

Short abstract
In the present study, we searched for mutant PTEN transcripts in aggressive rheumatoid arthritis synovial fibroblasts (RA-SF) and studied the expression of PTEN in RA. By automated sequencing, no evidence for the presence of mutant PTEN transcripts was found. However, in situ hybridization on RA synovium revealed a distinct expression pattern of PTEN, with negligible staining in the lining layer but abundant expression in the sublining. Normal synovial tissue exhibited homogeneous staining for PTEN. In cultured RA-SF, only 40% expressed PTEN. Co-implantation of RA-SF and normal human cartilage into severe combined immunodeficiency (SCID) mice showed only limited expression of PTEN, with no staining in those cells aggressively invading the cartilage. Although PTEN is not genetically altered in RA, these findings suggest that a lack of PTEN expression may constitute a characteristic feature of activated RA-SF in the lining, and may thereby contribute to the invasive behaviour of RA-SF by maintaining their aggressive phenotype at sites of cartilage destruction.
Keywords: rheumatoid arthritis, synovial membrane, fibroblasts, PTEN tumour suppressor, severe combined immunodeficiency (SCID) mouse model, cartilage destruction, in situ hybridization
Abstract
Aims:
PTEN is a novel tumour suppressor which exhibits tyrosine phosphatase activity as well as homology to the cytoskeletal proteins tensin and auxilin. Mutations of PTEN have been described in several human cancers and associated with their invasiveness and metastatic properties. Although not malignant, rheumatoid arthritis synovial fibroblasts (RA-SF) exhibit certain tumour-like features such as attachment to cartilage and invasive growth. In the present study, we analyzed whether mutant transcripts of PTEN were present in RA-SF. In addition, we used in situ hybridization to study the expression of PTEN messenger (m)RNA in tissue samples of RA and normal individuals as well as in cultured RA-SF and in the severe combined immunodeficiency (SCID) mouse model of RA.
Methods:
Synovial tissue specimens were obtained from seven patients with RA and from two nonarthritic individuals. Total RNA was isolated from synovial fibroblasts and after first strand complementary (c)DNA synthesis, polymerase chain reaction (PCR) was performed to amplify a 1063 base pair PTEN fragment that encompassed the coding sequence of PTEN including the phosphatase domain and all mutation sites described so far. The PCR products were subcloned in Escherichia coli, and up to four clones were picked from each plate for automated sequencing. For in situ hybridization, digoxigenin-labelled PTEN-specific RNA probes were generated by in vitro transcription. For control in situ hybridization, a matrix metalloproteinase (MMP)-2-specific probe was prepared. To investigate the expression of PTEN in the absence of human macrophage or lymphocyte derived factors, we implanted RA-SF from three patients together with normal human cartilage under the renal capsule of SCID mice. After 60 days, mice were sacrificed, the implants removed and embedded into paraffin.
Results:
PCR revealed the presence of the expected 1063 base pair PTEN fragment in all (9/9) cell cultures (Fig. 1). No additional bands that could account for mutant PTEN variants were detected. Sequence analysis revealed 100% homology of all RA-derived PTEN fragments to those from normal SF as well as to the published GenBank sequence (accession number U93051). However, in situ hybridization demonstrated considerable differences in the expression of PTEN mRNA within the lining and the sublining layers of RA synovial membranes. As shown in Figure 2a, no staining was observed within the lining layer which has been demonstrated to mediate degradation of cartilage and bone in RA. In contrast, abundant expression of PTEN mRNA was found in the sublining of all RA synovial tissues (Figs 2a and b). Normal synovial specimens showed homogeneous staining for PTEN within the thin synovial membrane (Fig. 2c). In situ hybridization using the sense probe gave no specific staining (Fig. 2d). We also performed in situ hybridization on four of the seven cultured RA-SF and followed one cell line from the first to the sixth passage. Interestingly, only 40% of cultured RA-SF expressed PTEN mRNA (Fig. 3a), and the proportion of PTEN expressing cells did not change throughout the passages. In contrast, control experiments using a specific RNA probe for MMP-2 revealed mRNA expression by nearly all cultured cells (Fig. 3b). As seen before, implantation of RA-SF into the SCID mice showed considerable cartilage degradation. Interestingly, only negligible PTEN expression was found in those RA-SF aggressively invading the cartilage (Fig. 3c). In situ hybridization for MMP-2 showed abundant staining in these cells (Fig. 3d).
Discussion:
Although this study found no evidence for mutations of PTEN in RA synovium, the observation that PTEN expression is lacking in the lining layer of RA synovium as well as in more than half of cultured RA-SF is of interest. It suggests that loss of PTEN function may not exclusively be caused by genetic alterations, yet at the same time links the low expression of PTEN to a phenotype of cells that have been shown to invade cartilage aggressively.
It has been proposed that the tyrosine phosphatase activity of PTEN is responsible for its tumour suppressor activity by counteracting the actions of protein tyrosine kinases. As some studies have demonstrated an upregulation of tyrosine kinase activity in RA synovial cells, it might be speculated that the lack of PTEN expression in aggressive RA-SF contributes to the imbalance of tyrosine kinases and phosphatases in this disease. However, the extensive amino-terminal homology of the predicted protein to the cytoskeletal proteins tensin and auxilin suggests a complex regulatory function involving cellular adhesion molecules and phosphatase-mediated signalling. The tyrosine phosphatase TEP1 has been shown to be identical to the protein encoded by PTEN, and gene transcription of TEP1 has been demonstrated to be downregulated by transforming growth factor (TGF)-β. Therefore, it could be hypothesized that TGF-β might be responsible for the downregulation of PTEN. However, the expression of TGF-β is not restricted to the lining but found throughout the synovial tissue in RA. Moreover, in our study the percentage of PTEN expressing RA-SF remained stable for six passages in culture, whereas molecules that are cytokine-regulated in vivo frequently change their expression levels when cultured over several passages. Also, cultured RA-SF that were implanted into SCID mice and deeply invaded the cartilage did not show significant expression of PTEN after 60 days. The drop in the percentage of PTEN expressing cells from the original cell cultures to the SCID mouse implants is of interest as this observation goes along with data from previous studies that have shown the prominent expression of activation-related molecules in the SCID mice implants that in vivo are found predominantly in the lining layer. Therefore, our data point to endogenous mechanisms rather than to the influence of exogenous human cytokines or factors in the downregulation of PTEN. Low expression of PTEN may belong to the features that distinguish between the activated phenotype of RA-SF and the sublining, proliferating but nondestructive cells.
Introduction
PTEN is a novel tumour suppressor that exhibits tyrosine phosphatase activity as well as homology to the cytoskeletal proteins tensin and auxilin [1,2]. Mutations in PTEN have been described in several human cancers, and have been associated with the invasiveness and metastatic properties of malignancies [1,2,3]. Although not malignant, rheumatoid arthritis (RA) synovial fibroblasts (SF) are imbued with certain tumour-like features such as attachment to cartilage and invasive growth [4]. Moreover, it has been suggested that tyrosine kinase activity, which counteracts the action of tyrosine phosphatases, is increased in RA [5] and may be involved in the activation of mitogen-activated protein kinase in human synovial cells [6].
In the present study, we analyzed whether mutant transcripts of PTEN were present in RA-SF. In addition, we studied the expression of PTEN messenger (m)RNA in tissue samples from seven RA patients and two normal individuals, as well as in cultured RA-SF and in the severe combined immunodeficiency (SCID) mouse co-implantation model of RA. Aggressively invading RA-SF expressed only low levels of PTEN, which showed no evidence for mutations. This lack of expression was maintained in cultured RA-SF over several passages, and when RA-SF were implanted into SCID mice together with normal human cartilage.
Methods
Tissue preparation and cell cultures
Synovial tissue specimens were obtained from seven patients with RA undergoing synovectomy or joint replacement and from two nonarthritic individuals. Immediately after surgery, one part of the tissue was embedded in Tissue-Tek OCT medium (Miles, Elkhart, IN, USA), snap-frozen and stored at -80°C, and a second was fixed in 4% buffered formalin for 6 h before embedding in paraffin. Another portion was digested enzymatically and the released cells were grown in Dulbecco's modified Eagle medium with 10% foetal calf serum [7]. At confluence, cells were harvested and half of them were used for complementary (c)DNA preparation. The remaining cells were used to maintain the culture, as well as for growing cells on chamber slides (Lab-Tek II; Nalge Nunc Int, Naperville, IL, USA) 48 h before in situ hybridization.
RNA isolation and reverse transcription polymerase chain reaction
Total RNA was isolated from cultured SA applying the TRIzol RNA isolation kit (Life Technologies, Basel, Switzerland) according to the manufacturer's protocol. After first strand cDNA synthesis using oligo-d(T)12-18 primers and Moloney murine leukemia virus (M-MuLV) reverse transcriptase (Boehringer-Mannheim, La Jolla, California, USA), a 1063 base pair PTEN cDNA fragment was amplified using polymerase chain reaction (PCR) with Pyrococcus furiosus (Pfu) DNA-polymerase (Stratagene, La Jolla, California, USA). This fragment encompassed the coding sequence of PTEN including the phosphatase domain and all mutation sites described so far [1,8,9,10]. The primer sequences were as follows: upper primer 5';-GAC AGC CAT CAT CAA AGA GA-3'; and lower primer, 5'-TGA CGG CTC CTC TAC TGT T-3'. The amplification was carried out for 32 cycles under annealing-extension conditions of 52°C for 1 min and 72°C for 2 min using a Perkin-Elmer (Foster City, California, USA) DNA-Thermocycler 480. To look for additional, low copy transcripts, the cycle number was increased stepwise up to 42 cycles and the annealing temperature was decreased to 48°C.
Cloning and sequencing of the PTEN fragments
The PCR products were then ligated into the PCR-Script Amp SK+ vector (Stratagene), and transformation of the vector into Epicurian Coli XL1-Blue MRF' Kan supercompetent cells was performed. After selection, up to four clones were picked from each plate and plasmid preparation of the PTEN-insert containing plasmids was performed using the Qiagen MiniPrep Kit (Qiagen, Basel, Switzerland). The sequences of the inserts were determined using automated, dideoxy sequencing.
Riboprobe preparation
The Qiagen MaxiPrep Kit was used for large scale preparation of PTEN-insert containing plasmids from two successfully transfected clones, and templates were prepared by linearization with BamH I or Not I (Life Technologies). Again, plasmid sequence was checked by automated sequencing, which confirmed the 100% identity of the PTEN fragment to the published GenBank sequence (accession number 193051). Antisense and sense RNA probes were then obtained by in vitro transcription using T3 and T7 RNA polymerase (Boehringer-Mannheim) with a commercially available transcription kit (Stratagene). For in situ hybridization, probes were labelled with digoxigenin-UTP (Boehringer-Mannheim). The RNA probe for control in situ hybridization to detect matrix metalloproteinase (MMP)-2 was prepared accordingly using a plasmid obtained from the American Type Culture Collection (ATCC, Rockville, Maryland, USA; ATCC number 79066).
In situ hybridization
In situ hybridization was performed as described by Kriegsmann et al [11]. Briefly, after fixation, tissue sections were hybridized with the digoxigenin-labelled riboprobes (either antisense or sense) in hybridization buffer containing 50% formamide, 1× Denhardt's solution, 10% dextran sulphate, 25 μ g/ml herring sperm DNA (Boehringer-Mannheim), 40 mg/ml yeast transfer RNA (Sigma Chemical Co, St Louis, Missouri, USA) for 16 h at 52°C. After hybridization, unbound probe was digested at 37°C for 45 min with 10 μ g/ml RNase A (Boehringer-Mannheim), and consecutive washing steps were performed at 50°C at the following stringencies: 50% formamide/2 × SSC (5 min); 1 × SSC + 1% sodium dodecyl sulphate (SDS; 15 min); 0.25 × SSC + 1% SDS (15 min); and 0.1% SSC + 1% SDS (15 min). Immunological detection was performed after blocking nonspecific binding sites with 2% horse serum (30 min at room temperature) by incubation with alkaline phosphatase-conjugated antidigoxigenin Fab fragments (Boehringer-Mannheim) for 1 h at room temperature, diluted 1/500 in Tris-NaCl, pH 7.6, containing 1% normal horse serum. After washing with Tris-NaCl (pH 7.6) and Tris-NaCl/MgCl2 (pH 9.5), the sections were incubated with 5-bromo-4-chloro-3-indolyl-phosphate/4-nitro blue tetrazolium chloride colour substrate solution (Boehringer Mannheim) containing 1 mmol/l levamisole (DAKO, Zug, Switzerland), and developed at room temperature in darkness. Colour development was stopped with Tris-NaCl (pH 7.6).
Severe combined immunodeficiency mouse co-implantation experiments
SCID mice were obtained from the Charles Rivers GmbH (Sulzfeld, Germany) and kept permanently in sterile conditions. Implantation of RA-SF together with normal human cartilage was performed as described previously [7]. RA-SF from three different patients were used for the SCID mouse experiments. Briefly, after trypsinization, washing and centrifugation, 105 cells were resuspended in 100 μl sterile culture medium and inserted into the cavity of an inert sponge (Gelfoam, Pharmacia & Upjohn, Dübendorf, Germany) together with an 1 mm3 piece of normal human articular cartilage. Mice were anaesthetized intraperitoneally with 0.014 mg/g Xylocain (Lidocain hydrochloride; Astra Pharmaceutica, Dieticon, Switzerland) and 0.09 mg/g Ketalar (Ketamin hydrochloride; Parke-Davis, Baar, Switzerland) in an isotonic solution, and a 1 cm incision was made on the left flank of the animals. The left kidney was exteriorized and, once a small incision was made, an implant was placed under the renal capsule. The peritoneal layer and the skin were closed using 5.0 prolene suture material. After 60 days, mice were sacrificed and the implants removed. Tissue preparation included fixation in 4% buffered formalin and paraffin embedding according to standard procedures.
Results
Using the specific primers, the expected 1063 base pair PTEN fragment was amplified from the total cDNA of all (all of nine) cell cultures by PCR. Moreover, no additional bands that could account for mutant PTEN variants were detected, even when PCR conditions were changed towards lower specificity (Fig. 1). PCR products were then subcloned into Escherichia coli, and up to four successfully transformed clones were picked for plasmid preparation from each culture plate (total number of samples 21). Sequence analysis revealed 100% homology of all RA-derived PTEN fragments to those obtained from normal SF as well as to the published GenBank sequence (accession number U93051).
Figure 1.
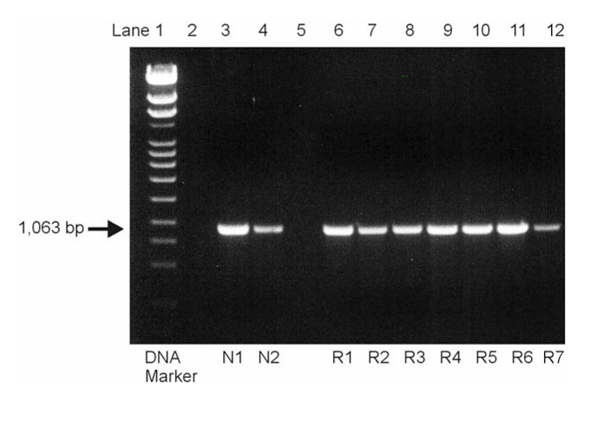
Analysis of the amplified polymerase chain reaction products by electrophoresis on a 1% agarose gel. The expected PTEN band was visible in all normal (N1, N2 in lanes 3 and 4) and rheumatoid arthritis (R1-R7 in lanes 6–12) specimens, and no additional transcripts could be detected, even at lower specificity.
In situ hybridization with digoxigenin-labelled RNA probes, however, demonstrated considerable differences in the expression of PTEN mRNA within the lining and the sublining layers of RA synovial membranes. As shown in Figure 2a, no staining was observed within the lining layer, which has been demonstrated to mediate degradation of cartilage and bone in RA [4]. In contrast, abundant expression of PTEN mRNA was found in the sublining layer of all RA synovial tissues (Figs 2a and b). Normal synovial specimens showed homogeneous staining for PTEN within the thin synovial membrane (Fig. 2c). Expression of PTEN mRNA was seen in the most superficial layer of normal synovium as well as in deeper regions, with most cells being of fibroblast shape (Fig. 2c). In situ hybridization using the sense probe gave no specific staining (Fig. 2d). We also performed in situ hybridization on four of the seven cultured RA-SF and followed one cell line from the first to the sixth passage. Interestingly, only 40% of cultured RA-SF expressed PTEN mRNA (Fig. 3a), and the proportion of PTEN-expressing cells did not change significantly throughout the passages. In contrast, control experiments using a specific RNA probe for MMP-2 revealed mRNA expression by nearly all cultured cells (Fig. 3b) indicating constitutive expression of MMP-2 but not of PTEN in the absence of macrophages and lymphocytes, and their locally derived factors.
Figure 2.
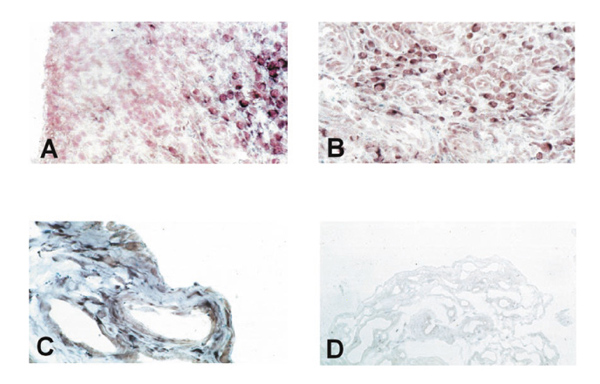
In situ hybridization on rheumatoid arthritis synovial tissue shows only negligible expression of PTEN in the lining layer (A) but abundant expression in the sublining layer (A, B). Normal synovium consisting of only two to three cell layers of synovial cells showed clear staining, both in the most superficial layers and in deeper regions (C). The sense probe gave no specific staining (D).
Figure 3.
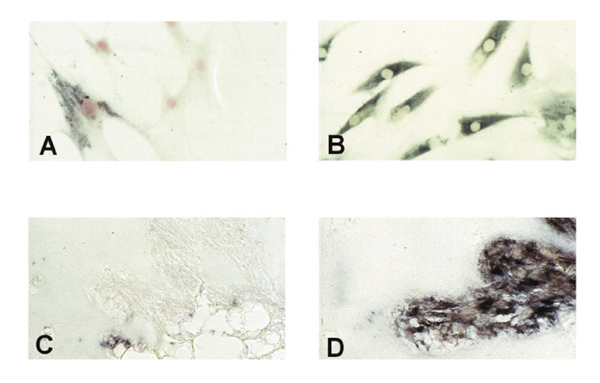
In cultured rheumatoid arthritis synovial fibroblasts (RA-SF) only about 40% expressed PTEN (A) whereas matrix metalloproteinase (MMP)-2 was expressed constitutively by almost every cell (B). PTEN expression (C) was negligible in RA-SF aggressively invading the co-implanted cartilage in the renal capsule of severe combined immunodeficiency mice. However, abundant expression of MMP-2 messenger RNA was found (D).
To test the hypothesis further that PTEN downregulation in RA-SF is not caused by such exogenous factors, we co-implanted RA-SF from three patients together with normal human cartilage under the renal capsule of SCID mice and maintained the implants for 60 days as described previously [7]. Before implantation, RA-SF showed the above described expression pattern for PTEN. Histological evaluation of the implants after the rats were killed revealed considerable cartilage degradation by the RA-SF [7]. Interestingly, by in situ hybridization with PTEN-specific RNA probes, only negligible PTEN expression was found in those RA-SF aggressively invading the cartilage (Fig. 3c). Again, control in situ hybridization with RNA probes for MMP-2 showed abundant staining in these cells (Fig. 3d).
Discussion
Although the present study found no evidence for mutations of PTEN in RA synovium, the observation that PTEN expression is lacking in the lining layer of RA synovium as well as in more than half of cultured RA-SF is of interest. This suggests that loss of PTEN function may not exclusively be caused by genetic alterations, but that it links the low expression of PTEN to a phenotype of cells that have been shown to invade cartilage aggressively. Moreover, the present results may also have an impact on further investigations of other tumour suppressors such as p53, which has been found to be genetically altered simultaneously with PTEN in several cancers [10], and somatic mutations of which have also been described in RA [12]. On the basis of the inconsistency and great variability of p53 mutations in RA, it has been proposed that these mutations, although contributing to the invasive behaviour of rheumatoid tissue, may occur secondary to other changes and may not represent the primary step in the activation of RA-SF [12]. These data, together with our observations of PTEN downregulation in nonmalignant but aggressively invading RA-SF, suggest that the lack of PTEN expression may be specifically associated with certain features of malignant cells.
It has been proposed that the tyrosine phosphatase activity of PTEN is responsible for its tumour suppressor activity [1,13] by counteracting the actions of protein tyrosine kinases. Because some studies have demonstrated an upregulation of tyrosine kinase activity in RA synovial cells, it might be speculated that the lack of PTEN expression in aggressive RA-SF contributes to the imbalance of tyrosine kinases and phosphatases in this disease [5]. The extensive amino-terminal homology of the predicted protein to the cytoskeletal proteins tensin and auxilin, however, suggests a complex regulatory function involving cellular adhesion molecules and phosphatase-mediated signalling [9].
Tamura et al [13] most recently demonstrated that PTEN interacts with the focal adhesion kinase, and negatively regulates cellular interactions with the extracellular matrix by inhibiting integrin-mediated cell spreading, as well as formation of focal adhesions. Also, the tyrosine phosphatase TEP1 has been shown to be identical to the protein encoded by PTEN, and gene transcription of TEP1 has been demonstrated to be downregulated by transforming growth factor (TGF)-β [14]. Therefore, it could be hypothesized that TGF-β, which is expressed abundantly in the RA synovial membrane [15], might be responsible for the downregulation of PTEN. The expression of TGF-β is not restricted to the lining, however, but is found throughout the synovial tissue in RA [15]. Moreover, in the present study the percentage of PTEN-expressing RA-SF remained stable for six passages in culture, whereas molecules that are cytokine-regulated in vivo frequently change their expression levels when cultured over several passages. Also, cultured RA-SF that were implanted into SCID mice and deeply invaded the cartilage did not show significant expression of PTEN after 60 days.
The drop in the percentage of PTEN-expressing cells from the original cell cultures to the SCID mouse implants is of interest, because this observation is in accord with data from previous studies [16] that showed the prominent expression of activation related molecules in the SCID mice implants that are found predominantly in the lining layer in vivo. It may be speculated that activated, aggressive RA-SF are selected positively during the implantation by apoptosis of the nonaggressive cells.
With regard to PTEN, the present data point to endogenous mechanisms rather than to the influence of exogenous human cytokines or factors in the downregulation of PTEN. In this context, the question of whether the PTEN low-expressing phenotype constitutes a subset of RA-SF that are identical to previously described activated RA-SF is of critical importance. Some recent studies [16] searching for apoptosis regulating molecules as well as adhesion molecules and signalling cascades in RA-SF have provided novel insights into the nature of these aggressive RA-SF and have helped to characterize them on a molecular level. Thus far, though, there is no specific marker for the activated phenotype of RA-SF found in the lining layer of RA patients. Low expression of PTEN may be among the features that distinguish between the activated phenotype of RA-SF and the sublining, proliferating but nondestructive cells. It needs to be stressed, however, that the association between the lack of PTEN expression and the aggressive phenotype of RA-SF is, at this point, only phenomenological. A comprehensive analysis of different markers and pathways including functional analysis will be needed to clearly identify and specifically distinguish the activated phenotype of RA-SF that aggressively invade the cartilage in RA on a molecular level.
References
- Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nature Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Teng DH, Hu R, Lin H, et al. MMAC1/PTEN mutations in primary tumor specimens and tumor cell lines. Cancer Res. 1997;57:5221–5225. [PubMed] [Google Scholar]
- Muller-Ladner U, Gay RE, Gay S. Cellular pathways of joint destruction. Curr Opin Rheumatol. 1997;9:213–220. doi: 10.1097/00002281-199705000-00007. [DOI] [PubMed] [Google Scholar]
- Williams WV, VonFeldt JM, Ramanujam T, Weiner DB. Tyrosine kinase signal transduction in rheumatoid synovitis. Semin Arthritis Rheum . 1992;21:317–329. doi: 10.1016/0049-0172(92)90025-9. [DOI] [PubMed] [Google Scholar]
- Migita K, Eguchi K, Tsukada T, et al. The role of protein kinase in human synovial fibroblast growth. Biochem Biophys Res Commun. 1995;210:1066–1075. doi: 10.1006/bbrc.1995.1765. [DOI] [PubMed] [Google Scholar]
- Muller-Ladner U, Kriegsmann J, Franklin BN, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. . Am J Pathol. 1996;149:1607–1615. [PMC free article] [PubMed] [Google Scholar]
- Furnari FB, Lin H, Huang HS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci USA. 1997;94:12479–12484. doi: 10.1073/pnas.94.23.12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Freije D, Nusskern DR, et al. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998;58:204–209. [PubMed] [Google Scholar]
- Rasheed BK, Stenzel TT, McLendon RE, et al. PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res. 1997;57:4187–4190. [PubMed] [Google Scholar]
- Kriegsmann J, Keyszer GM, Geiler T, et al. Expression of vascular cell adhesion molecule-1 mRNA and protein in rheumatoid synovium demonstrated by in situ hybridization and immunohistochemistry. Lab Invest. 1995;72:209–214. [PubMed] [Google Scholar]
- Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. . Proc Natl Acad Sci USA. 1997;94:10895–10900. doi: 10.1073/pnas.94.20.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M, Gu J, Matsumoto K, et al. Inhibition of cell migration, and focal adhesion by tumor suppressor PTEN. Science . 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- Lafyatis R, Thompson NL, Remmers EF, et al. Transforming growth factor-beta production by synovial tissues from rheumatoid patients and streptococcal cell wall arthritic rats. Studies on secretion by synovial fibroblast-like cells and immunohistologic localization. J Immunol. 1989;143:1142–1148. [PubMed] [Google Scholar]
- Franz JK, Pap T, Hummel KM, et al. Expression of sentrin, a novel anti-apoptotic molecule at sites of synovial invasion in rheumatoid arthritis. Arthritis Rheum. 2000;43 doi: 10.1002/1529-0131(200003)43:3<599::AID-ANR17>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]