM-1 is constitutively expressed in BM microvessels and mediates firm adherence of hematopoietic progenitors (HPC) in skull BM21. The main ligand for VCAM-1, the integrin α4β1 (VLA-4), is expressed on immature and mature DC (Table 1). DC sticking was significantly reduced by anti-α4 treatment and in VCAM-1 deficient mice (p<0.01; Fig 3d). Thus, circulating DC interact with the two endothelial selectins to roll and with VCAM-1 to arrest in normal BM microvessels. Interestingly, although firm arrest on VCAM-1 presumably required that the rolling DC receive a chemoattractant signal that activates α4β1, DC sticking was unaffected after Gαi protein blockade with pertussis toxin (data not shown). Moreover, DC sticking was not reduced by anti-LFA-1 (αLβ2), which mediates leukocyte arrest in other microvascular beds16 and is abundantly expressed on DC (Table 1). The recruitment signals for BM-tropic DC thus are very similar to the multi-step adhesion cascade that mediates homing of HPC and TCM to the BM12,21.
DC can activate BM-resident TCM
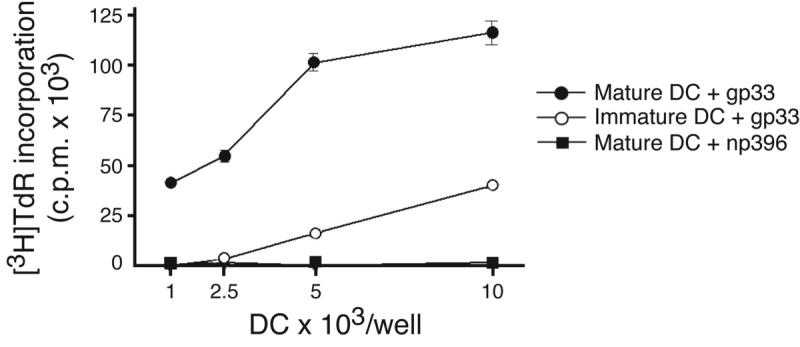
To determine whether blood-borne DC could trigger immune responses after homing to the BM, CFSE-labeled DC were i.v. injected and then sorted from recipient organs 24h later. Recovered DC from BM and spleen were as potent as freshly-isolated splenic DC at stimulating allogeneic T cells (data not shown). Thus, homing does not alter the immunostimulatory capacity of DC. Next, we asked whether DC can transport and present Ag to TCM, which represent a major subset among human and murine BM-resident T cells12. TCR transgenic CD8+ T cells from P14 mice were differentiated into TCM-like cells using established tissue culture methods22. In vitro differentiated TCM have equivalent homing characteristics as endogenous TCM23, migrate avidly to the BM12 and are readily restimulated by recall Ag22. Accordingly, P14 TCM proliferated vigorously during co-culture with mature and, to a lesser extent, immature DC that had been pulsed with specific (gp33), but not irrelevant (np396) peptide Ag (Fig. 4a).
Figure 4.



Homed DC induce antigen-specific TCM proliferation in the BM. (a) In vitro proliferation of TCM in response to peptide-pulsed immature and mature DC. Proliferation of T cells in response to different numbers of gp33-pulsed mature (filled circles) or immature DC (empty circles) or np396-pulsed control DC (filled squares) was measured by [3H]thymidine incorporation after 3d of coculture. (b) In vivo proliferation of CFSE-labeled TCM 42h after injection of immature DC pulsed with gp33 or np396, or mature DC pulsed with gp33. Histograms are gated on Vα2+CFSE+ T cells. (c) T cell proliferation in spleen, BM and liver induced by immature DC (empty bars) and mature DC (solid bars; n = 2-4/group). (d) Mice harboring CFSE-labeled TCM were injected with mature gp33-pulsed or control DC. Spleen, BM and LN were removed 17h later and single-cell suspensions were cultured for 2 more days (66h). Cultures contained single-cell suspensions prepared from tissues without any addition of cytokines.
To determine whether DC could also stimulate BM-resident TCM in vivo, 5-7×106 CFSE-labeled P14 TCM were injected i.v. into recipient mice, then rested for 24h. Subsequently, 3-5×106 immature or mature DC pulsed with gp33 or control peptide were injected i.v. Two days later, TCM were harvested from different organs and analyzed for CFSE dilution, a measure of proliferation. As expected, T cells did not divide in mice injected with np396-pulsed DC (Fig. 4b), whereas gp33-pulsed DC elicited vigorous T cell proliferation in spleen, liver and BM (Fig. 4c). By 42h after mature DC injection, TCM had divided 4-5 times in these organs; in recipients of immature DC TCM had also divided, but lagged behind those stimulated with mature DC by ∼half a division. Irrespective of the maturation state of injected DC, few TCM proliferated in LN, which is consistent with the inability of circulating DC to home to these organs24.
To exclude the possibility that the divided TCM found in DC-challenged BM had homed there after having received activation signals elsewhere, we harvested organs early (17h) after DC injection before any TCM had a chance to divide (Fig. 4d). Single cell suspensions of collected tissues were then maintained in vitro. After 2d in culture, vigorous T cell proliferation was apparent in both spleen and BM cultures from recipients of gp33-pulsed DC. This effect was Ag-specific because tissues from animals that had received control peptide-pulsed DC contained only undivided TCM. LN cultures from recipients of gp33-pulsed DC showed no TCM proliferation, suggesting that the few divided cells in LN that were harvested 42h after DC injection had been stimulated elsewhere and subsequently migrated to LN via the blood. Conversely, TCM proliferation in early BM cultures demonstrated that BM-tropic DC are immunocompetent. Thus, at least a fraction of the divided TCM observed in BM in vivo (Fig. 4b) was most likely activated by homed DC in situ.
Visualization of DC-T cell interactions in BM
Having determined that circulating DC can carry Ag to the BM and potently stimulate Ag-specific responses by TCM, we asked whether and how DC and TCM interact in the BM. To this end, we conducted 2-photon IVM in mouse skull BM cavities12. P14 TCM were labeled with the blue nuclear dye Hoechst 33342, and injected i.v. into recipient mice. Peptide-pulsed DC were labeled with CMTMR, a red-orange fluorophor, and injected 12-16h later. After 2h, the mice were prepared for 2-photon IVM as described12. The luminal compartment of BM microvessels was visualized with FITC-dextran, and three-color fluorescence image stacks were acquired over 30min (1 stack/min) to record 3D time-lapse videos of extravascular TCM-DC interactions.
With no Ag present, both DC and TCM exhibited continuous random movements throughout the BM cavity (supplemental video 1), and the majority of contacts between DC and TCM were short-lived, although about one in four TCM remained associated with DC throughout the observation period (Fig. 5a,c). However, most Ag-independent prolonged contacts were not associated with a shape change of the T cells, whereas P14 TCM polarized to undergo tight, long-lasting contacts with a broadened interface with gp33-pulsed DC (Fig. 5b,c supplemental video 2). The presence of Ag also resulted in a decrease of TCM instantaneous velocity, whereas DC migrated slowly whether they presented cognate Ag to TCM or not (Fig. 5d). This was also reflected in the mean square displacement plots (a measure of cell motilty25), which showed a marked Ag-dependent suppression in the motility of TCM (Fig. 5e), but not of DC (Fig. 5f).
Figure 5.
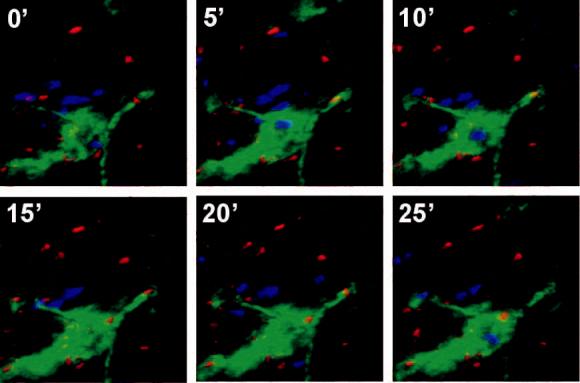
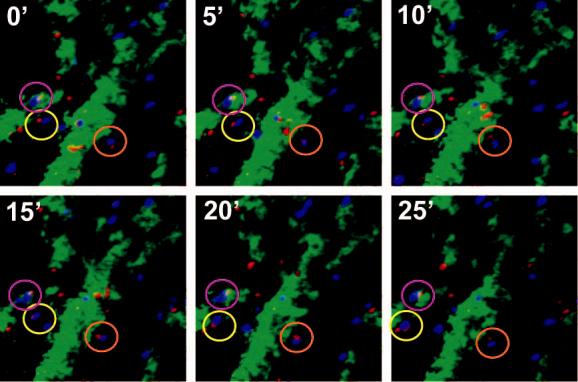

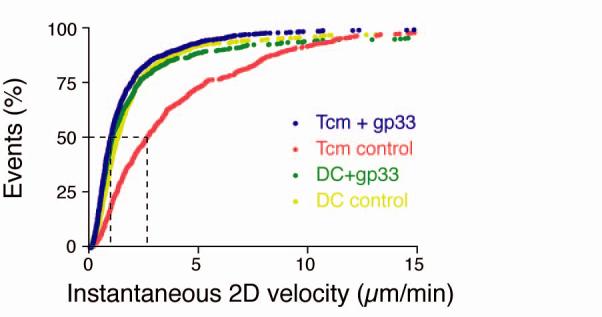

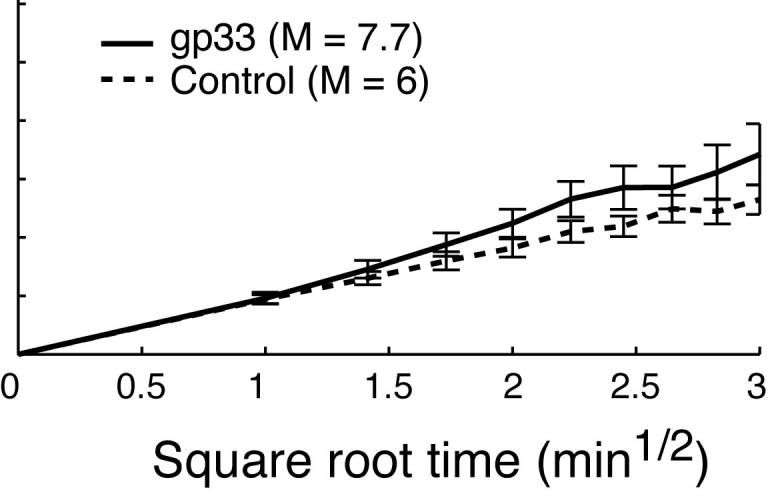
Two-photon microscopy analysis of DC-T cell interactions and motility in skull BM. (a, b) Intravital multiphoton micrographs of a BM cavity in mouse skull after injection of TCM (blue) and mature DC (red) that were either unpulsed (a) or pulsed with gp33 peptide (b). Vessels were delineated by injection of 2MDa FITC-dextran (green). Time-lapse images at 5 min intervals illustrate contacts between T cells and DC in the absence, or presence of Ag. (c) Duration of T cell-DC conjugates with or without gp33 antigen was determined manually and is displayed as cumulative dissociation curve. (d) Instantaneous 2D velocities for individual DC and T cells were determined using automatic tracking software and are displayed as cumulative velocity curves. The red (T cell control) and blue (T cell + gp33) are significantly different as determined by Mann Whitney t test for non-gaussian distributions (p<0.001). (e, f) Mean displacement plots for T cells (e) and DC (f), in the presence (solid line) or absence (dotted line) of antigen. The motility coefficient (M) for each population is given in parenthesis.
Discussion
This study was conceived with the objective to better define the target organs of circulating DC. It is already well established that tissue-resident DC continuously sample Ag in peripheral tissues and carry it to draining LN where they present it to recirculating T cells1. Earlier studies have noted that a small fraction of DC from certain peripheral tissues can relocate to distant SLO that are not connected by lymphatics, indicating that some tissue-resident DC can return to the blood5-7. These observations imply that blood-borne DC may be highly migratory and composed of more than one subset distinguished by different histories and perhaps destined for distinct target tissues18,26,27. However, our understanding of blood-borne DC trafficking is largely based on comparisons of DC vaccines that were administered by different routes8. In addition, it is known that blood-borne DC migrate to inflamed tissues28, but the path(s) taken by circulating DC in the absence of inflammation are still poorly understood.
Our initial strategy was to inject labeled primary DC into mice to track their migration. However, the number of bona fide DC that can be routinely harvested from normal mice is prohibitively small for in vivo trafficking studies. To overcome this obstacle, donor mice were implanted with a melanoma secreting Flt3-ligand, which dramatically increased production of all DC subsets without perturbing their function14,15. When purified Flt3-ligand-expanded DC were injected i.v. they migrated preferentially to the liver, lung and spleen, but were excluded from all other SLO, consistent with earlier observations24,29-31. The inability of circulating DC to home to LN and PP is due to insufficient expression of essential homing receptors, especially L-selectin and (on immature DC) CCR724. By contrast, DC express abundant ligands for selectins and α4β1, the VCAM-1 receptor15,24. Both endothelial selectins as well as VCAM-1 are constitutively expressed in BM where they mediate HPC and TCM recruitment12,21,32. Indeed, DC homing to the BM depended on P- (and E-)selectin-mediated rolling followed by VCAM-1-dependent sticking. Remarkably, the number of homed DC in BM was comparable to the spleen, and the BM retained DC even better than the spleen after 24h.
One caveat was that the injection of supraphysiologic DC numbers could have saturated the sites at which DC normally exit the circulation resulting in abnormal spillover to the BM. Therefore, we studied DC migration in parabiotic mice. In these experiments partner-derived DC were detectable in the BM with kinetics that were much more rapid than the slow differentiation of DC from progenitors26. We conclude that substantial numbers of full-fledged DC continuously access normal BM from the blood.
What are the functional consequences of this migratory pathway? The BM can function as a SLO in virus-infected mice in which lymphocytes cannot traffic to SLO33. Thus, the BM provides a suitable microenvironment for T cell priming. Homing of immature DC to the BM could contribute to this function by supplying highly phagocytic cells that may collect Ag locally. Additionally, DC can capture Ag in the blood and subsequently migrate to extravascular sites to prime lymphocytes4.
Although the physiological role of the BM during T cell priming is still being debated, there is mounting evidence that the BM plays a role in recall responses and memory T cell homeostasis33. Indeed, viral infections induce the formation of TCM that are particularly important for long-term protection34. These cells preferentially accumulate and proliferate in the BM and mount stronger effector responses than the corresponding population in the blood33. Moreover, recent evidence suggests that optimal preservation of CD8 memory requires the presence of DC35. Given these findings, we hypothesized that DC could traffic to the BM to trigger recall responses by BM-resident TCM.
As predicted, homed DC efficiently stimulated BM-resident TCM. In contrast to naïve T cells, which are only activated by mature DC, TCM proliferation was readily observed with immature DC. This is consistent with the lower costimulation requirements for memory T cell activation9. Another difference between naïve T cells and TCM is suggested by our 2-photon microscopy experiments; as early as two hours after DC injection virtually all TCM-DC interactions were manifest as tight, long-lasting conjugates. By contrast, 2-photon IVM of naïve T cell priming in LN show that there is an initial phase of ∼8h during which T cells touch Ag-presenting DC only briefly. Tight contacts are only observed thereafter36. The faster kinetics of stable conjugation in the BM might reflect the enhanced susceptibility of TCM to recall Ag. Alternatively, the BM microenvironment may foster different T cell-APC interaction kinetics compared to PLN.
An important question was whether tissue-resident DC can collect Ag and then migrate to the BM via the blood. It seemed plausible that such peripheral DC might enter local lymphatics and bypass or traverse downstream LN to reach the circulation via the thoracic duct. Consistently, we found that thoracic duct lymph fluid in normal mice contained a small DC population that constituted 0.013±0.004% of viable cells. Assuming a leukocyte flux of ∼7×106/h through the thoracic duct of an adult mouse37, we calculate that at least 22,000 peripheral DC return to the circulation via this route every day. Only a fraction of this population is likely to migrate to the BM, whereas our results in parabiotic mice indicate that at least 30,000 partner-derived DC enter the BM every day. Therefore, DC can probably reach the blood-BM conduit also via routes other than lymphatics.
Indeed, while the mechanisms that govern interstitial DC migration into draining lymph vessels are partially understood, it is unclear whether this is the only route by which DC leave tissues. For example, high endothelial venules and chronically inflamed microvessels express CCL21, a ligand for CCR7, which mediates lymphocyte traffic in SLO and is also required for DC migration into lymph vessels38. It is conceivable that CCR7+ DC could enter the bloodstream by migrating across CCL21+ venules. Indeed, monocyte-derived DC can undergo reverse transmigration, whereby they cross an endothelial monolayer in abluminal-to-luminal direction39.
The suggestion that tissue-resident DC could reach the blood, and then migrate to other organs has surfaced repeatedly in the literature, but has not been addressed systematically. DC carry fluorescent beads, dyes or antigens to the spleen after either i.c. injection or instillation into the lung5-7,40,41. These cells could only have reached the spleen by re-entering the circulation. Similarly, after solid organ transplantation, graft-derived DC appear rapidly in the recipient’s blood and spleen42,43. Finally, CD18+ leukocytes, including CD11c+ DC, collect S. typhimurium from the intestinal lumen and then appear in the blood as early as 15 min after intragastric infection44. We have performed similar experiments by infecting mice orally with invasion-deficient S. typhimurium. At 30-60 min after infection we detected small numbers (10-20 cfu/ml) of bacteria in peripheral blood. By 24-36 h after infection, bacteria could be cultured from the BM (30-1120 cfu/whole BM) and the spleen (150-9700 cfu/spleen). Indeed, using GFP-tagged S. typhimurium we could detect GFP+ DC in spleen and BM of infected mice (unpublished data).
Our results provide evidence for a migratory route that channels DC from peripheral tissues. While the number of DC that could be recovered from the BM was 10- to 30-fold lower than what was found in the draining LN, the relatively small number of migrating DC might well be sufficient to elicit substantial immune responses by the highly reactive memory T cell population that is prevalent in the BM. Following deposition in the footpad, equivalent numbers of immature and mature DC were recovered from the BM, although the former migrated more slowly than the latter. This maturation-dependent difference in migratory kinetics indicates that the departure of DC from peripheral tissues is an active process and not simply a consequence of the sudden increase in interstitial pressure caused by cell injection.
Our findings demonstrate that small numbers of DC continuously leave peripheral tissues and gain access to the bloodstream via the thoracic duct. Using adoptive transfer protocols and several approaches to trace endogenous DC, we show that blood-borne mature and immature DC home to normal BM through a multi-step cascade that involves P- and E-selectin as well as VCAM-1. Our experimental data suggest that DC can take this novel migration route to induce rapid Ag-specific activation of BM-resident CD8+ TCM as a truly central memory response.
Materials and Methods
Mice
C57Bl/6 (CD45.2/Ly5.2), congenic CD45.1/Ly5.1 (B6.SJL-PtprcaPep3b/BoyJ), FVB and BALB/c mice were purchased from Jackson Labs (Bar Harbor, ME), Taconic (Germantown, NY) or Charles River Laboratories (Wilmington, MA). P14 transgenic mice45 were obtained from Jackson Labs. Conditional endothelial VCAM-1-deficient mice were generated as described46. All mice were housed and bred in a specific pathogen free/viral antibody free animal facility. All experiments were performed in accordance with NIH guidelines and approved by Committees on Animals at both Harvard Medical School and the CBRI.
Reagents
The LCMV gp33 (KAVYNFATC) and np396 (FQPQNGQFI) peptides were synthesized at Biosource International (Camarillo, CA).
Antibodies
For intravital microscopy mAbs against murine P-selectin (clone 5H1) and E-selectin (9A9) were provided by B. Wolitzky (Hoffman LaRoche, Nutly, NJ); anti-CD11a (TIB213), anti-α4 (PS/2) and anti-β7 (FIB504) mAbs were a gift from E. Butcher (Stanford University, CA). Anti-β2 mAb (GAME-46) was purchased from Pharmingen (San Diego, CA).
For flow cytometry, fluorochrome-labeled mAbs were purchased from Pharmingen. Expression of P-selectin ligands was detected with P-selectin-Ig (Pharmingen). CCR7 expression was measured by the binding of CCL19-Ig23. Data acquisition was performed using a FACScalibur flow cytometer, and analyzed using CellQuest software (BD Biosciences, San Jose, CA).
Cell preparation
Splenic DC were isolated from C57Bl/6 mice injected subcutaneously with 4×106 Flt3-ligand-secreting B16 tumor cells 11-14 d previously. For homing assays, DC were enriched from total splenocytes by density gradient centrifugation over Optiprep (Sigma-Aldrich), and the low density cells collected. DC-enriched preparations routinely contained 75-85% CD11c+ DC. For IVM experiments, CD11c+ DC were purified by positive selection with anti-CD11c-microbeads (>95% CD11c+; Miltenyi Biotec, Auburn, CA). DC maturation was induced by culture in the presence of 1 μg/ml LPS (E.coli 0.26:B6; Sigma) for 24-48 h. TCM were generated in vitro using established techniques22.
Homing assays
DC were labeled for 20 min at 37°C with 30 μM CFSE (5(6)-FAM, SE (5(6)-carboxyfluorescein succinimidyl ester; Molecular Probes, Inc., Eugene, OR). Recipient mice were injected i.v. with 2-5×107 DC. In some experiments 2-8×106 DC were injected s.c. into both footpads instead. After 2-48 h, recipient mice were sacrificed, the blood and other organs harvested and processed to single cell suspension. Livers and lungs were first digested with collagenase type 2 (0.5%; Worthington Biochemical Corp., Lakewood, NJ). BM was collected from tibias and femora of hind legs, which accounts for ∼20% of total body BM47. Homed cells were identified as DC by CD11c staining.
Cannulation of the thoracic duct
C57BL/6 mice were fed with 700 μl of olive oil by gavage. 45 minutes later, mice were anaesthetized by intraperitoneal injection of ketamine (50 mg kg-1) and xylazine (10 mg kg-1) and laparotomized. A heparinized PE-10 polyethylene catheter was inserted into the cysterna chyli48, lymph was collected for 30-45 min, stained with mAbs and analyzed by FACS.
Intravital microscopy
IVM of the murine cranial BM microcirculation was performed as described21. Boluses of calcein-AM (Molecular Probes)-labeled DC were retrogradely injected into the carotid artery. Cells that entered the BM microvasculature were visualized by video-triggered stroboscopic epi-illumination (Chadwick Helmuth, El Monte, CA) through a FITC filter set. Video images were recorded using a low-lag silicon-intensified target camera (VE1000-SIT; Dage MTI, Michigan City, IN), a time base generator (For-A Corp. Ltd, Montvale, NJ) and a Hi8 VCR (Sony, Boston, MA). Cell behavior was determined by off-line analysis. The rolling fraction per venule was measured as the percentage of DC that interacted with the vascular wall within the total number of fluorescent cells that passed through the vessel during the observation period. The sticking fraction was defined as the percentage of rolling cells that became firmly adherent for ≥30 s.
Parabiotic mice
Pairs of parabiotic mice, consisting of one CD45.1 and one CD45.2 congenic mouse, were prepared as described19. Pairs of mice were analyzed between 3 and 28 d after parabiosis. One pair of mice was surgically separated after 28d, and analyzed 28 d later.
In vivo antigen presentation
Mice were injected intravenously with 5-15×106 CFSE-labeled P14 TCM cells. 24 h later, the same mice were injected intravenously with 5-10×106 peptide-pulsed DC (5 μg/ml for 2 h at 37°C). After a further 42-48 h, organs were harvested and single cell suspensions stained for the P14-specific TCR Vα2. In some experiments mice were sacrificed 17-18 h after DC injection, organs harvested and unpurified single-cell suspensions cultured without additional cytokines for up to 3 d. Proliferation was assessed by CFSE dilution as described 49.
Two photon microscopy
For 2-photon imaging, P14 TCM were labeled with Hoechst 33342 (10 μg/ml; Molecular Probes) for 20 min at 37°C prior to i.v. injection into recipient mice. 16-18 h later, the mice were injected i.v. with DC labeled with 10 μM; 5-and-6, 4-chloromethylbenzoylamino tetramethylrhodamine (CMTMR; Molecular Probes). Imaging commenced approximately 2-3 h after injection of DC. Mice were prepared for IVM imaging of the skull bone marrow as described12. Immediately prior to imaging, mice were injected with FITC-dextran (2 MDa; Sigma) to delineate BM vasculature. Two-photon imaging was performed using an Olympus BX50WI fluorescence microscope equipped with a 20X/NA 0.95 objective and a Radiance 2100MP Confocal/Multiphoton microscopy system, controlled by Lasersharp software (BioRad, Hercules, CA). For two-photon excitation, a MaiTai Broad-band Ti:S laser (Spectra Physics, Mountain View, CA) was tuned to 800 nm.
3D analysis of cell migration was previously described12,36. Briefly, stacks of x-y sections were acquired every 30-60s. Sequences of image stacks were transformed into volume-rendered four-dimensional movies using Volocity software (Improvision), which was also used for semi-automated tracking of moving objects. From centroid coordinates, parameters of cellular motility were calculated by using custom scripts in Matlab (MathWorks)50.
Statistical analysis
All data are presented as mean ± SEM. The Students’ t test was used for comparison of two groups. In all cases, significance was determined at p<0.05.
Supplementary Material
Acknowledgments
We thank A. Vazquez-Torres (University of Colorado) for mutant strains of Salmonella; and G. Cheng and B. Reinhardt for technical support. Special thanks to L. Scimone, for help with intravital microscopy analysis. Supported by NIH grants AI061663, HL62524, HL54936, HL56949 to (U.H.v.A). I.B.M. was recipient of the Amy Potter fellowship and grants from the Charles Hood Foundation and the Multiple Myeloma Foundation. R.B. was supported, in part, by a fellowship from Federazione Italiana Ricerca sul Cancro.
References
- 1.Banchereau J, et al. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 2.Cavanagh LL, von Andrian UH. Travellers in many guises: The origins and destinations of dendritic cells. Immunol. Cell. Biol. 2002;80:448–62. doi: 10.1046/j.1440-1711.2002.01119.x. [DOI] [PubMed] [Google Scholar]
- 3.Sallusto F, et al. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur. J. Immunol. 1998;28:2760–2769. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 4.Balazs M, Martin F, Zhou T, Kearney J. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity. 2002;17:341–352. doi: 10.1016/s1074-7613(02)00389-8. [DOI] [PubMed] [Google Scholar]
- 5.Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–761. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- 6.Legge KL, Braciale TJ. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 2003;18:265–77. doi: 10.1016/s1074-7613(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 7.Mullins DW, et al. Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J. Exp. Med. 2003;198:1023–1034. doi: 10.1084/jem.20021348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr. Opin. Immunol. 2003;15:138–47. doi: 10.1016/s0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 9.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 2004;22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 10.Di Rosa F, Santoni A. Bone marrow CD8 T cells are in a different activation state than those in lymphoid periphery. Eur. J. Immunol. 2002;32:1873–1880. doi: 10.1002/1521-4141(200207)32:7<1873::AID-IMMU1873>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 11.Becker TC, Coley SM, Wherry EJ, Ahmed R. Bone marrow is a preferred site for homeostatic proliferation of memory CD8 T cells. J. Immunol. 2005;174:1269–73. doi: 10.4049/jimmunol.174.3.1269. [DOI] [PubMed] [Google Scholar]
- 12.Mazo IB, et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity. 2005;22:259–70. doi: 10.1016/j.immuni.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 13.Feuerer M, et al. Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat. Med. 2001;7:452–458. doi: 10.1038/86523. [DOI] [PubMed] [Google Scholar]
- 14.Maraskovsky E, et al. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J. Exp. Med. 1996;184:1953–62. doi: 10.1084/jem.184.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mora JR, et al. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 16.von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N. Engl. J. Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 17.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nature Rev. Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 18.Donnenberg VS, et al. Rare-event analysis of circulating human dendritic cell subsets and their presumptive mouse counterparts. Transplantation. 2001;72:1946–51. doi: 10.1097/00007890-200112270-00014. [DOI] [PubMed] [Google Scholar]
- 19.Wright DE, Wagers AJ, Gulati AP, Johnson FL, Weissman IL. Physiological migration of hematopoietic stem and progenitor cells. Science. 2001;294:1933–1936. doi: 10.1126/science.1064081. [DOI] [PubMed] [Google Scholar]
- 20.Kamath AT, et al. The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J. Immunol. 2000;165:6762–6770. doi: 10.4049/jimmunol.165.12.6762. [DOI] [PubMed] [Google Scholar]
- 21.Mazo IB, et al. Hematopoietic progenitor cell rolling in bone marrow microvessels: Parallel contributions by endothelial selectins and VCAM-1. J. Exp. Med. 1998;188:465–474. doi: 10.1084/jem.188.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manjunath N, et al. Effector differentiation is not prerequisite for generation of memory cytotoxic T lymphocytes. J. Clin. Invest. 2001;108:871–878. doi: 10.1172/JCI13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weninger W, Crowley MA, Manjunath N, von Andrian UH. Migratory properties of naive, effector, and memory CD8(+) T cells. J. Exp. Med. 2001;194:953–966. doi: 10.1084/jem.194.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robert C, et al. Gene therapy to target dendritic cells from blood to lymph nodes. Gene Ther. 2003;10:1479–1486. doi: 10.1038/sj.gt.3302008. [DOI] [PubMed] [Google Scholar]
- 25.Sumen C, Mempel TR, Mazo IB, Von Andrian UH. Intravital microscopy; visualizing immunity in context. Immunity. 2004;21:315–329. doi: 10.1016/j.immuni.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 26.del Hoyo GM, et al. Characterization of a common precursor population for dendritic cells. Nature. 2002;415:1043–1047. doi: 10.1038/4151043a. [DOI] [PubMed] [Google Scholar]
- 27.O’Keeffe M, et al. Dendritic cell precursor populations of mouse blood: identification of the murine homologues of human blood plasmacytoid pre-DC2 and CD11c+ DC1 precursors. Blood. 2003;101:1453–9. doi: 10.1182/blood-2002-03-0974. [DOI] [PubMed] [Google Scholar]
- 28.Robert C, et al. Interaction of dendritic cells with skin endothelium: A new perspective on immunosurveillance. J. Exp. Med. 1999;189:627–636. doi: 10.1084/jem.189.4.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fossum S. Lymph-borne dendritic leucocytes do not recirculate, but enter the lymph node paracortex to become interdigitating cells. Scand. J. Immunol. 1988;27:97–105. doi: 10.1111/j.1365-3083.1988.tb02326.x. [DOI] [PubMed] [Google Scholar]
- 30.Kupiec-Weglinski JW, Austyn JM, Morris PJ. Migration patterns of dendritic cells in the mouse. Traffic from the blood, and T cell-dependent and -independent entry to lymphoid tissues. J. Exp. Med. 1988;167:632–45. doi: 10.1084/jem.167.2.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lappin MB, et al. Analysis of mouse dendritic cell migration in vivo upon subcutaneous and intravenous injection. Immunology. 1999;98:181–8. doi: 10.1046/j.1365-2567.1999.00850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frenette PS, Subbarao S, Mazo IB, von Andrian UH, Wagner DD.Endothelial selectins and vascular cell adhesion molecule-1 promote hematopoietic progenitor homing to bone marrow Proc. Natl. Acad. Sci. U S A 19989514423–14428.see comments [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Rosa F, Pabst R. The bone marrow: a nest for migratory memory T cells. Trends Immunol. 2005;26:360–6. doi: 10.1016/j.it.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 34.Wherry EJ, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 2003;4:225–34. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 35.Zammit DJ, Cauley LS, Pham QM, Lefrancois L. Dendritic cells maximize the memory CD8 T cell response to infection. Immunity. 2005;22:561–70. doi: 10.1016/j.immuni.2005.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mempel TR, Henrickson SE, von Andrian UH. T cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 37.Gesner BM, Gowans JL. The output of lymphocytes from the thoracic duct of unanaesthetized mice. Br. J. Exp. Path. 1962;43:424. [PMC free article] [PubMed] [Google Scholar]
- 38.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nature Reviews Immunology. 2003;3:867–878. doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
- 39.Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, Muller WA. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998;282:480–483. doi: 10.1126/science.282.5388.480. [DOI] [PubMed] [Google Scholar]
- 40.Enioutina EY, Visic D, Daynes RA. The induction of systemic and mucosal immune responses to antigen-adjuvant compositions administered into the skin: alterations in the migratory properties of dendritic cells appears to be important for stimulating mucosal immunity. Vaccine. 2000;18:2753–2767. doi: 10.1016/s0264-410x(00)00059-1. [DOI] [PubMed] [Google Scholar]
- 41.Racanelli V, Behrens SE, Aliberti J, Rehermann B. Dendritic cells transfected with cytopathic self-replicating RNA induce crosspriming of CD8+ T cells and antiviral immunity. Immunity. 2004;20:47–58. doi: 10.1016/s1074-7613(03)00353-4. [DOI] [PubMed] [Google Scholar]
- 42.Larsen CP, Morris PJ, Austyn JM. Migration of dendritic leukocytes from cardiac allografts into host spleens. A novel pathway for initiation of rejection. J. Exp. Med. 1990;171:307–14. doi: 10.1084/jem.171.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saiki T, Ezaki T, Ogawa M, Matsuno K. Trafficking of host- and donor-derived dendritic cells in rat cardiac transplantation: allosensitization in the spleen and hepatic nodes. Transplantation. 2001;71:1806–15. doi: 10.1097/00007890-200106270-00017. [DOI] [PubMed] [Google Scholar]
- 44.Vazquez-Torres A, et al. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature. 1999;401:804–8. doi: 10.1038/44593. [DOI] [PubMed] [Google Scholar]
- 45.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–61. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 46.Koni PA, et al. Conditional Vascular Cell Adhesion Molecule 1 Deletion in Mice. Impaired lymphocyte migration to bone marrow. J. Exp. Med. 2001;193:741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boggs DR. The total marrow mass of the mouse: a simplified method of measurement. Am. J. Hematol. 1984;16:277–286. doi: 10.1002/ajh.2830160309. [DOI] [PubMed] [Google Scholar]
- 48.Boak JL, Woodruff MFA. Modified technique for collecting mouse thoracic duct lymph. Nature. 1965;205:396. doi: 10.1038/205396a0. [DOI] [PubMed] [Google Scholar]
- 49.Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J. Immunol. Methods. 2000;243:147–54. doi: 10.1016/s0022-1759(00)00231-3. [DOI] [PubMed] [Google Scholar]
- 50.Mempel TR, Scimone ML, Mora JR, von Andrian UH. In vivo imaging of leukocyte trafficking in blood vessels and tissues. Curr. Opin. Immunol. 2004;16:406–17. doi: 10.1016/j.coi.2004.05.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.