

Short abstract
IFN-γ was measured in supernatants after in vitro stimulation of peripheral blood mononuclear cells with collagen type II (CII), purified protein derivative or influenza virus. IFN-γ production in response to CII was similar in rheumatoid arthritis (RA) patients and healthy control individuals. The IFN-γ response to purified protein derivative and influenza virus was lower in RA patients, reflecting a general T-cell hyporesponsiveness in RA. After recalculating the response to CII taking this hyporesponsiveness into account the CII response was higher in RA patients, and was associated with human leucocyte antigen (HLA)-DRB1*0401 and HLA-DQA1*0301-DQB1*0302 (HLA-DQ8). Rheumatoid arthritis patients with elevated serum levels of immunoglobulin (Ig)G anti-CII antibodies had lower CII-induced IFN-γ production than patients with low anti-CII levels. The relative increase in CII-reactivity in RA patients as compared with healthy control individuals, and the association of a higher response with RA-associated HLA haplotypes, suggest the existence of a potentially pathogenic cellular reactivity against CII in RA.
Keywords: collagen type II, human leucocyte antigen-DR, IFN-γ, rheumatoid arthritis, T cell
Abstract
Introduction:
Despite much work over past decades, whether antigen-specific immune reactions occur in rheumatoid arthritis (RA) and to what extent such reactions are directed towards joint-specific autoantigens is still questionable. One strong indicator for antigenic involvement in RA is the fact that certain major histocompatibility complex (MHC) class II genotypes [human leucocyte antigen (HLA)-DR4 and HLA-DR1] predispose for the development of the disease [1]. In the present report, collagen type II (CII) was studied as a putative autoantigen on the basis of both clinical and experimental data that show an increased frequency of antibodies to CII in RA patients [2,3,4] and that show that CII can induce experimental arthritis [5].
It is evident from the literature that RA peripheral blood mononuclear cells (PBMCs) respond poorly to antigenic stimulation [6,7,8], and in particular evidence for a partial tolerization to CII has been presented [9]. The strategy of the present work has accordingly been to reinvestigate T-cell reactivity to CII in RA patients, to relate it to the response to commonly used recall antigens and to analyze IFN-γ responses as an alternative to proliferative responses.
Aims:
To study cellular immune reactivity to CII in patients with RA and in healthy control individuals and to correlate this reactivity to HLA class II genotypes and to the presence of antibodies to CII in serum.
Methods:
Forty-five patients who met the 1987 American College of Rheumatology classification criteria for RA [10] and 25 healthy control individuals of similar age and sex were included. Twenty-six of these patients who had low levels of anti-CII in serum were randomly chosen, whereas 19 patients with high anti-CII levels were identified by enzyme-linked immunosorbent assay (ELISA)-screening of 400 RA sera.
Heparinized blood was density gradient separated and PBMCs were cultured at 1 × 106/ml in RPMI-10% fetal calf serum with or without antigenic stimulation: native or denatured CII (100 μ g/ml), killed influenza virus (Vaxigrip, Pasteur Mérieux, Lyon, France; diluted 1 : 1000) or purified protein derivative (PPD; 10 μ g/ml). CII was heat-denatured in 56°C for 30 min.
Cell supernatants were collected after 7days and IFN-γ contents were analyzed using ELISA. HLA-DR and HLA-DQ genotyping was performed utilizing a polymerase chain reaction-based technique with sequence-specific oligonucleotide probe hybridization. Nonparametric statistical analyses were utilized throughout the study.
Results:
PBMCs from both RA patients and healthy control individuals responded with inteferon-γ production to the same degree to stimulation with native and denatured CII (Fig. 1a), giving median stimulation indexes with native CII of 4.6 for RA patients and 5.4 for healthy control individuals, and with denatured CII of 2.9 for RA patients and 2.6 for healthy control individuals. RA patients with elevated levels of anti-CII had a weaker IFN-γ response to both native and denatured CII than did healthy control individuals (P = 0.02 and 0.04, respectively).
Stimulation with the standard recall antigens PPD and killed influenza virus yielded a median stimulation index with PPD of 10.0 for RA patients and 51.3 for healthy control individuals and with influenza of 12.3 for RA patients and 25.7 for healthy, control individuals. The RA patients displayed markedly lower responsiveness to both PPD and killed influenza virus than did healthy control individuals (Fig. 1b). IFN-γ responses to all antigens were abrogated when coincubating with antibodies blocking MHC class II.
The low response to PPD and killed influenza virus in RA patients relative to that of healthy control individuals reflects a general downregulation of antigen-induced responsiveness of T cells from RA patients [6,7,8]. That no difference between the RA group and the control group was recorded in CII-induced IFN-γ production therefore indicates that there may be an underlying increased responsiveness to CII in RA patients, which is obscured by the general downregulation of T-cell responsiveness in these patients. In order to address this possibility, we calculated the fraction between individual values for the CII-induced IFN-γ production and the PPD-induced and killed influenza virus-induced IFN-γ production, and compared these fractions. A highly significant difference between the RA and healthy control groups was apparent after stimulation with both native CII and denatured CII when expressing the response as a fraction of that with PPD (Fig. 2a). Similar data were obtained using killed influenza virus-stimulated IFN-γ values as the denominator (Fig. 2b).
When comparing the compensated IFN-γ response to denatured CII stimulation between RA patients with different HLA genotypes, highly significant differences were evident, with HLA-DRB1*0401 patients having greater CII responsiveness than patients who lacked this genotype (Fig. 3a). HLA-DQ8 positive patients also displayed a high responsiveness to CII as compared with HLA-DQ8 negative RA patients (Fig. 3b). These associations between the relative T-cell reactivity to denatured CII and HLA class II genotypes were not seen in healthy control individuals. Similar results were achieved using influenza as denominator (P = 0.02 for HLA-DRB1*0401 and P = 0.01 for HLA-DQ8).
Discussion:
No reports have previously systematically taken the general T-cell hyporesponsiveness in RA into account when investigating specific T-cell responses in this disease. In order to address this issue we used the T-cell responses to PPD and killed influenza virus as reference antigens. This was made on the assumption that exposure to these antigens is similar in age-matched and sex-matched groups of RA patients and healthy control individuals. The concept of a general hyporesponsiveness in RA T cells has been documented in several previous reports, in which both nominal antigens [6,7,8] and mitogens [11,12,13] have been used. The fact that a similar functional downregulation in RA PBMCs was obtained with both PPD and killed influenza virus as reference antigens strengthens the validity of our approach.
We identified an association between the IFN-γ response to CII and HLA-DRB1*0401 and HLA-DQ8 in the RA patient group, which is of obvious interest because both these MHC class II alleles have been associated with high responsiveness to CII in transgenic mice that express these human MHC class II molecules [14,15]. There was no association between high anti-CII levels and shared epitope (HLA-DRB1*0401 or HLA-DRB1*0404).
Conclusion:
CII, a major autoantigen candidate in RA, can elicit an IFN-γ response in vitro that is associated with HLA-DRB1*0401 and HLA-DQ8 in RA patients. This study, with a partly new methodological approach to a classical problem in RA, has provided some additional support to the notion that CII may be a target autoantigen of importance for a substantial group of RA patients. Continued efforts to identify mechanisms behind the general hyporesponsiveness to antigens in RA, as well as the mechanisms behind the potential partial anergy to CII, may provide us with better opportunities to study the specificity and pathophysiological relevance of anti-CII reactivity in RA.
Introduction
Despite much work over past decades, whether antigen-specific immune reactions occur in rheumatoid arthritis (RA) and to what extent such reactions are directed towards joint-specific autoantigens is still questionable. The fact that certain major histocompatibility complex (MHC) class II genotypes [human leucocyte antigen (HLA)-DR4 and HLA-DR1] predispose to the development of RA [1] points to the possibility of antigenic involvement. The presence of an increased frequency of autoantibodies to cartilage-specific molecules such as collagen type II (CII) in RA [2,3,4] and the fact that CII and other joint-derived proteins can cause arthritis in experimental animals after immunization [5] indicate a possible pathogenic role of autoimmune reactions towards these molecules. It has been difficult, however, to convincingly demonstrate cellular reactivity against joint-derived autoantigens such as CII in RA. This has contributed to the widespread notion that specific T-cell reactivities may not be all that important in RA, and that autoimmunity to joint antigens such as CII may only be of importance in very limited subgroups of RA patients.
A few lines of partly new evidence have now led us to reinvestigate the issue of cellular reactivity to CII in RA. First, there is increasing evidence that immunoreactivity to CII can indeed be associated with the RA-associated HLA allele DRB1*0401 and the closely linked allele DQA1*0301-DQB1*0302 (HLA-DQ8). The best evidence comes from studies in DRB1*0401 [14,16] or HLA-DQ8 [15] transgenic mice, which have a high susceptibility to CII-induced arthritis. There are also studies of antibody production to CII in joints that indicate that this is a relatively common feature among RA patients [17,18] and is associated with the presence of HLA-DR4 [18,19]. Second, at least two possible mechanisms by which an existing T-cell responsiveness to CII may have remained undetected using conventional methods (such as determination of proliferation in response to CII in vitro) have been suggested. One mechanism may reside in the low T-cell reactivity to recall antigens in RA patients as compared with healthy control individuals [6,7,8]. Another mechanism may reside in the partial tolerization to CII that can occur in vivo, as reported in transgenic mice in which the DNA sequence of an immunodominant peptide from rat CII was expressed in the mouse CII gene [9]. In that study, T-cell responses to the rat CII peptide was measured after immunization of rat CII. Proliferative responses were not detected but peptide-induced IFN-γ production was evident in vitro, indicating that the mouse was tolerized to the antigen but that this tolerization only affected the proliferative response.
The strategy behind the present work has accordingly been to reinvestigate T-cell reactivity to CII in RA patients, taking the potential partial tolerization of CII-specific T cells and the general hyporesponsiveness of RA T cells into consideration. We investigated IFN-γ responses after CII stimulation of peripheral blood mononuclear cells (PBMCs) in vitro and compensated for the general T-cell hyporesponsiveness by expressing the CII response as a fraction of the response to standard recall antigens. We also investigated possible associations of the recorded cellular reactivity to CII with HLA genotypes. Finally, we compared CII-induced IFN-γ responses in RA patients with and without antibodies to CII.
Materials and methods
Patients
Initially 400 RA patients, including both those with early arthritis and those with more advanced stages of disease, were screened for the occurrence of high levels of anti-CII antibodies. From this screening 23 patients were identified as having more than 15U/ml anti-CII antibodies [for definition of these units, see separate method description of CII enzyme-linked immunosorbent assay (ELISA)]. These patients were asked to return and participate in the present study. Of these 19 also had elevated anti-CII levels (> 9U/ml) at inclusion into the study and comprised the group of RA patients with high anti-CII levels (median age 54 years, range 31–86 years). In addition, 26 RA patients (median age 55 years, range 23–78 years) with low anti-CII levels were randomly chosen. All patients met the 1987 American College of Rheumatology classification criteria for RA [10]. Twenty-five healthy staff members of similar age and sex from the Department of Rheumatology, Karolinska Hospital, served as healthy control individuals. Informed consent was obtained from all patients and control individuals included in the study. Patient characteristics are given in Table 1.
Table 1.
Patient characteristics
| Number | Age | Sex | Disease | Erosive | |||
| of subjects | (years) | (F/total) | duration (years) | CRP | disease | ||
| All patients | 45 | 55 | 34/45 | 10 | 15 | 27/42 | |
| (23–86) | (76%) | (0.5–30) | (0–95) | (64%) | |||
| Patients with | 54 | 15/19 | 8 | 10 | 12/18 | ||
| high anti-CII levels | 19 | (31–86) | (79%) | (0.5–30) | (0–95) | (67%) | |
| Patients with low | 26 | 55 | 19/26 | 12.5 | 20 | 15/24 | |
| anti-CII levels | (23–78) | (73%) | (2.0–30) | (0–62) | (63%) | ||
| Healthy controls | 25 | 51 | 22/25 | – | ND | – | |
| (36–61) | (88%) | ||||||
| Number of | Number of | Mean daily | |||||
| swollen | tender | NSAID | Steroid | prednisolone | MTX | DMARD | |
| joints | joints | therapy | therapy | dosage | therapy | therapy | |
| All patients | 2 | 4 | 26/42 | 13/42 | 2.0 | 13/42 | 30/42 |
| (0–17) | (0–17) | (62%) | (31%) | (31%) | (71%) | ||
| Patients with | 9 | 6 | 10/18 | 4/18 | 1.1 | 5/18 | 9/18 |
| high anti-CII levels | (0–17) | (0–17) | (56%) | (22%) | (28%) | (50%) | |
| Patients with low | 2 | 3 | 16/24 | 9/24 | /2.7 | 8/24 | 21/24 |
| anti-CII levels | (0-15) | (0–16) | (67%) | (38%) | (33%) | (88%) | |
| Healthy controls | – | – | – | – | – | – | – |
Data are given as median values and ranges for the various groups. Daily prednisolone dosages are presented as mean values in mg. Disease Modifying antirheumatic drugs (DMARDs) include sulfasalazin (0/18 high anti-CII patients treated, 6/24 low anti-CII patients treated,), cyklosporine (1/18 high anti-CII, 1/24 low anti-CII), Reumacon (a podophyllin derivative, 1/18 high anti-CII, 1/24 low anti-CII), chloroquine (0/18 high anti-CII, 2/24 low anti-CII), aurathiomalase (0/18 high anti-CII, 3/24 low anti-CII), auranofin (1/18 high anti-CII, 0/24 low anti-CII) and azatioprin (1/18 high Anti-CII, 1/24 low anti-CII). Figures for sex distribution, erosiveness and medications are given both as fraction of number positive/number Investigated, and as percentage. Data on erosive disease are based on X-ray evaluations. Patient charts were not available for three patients, one With high anti-CII levels and two with low anti-CII levels. F, female; CRP, C-reactive protein; NSAID, nonsteroidal anti-inflammatory drug; MTX, Methotrexate; ND, not done; CII, collagen type II.
Cell separation and stimulation
Peripheral blood was collected into heparinized tubes and diluted 1 : 2 with phosphate buffered saline (PBS). Mononuclear cells were isolated by density gradient centrifugation and diluted to 1 × 106/ml in RPMI-1640 (Flow Laboratories, Irvine, Scotland, UK) supplemented with glutamine, HEPES buffer (Life Technologies, Paisley, Scotland, UK), penicillin, streptomycin and 10% of a defined batch of fetal calf serum (Flow Laboratories). Chick CII (Sigma, St Louis, MO, USA) or human CII [a kind gift from Alvar Grönberg (formerly Pharmacia), Uppsala, Sweden] diluted in 0.1 mol/l acetic acid, or just 0.1 mol/l acetic acid as a buffer control, was added to 1 ml of the cell suspension, giving a final concentration of 100 μ g/ml of CII and 2 mmol/l acetic acid. When stimulating with denatured CII, a stock solution of CII was incubated at 56°C for 30 min and then added to the cell suspension.
As standard recall antigens, purified protein derivative (PPD; Statens Smittskyddsinstitut, Solna, Sweden; final concentration 10 μ g/ml) and killed influenza virus (Vaxigrip, Pasteur Mérieux, Lyon, France; diluted 1:1000) was added to 1 ml of the cell suspension.
For blocking of HLA class II antigens, three different sets of antibodies were used in parallel: a mixture of 1 μ g/ml of each of three monoclonal antibodies: antibody 2.06 [anti-DR, mouse immunoglobulin (Ig)G1], IVA12 (anti-DR, DP, mouse IgG1) and 9.3F10 (anti-DR, DQ, mouse IgG2a; all from ATCC, Rockville, Maryland, USA). This mixture of anti-class II antibodies has earlier been described to specifically block antigen-induced cytokine responses [20]. As control, an isotype-weighted mixture of the antikeyhole limpet haemocyanin monoclonal antibodies HS (mouse IgG1 [21]) and 7B4 (mouse IgG2a [21]) were used. In a second set of experiments, 10 μ g/ml of L243 (mouse IgG1; ATCC [22]) were used with HS as control antibody. In some confirmatory experiments 10 μ g/ml of a F(ab)' 2 rabbit polyclonal antibody against human class II^ antigens were used with rabbit F(ab)' 2 antibody against human IgG as control [23].
Cells were incubated in round bottomed 96-well plates (NUNC A/S, Roskilde, Denmark) for 7 days using a protocol that has been optimized for antigen induced IFN-γ detection in supernatants ([24], our unpublished data). Supernatants were collected and frozen at -20°C for later analyses of cytokine content.
IFN-γ measurements
ELISA plates (Maxisorp; NUNC A/S) were coated with 50 μ l of 2 μ g/ml catcher antibody (1-D1K; MabTech, Stockholm, Sweden) in PBS overnight at 4°C. After blocking the plates with 100 μ l PBS + 1% bovine serum albumin for 1 h at room temperature, the plates were washed with PBS + 0.05% Tween. Recombinant IFN-γ (R&D Systems, Minneapolis, MN, USA) was diluted in medium and 50 μ l of samples and cytokine standards were added in duplicates. The plates were incubated for 4 h at room temperature, washed and 50 μ l/well of a secondary biotinylated antibody (7-B6-1; MabTech) diluted to 1 μ g/ml in PBS + 1% bovine serum albumin + 0.05% Tween was added. Incubation was done at 4°C overnight. After washing, the plates were incubated for 1 h at room temperature with 50 μ l/well avidine-alkaline phosphatase (Dakopatts, Glostrup, Denmark) diluted 1 : 1000 in PBS + 0.05% Tween. After washing, 50 μ l/well substrate [p-nitrophenyl-phosphate tablets (Sigma) 1 mg/ml in diethanolamine buffer, pH9.8] was added and the reaction was read at 405 nm in a spectrophotometer. To correct for the general low T-cell reactivity seen in RA PBMCs (Fig. 1a), the CII-induced IFN-γ production was expressed as a fraction of the PPD or killed influenza virus-induced IFN-γ production obtained in parallel PBMC cultures.
Figure 1.
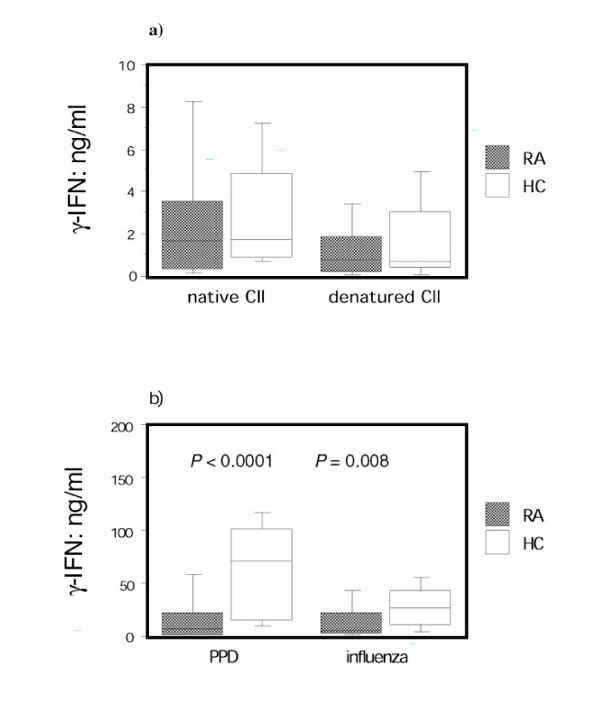
Rheumatoid arthritis (RA) peripheral blood mononuclear cells (PBMCs) respond similarly to collagen type II (CII) as healthy control individual PBMCs, but they are hyporesponsive to other antigens. PBMCs from RA patients (n = 45) and healthy control individuals (n = 25) were stimulated with antigen for 7 days, and supernatants were analyzed by enzyme-linked immunosorbent assay (ELISA) for concentration of IFN-γ . The background IFN-γ production in wells without added antigen was subtracted in each case. (a) Levels of IFN-γ after stimulation with native or denatured CII, and (b) after stimulation with purified protein derivative (PPD) or killed influenza virus are shown. The box plots show the median as a line and the 25th and 75th centiles limiting the box, with the 10th and 90th centiles indicated with bars.
Serum anticollagen type II detection by enzyme-linked immunosorbent assay
ELISA plates (Maxisorp; NUNC S/A) were coated with 100 μ l/well with native chick CII (Sigma) diluted to 5 μ g/ml in ice-cold PBS overnight at 4°C. After blocking with PBS + 1% bovine serum albumin and washing with PBS + 0.05% Tween, 100 μ l/well of a high-titre standard serum (added in a dilution series) and the samples, diluted at least 1 : 5 in PBS + 1% bovine serum albumin, were added in duplicate and incubated for 2 h at room temperature. After washing, 100 μ l of a biotinylated goat antihu-man IgG antibody (Tago, Burlingame, CA, USA) diluted to 1 μ g/ml in PBS + 1% bovine serum albumin was added and incubated overnight at 4°C. The ELISA was developed as above. Elevated levels of IgG anti-CII were defined as mean ± two standard deviations of sera from 39 healthy controls (> 9U/ml). The standard serum was arbitrarily defined as having 1500 U/ml.
Human leucocyte antigen genotyping
HLA-DR and HLA-DQ genotyping was performed utilizing a polymerase chain reaction-based technique with sequence-specific oligonucleotide probes hybridization [25,26]. DRB1*01 subtypes were identified by allele-specific polymerase chain reaction primers [27].
Statistical analysis
Nonparametric methods were used throughout the study. Differences between groups were analyzed using the Mann–Whitney U-test, and analyses for matched pairs were performed using Wilcoxon's signed-rank test. When analyzing correlations, Spearman rank correlation was used. P <0.05 was considered statistically significant.
Results
PBMCs from RA patients and healthy control individuals were challenged in vitro with native and denatured CII, and supernatants from these cultures were investigated for IFN-γ content. In both groups, an increased IFN-γ production was recorded after in vitro culture with both native and denatured CII as compared with cultures with no added antigen, giving a median stimulation index with native CII of 4.6 for RA patients and 5.4 for healthy control individuals, and with denatured CII of 2.9 for RA patients and 3.0 for healthy control individuals. No difference in IFN-γ production in response to CII could be demonstrated between RA patients and healthy control individuals (Fig. 1a). As control, PBMCs from 12 RA patients and seven healthy control individuals were stimulated with pepsin (10 μg/ml). No IFN-γ induction was evident with this stimulation (data not shown).
Parallel cultures were also stimulated with the standard recall antigens PPD and killed influenza virus. These antigens also induced the production of IFN-γ in PBMCs from both RA patients and healthy control individuals, giving median stimulation indexes with PPD of 10.0 (RA patients) and 51.3 (healthy control individuals), and with killed influenza virus of 12.3 (RA patients) and 25.7 (healthy control individuals). For both PPD and influenza stimulations there was a significant difference between the RA group and the healthy control group in that the RA patients displayed a markedly lower responsiveness to PPD and to influenza (Fig. 1b).
In order to investigate whether the observed production of IFN-γ in response to stimulation with CII, PPD and influenza was mediated by MHC class II-dependent T-cell activation, blocking antibodies were added to the cell cultures. As shown in Table 2 these antibodies inhibited the CII as well as the PPD-induced and killed influenza virus-induced IFN-γ production. No such inhibitory effects were observed after incubation with isotype-matched control monoclonal antibodies.
Table 2.
CII-induced IFN-γ response is major histocompatibility Complex (MHC) class II restricted
| No ab | Class II ab | Control ab | |
| CII | 100 | 10 (6–22) | 75 (66–125) |
| PPD | 100 | 13 (8–32) | 109 (63–123) |
| influenza | 100 | 7 (4–15) | 95 (68–125) |
Peripheral blood mononuclear cells from two rheumatoid arthritis Patients and three healthy controls were stimulated with native Collagen type II (CII), influenza virus or purified protein derivative in the Presence of antibodies (ab) specific for major histocompatibility Complex-class II or isotype-matched control antibodies. Supernatants Were collected after 7 days of culture and the concentration of IFN-γ was analysed by enzyme-linked immunosorbent assay. Values are expressed as median % (range) of cytokine present in Supernatants of which no antibodies were added.
As stated in the introduction to this report, the lower responses to PPD and killed influenza virus in RA patients than in healthy control individuals reflect a general down-regulation of antigen-induced responsiveness of T cells from RA patients [6,7,8]. That no difference between the RA group and the control group was recorded in CII-induced IFN-γ production therefore indicates that there may be an underlying increased specific and MHC class II-dependent responsiveness to CII in the RA patients, which is obscured by the general downregulation of T-cell responsiveness. In order to address this possibility, we calculated the fraction between individual values for the CII-induced IFN-γ production and the PPD-induced and killed influenza virus-induced IFN-γ production, and compared these fractions.
When compensating for the general T-cell hyporesponsiveness in RA-derived T cells in this manner, using the PPD response as the denominator, a highly significant difference between the RA group and the healthy control individuals was apparent after stimulation with both native CII and denatured CII (Fig. 2a). Similar data were obtained using killed influenza virus-stimulated IFN-γ values as the denominator, but no statistical difference between RA patients and healthy control individuals could be determined for native CII (Fig. 2b).
Figure 2.
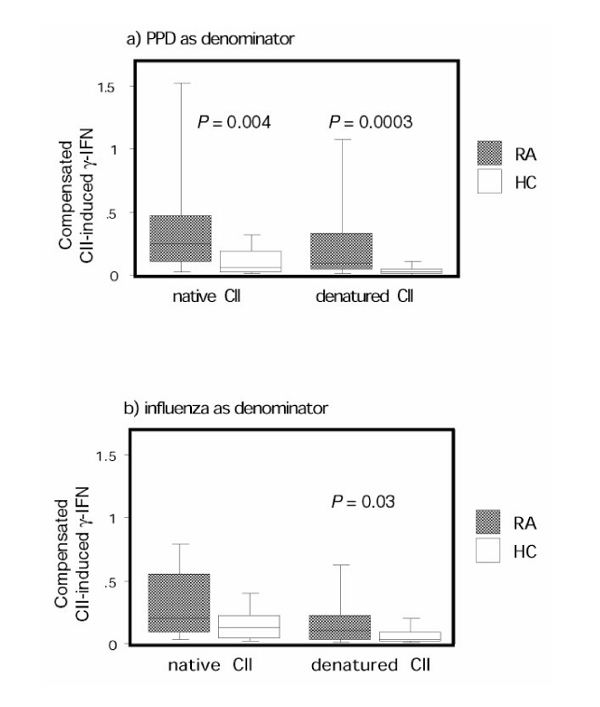
After compensating for the antigen hyporesponsiveness, rheumatoid arthritis (RA) peripheral blood mononuclear cells (PBMCs) had a higher response to collagen type II (CII) than did healthy control individual PBMCs. Production of IFN-γ in RA patients (n = 45) and healthy control individuals (n = 25) in response to stimulation with native or denatured CII after compensation for the diminished responsiveness in RA patients (a) for purified protein derivative (PPD) and (b) for killed influenza virus are shown. On the y-axis the quotient between the CII-induced IFN-γ production and (a) the PPD-induced IFN-γ production and (b) the killed influenza virus-induced IFN-γ production is given. The box plots show the median as a line and the 25th and 75th centiles limiting the box, with the 10th and 90th centiles indicated with bars.
We next investigated whether there was a difference between patients with elevated serum levels of anti-CII antibodies as compared with other RA patients with regard to the T-cell response to CII. Analyzing uncompensated IFN-γ production in response to CII stimulation revealed, contrary to our expectations, that RA patients with high levels of anti-CII had lower IFN-γ responses to denatured CII compared with those of RA patients with low levels of anti-CII (Fig. 4a). A similar tendency, but not statistically significant, could also be demonstrated using native CII. There was no difference in the PPD-induced or killed influenza virus-induced IFN-γ responses between RA patients with high and low anti-CII levels (data not shown).
Figure 4.

Rheumatoid arthritis (RA) patients with low levels of anticollagen type II (CII) had a higher cellular reactivity to denatured CII compared with RA patients with high levels of anti-CII. Production of IFN-γ in RA patients with high serum levels of anti-CII antibodies (n = 19) or low anti-CII levels (n = 26) and healthy control individuals (n = 25) is shown. In (a), IFN-γ induction after stimulation with native or denatured CII is shown. In (b), the CII response has been expressed as a fraction of the purified protein derivative (PPD) response showing, on the y axis, the quotient between the CII-induced IFN-γ production and the PPD-induced IFN-γ production. Shaded boxes represent RA patients with high serum levels of anti-CII, striped boxes represent RA patients with low levels of anti-CII and open boxes represent healthy control individuals. The box plots show the median as a line and the 25th and 75th centiles limiting the box, with the 10th and 90th centiles indicated with bars.
The results obtained after compensating for the T-cell hyporesponsiveness using PPD as the denominator demonstrate that both the RA patients with low anti-CII antibody levels and those with high levels had significantly greater responses to native CII than did healthy control individuals (Fig. 4b). For the group with low anti-CII levels there was also a highly significant increased response to denatured CII as compared with healthy control individuals, which could not be observed for patients with high anti-CII levels. Similar results were observed using influenza-induced IFN-γ as the denominator (data not shown).
We also correlated the responsiveness to CII to various clinical parameters (C-reactive protein levels, presence of erosions as measured by radiography, numbers of swollen and tender joints, and disease duration). No correlations between C-reactive protein, presence of erosions or number of swollen joints and the compensated IFN-γ response to CII could be demonstrated; the only significant association noted was a weak negative relationship between number of tender joints and the PPD-compensated CII-reactivity (P = 0.009, rs = -0.46, data not shown). The RA group with high levels of anti-CII had a significantly higher number of swollen joints than the RA group with low levels of anti-CII (P = 0.029, data not shown). A positive correlation was noted between disease duration and responsiveness to native or denatured CII (compensated values; P = 0.036, rs = 0.33 for native CII and P = 0.002, rs = 0.49 for denatured CII).
Finally, we compared the compensated IFN-γ response to denatured CII stimulation between RA patients with different HLA genotypes. Of the RA patients included in this study, 13 were DRB1*0401 positive and 15 were DQ8 positive. Out of these patients, 12 were DRB1*0401/DQ8 double positive. Of the healthy control individuals, nine were DRB1*0401 positive and five were DQ8 positive (four were not genotyped for HLA-DQ). Out of these healthy control individuals, four were DRB1*0401/DQ8 double positive. Highly significant differences were evident, with HLA-DRB1*0401 patients having higher CII responsiveness than patients lacking this genotype (Fig. 3a). HLA-DQ8-positive patients also displayed a high responsiveness to CII as compared with HLA-DQ8-negative RA patients (Fig. 3b). Similar results were achieved using killed influenza virus as the denominator (P = 0.02 for HLA-DRB1*0401 and P = 0.01 for HLA-DQ8). These associations between the relative T-cell reactivity to denatured CII and HLA class II genotypes were not seen in healthy control individuals. No significant relationship between the analyzed HLA-DR or DQ haplotypes and responsiveness to CII remained when the two different groups of RA patients were analyzed separately. No association was detected between serum levels of anti-CII and the shared epitope (DRB1*0401 or DRB1*0404).
Figure 3.
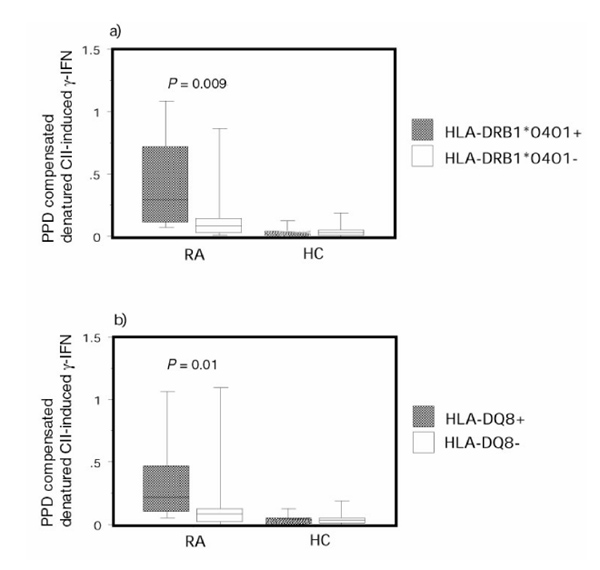
Rheumatoid arthritis (RA) patients with disease-associated human leucocyte antigen (HLA) genotypes had a higher relative reactivity to CII than RA patients with other HLA genotypes. Purified protein derivative (PPD) compensated IFN-γ production in response to denatured type II collagen are shown: (a) HLA-DRB1*0401-positive (n =13) or DRB1*0401-negative (n = 32) RA patients and healthy control individuals (n = 9 and 15, respectively); and (b) HLA-DQA1*0301-DQB1*0301 (HLA-DQ8)-positive (n = 15) and HLA-DQ8 negative (n = 30) RA patients and healthy control (HC) individuals (n =5 and 15, respectively). The box plots show the median as a line and the 25th and 75th centiles limiting the box, with the 10th and 90th centiles indicated with bars.
Discussion
The major findings reported in this paper are as follows. First, an increased IFN-γ production in response to in vitro stimulation with CII in RA patients as compared with age-matched and sex-matched healthy control individuals when compensation was made for the general hyporesponsiveness of RA patient PBMCs to nominal antigens. Second, the response to CII was dependent on MHC class II molecules, as demonstrated by the blockade of the response with anti-MHC class II antibodies. Finally, RA patients positive for the genotypes HLA-DRB1*0401 or HLA-DQ8 exhibited a higher IFN-γ response than did RA patients without these alleles.
The choice of IFN-γ production as the parameter for registration of the cellular response to CII was made both from experience in mice immunized with CII and from multiple sclerosis. In mice immunized with autologous CII, an autoimmune arthritis-provoking response may occur that is associated with in vivo antibody production to CII and with CII-induced IFN-γ production in vitro, but not with any proliferative response to CII [9]. In multiple sclerosis patients, in vitro production of T-cell cytokines such as IFN-γ is increased after in vitro stimulation with myelin antigens such as myelin basic protein and myelin-oligodendrocyte glyco-protein, despite the difficulties in detecting proliferative responses towards these antigens [28].
Although there is much previous support for the choice of IFN-γ as a readout for T-cell responsiveness to antigens, thus taking the potential partial anergy to CII into account, no reports have previously systematically taken the general T-cell hyporesponsiveness in RA into account when investigating specific T-cell responses in this disease. In the present study we used the T-cell responses to PPD and killed influenza virus as reference antigens for making this compensation. This was based on the assumption that exposure to these antigens is similar in age-matched and sex-matched groups of RA patients and healthy control individuals. The concept of a general hyporesponsiveness of RA T cells has been documented in several previous reports in which both nominal antigens [6,7,8] and mitogens [11,12,13] were used. Even though the mechanisms behind this defective T-cell response are not clear, it appears that the hyporesponsiveness is a feature of the disease itself because it has been observed also in patients who are not on disease-modifying antirheumatic drug therapy [6]. The fact that a similar functional down-regulation in RA PBMCs was obtained with both PPD and killed influenza virus as reference antigens strengthens the validity of our approach.
Both native and heat-denatured CII were used in the present study on the basis that native and denatured CII may be processed in different ways by antigen-presenting cells. As shown in Figures 1a and 4a, native CII induced more IFN-γ than did denatured CII, indicating an important role for the structure of CII in cellular stimulation. The difference in the amount of IFN-γ induced by native and denatured CII might depend on differences in uptake or processing of these structurally different CII molecules.
Proliferation in response to in vitro CII stimulation of cells from both immunized animals and in human studies have in some instances been found to be a response to contaminating pepsin in the CII preparation [29]. This possibility was excluded in the present study by control experiments that failed to demonstrate any IFN-γ production in response to pepsin (data not shown).
RA patients as well as healthy control individuals responded with IFN-γ production in vitro when PBMCs were stimulated with chick or human CII. It has not directly been demonstrated that the IFN-γ-producing cells are T cells, but the fact that blocking of MHC class II blocks the IFN-γ response by RA PBMCs as well as by control PBMCs indicates that this is indeed the case. We also attempted to identify induction of a type 2 cytokine, but failed to detect interleukin-4 in CII stimulated cultures using methods that are capable of detecting inter-leukin-4 in cultures stimulated with a recall antigen.
We identified an association between the magnitude of the IFN-γ response to CII and HLA-DRB1*0401 and HLA-DQ8 in the RA group, which is of obvious interest because both these MHC class II alleles have been associated with high responsiveness to CII in transgenic mice that express these human MHC class II molecules [14,15,16]. Thus far, susceptibility to RA has mainly been associated with HLA-DRB1*0401 and with other MHC class II molecules carrying the 'shared epitope' [30,31], whereas it remains unclear whether HLA-DQ8 has an independent association with RA. In in vitro studies of cellular responses to CII in RA patients, some reports have claimed that cellular reactivity to CII (measured as CII-induced proliferation or production of factors capable of stimulation of leucocyte functions) is associated with HLA-DR4 [32,33,34]. Others, however, have not found this association [35,36]. Anti-CII-producing B cells have been reported to be present in the joint [17,18] and have in some instances been associated with HLA-DR4 [18,19]. It is difficult to separate the influences of HLA-DR4 and HLA-DQ8 in studies of the association between MHC class II alleles and RA or in in vitro associations between cellular reactivity to CII and MHC class II alleles, because of the strong linkage disequilibrium between DRB1*0401 and HLA-DQ8. Finally, it should be noted that the RA patients in the present study were recruited in two different ways; 19 patients were selected on the basis of their high anti-CII antibody levels and 26 patients with low anti-CII levels were randomly recruited from our outpatient clinic. The HLA associations to cellular CII reactivity were only seen when both groups were combined, probably because of the rather low numbers of patients in each group. It appears unlikely to us that the association between HLA haplotype and CII-induced T-cell responsiveness seen in the combined group should be due to a bias introduced by selection of patients with high anti-CII levels for the following reasons: the high anti-CII group had a lower responsiveness to denatured CII than the randomly chosen group of RA patients; and there was no association between anti-CII levels and shared epitope (HLA-DRB1*0401 or HLA-DRB1*0404).
The IFN-γ reactivity to CII seen in most individuals implies that T-cell tolerance to CII is incomplete. The finding that high cellular response to CII associates with HLA-DRB1*0401 and HLA-DQ8 could mean that selection in thymus favours a T-cell repertoire containing T-cell receptors with high specificity for some CII epitope(s). Another explanation for the HLA-associated cellular response to CII is that these class II molecules could be able to present some immunodominant peptides more efficiently than other class II molecules.
One recent study [37] has investigated CII-specific proliferative responses and their relation to the presence of circulating anti-CII antibodies in RA patients. That study could not demonstrate a difference in proliferative response to CII between RA patients and healthy control individuals. In contrast to the present results, however, a lower frequency of responsive RA patients without anti-CII compared with that in RA patients with anti-CII was recorded, which could not be explained by a difference in disease-modifying antirheumatic drug therapy between the two patient groups. This contradiction could be explained by the definition of anergy, as suggested by Mueller et al [38], that proliferative responses are lost whereas effector functions are retained in anergized T cells. Hence, T cells in RA patients without anti-CII might be anergic in the proliferative aspect but are still able to respond by cytokine production.
It has been shown by Cook et al [39] that levels of anti-CII in serum of RA patients decreases over time. This tendency was also apparent in the present study, but it was not designed to address this question (data not shown). We demonstrated a positive correlation between disease duration and IFN-γ response to CII, indicating that the T-cell response to CII (expressed as a fraction of the PPD-induced IFN-γ response) increases with time. As the PPD-induced IFN-γ production decreases with disease duration (data not shown), this implies that general T-cell reactivity decreases gradually during the course of RA. CII-induced IFN-γ production remains stable, however, and it increases when expressing the CII cellular reactivity as a fraction of the PPD cellular reactivity. Consequently, although humoral CII reactivity decreases with time [39], cellular CII reactivity increases. It is thus plausible that serum antibody levels to CII constitute a relatively poor mirror of T-cell responsiveness to CII. Because anti-CII antibodies and T cells reactive with CII may synergize in causing arthritis [40] (at least in rodents), there is an obvious need for further detailed parallel studies of B-cell and T-cell reactivity to CII in RA, taking the fine specificity (not addressed in the present study) into account.
In conclusion, CII, a major autoantigen candidate in RA, can elicit an IFN-γ response in vitro that is associated with HLA-DRB1*0401 and HLA-DQ8 in RA patients. The present study, with a partly new methodological approach to a classical problem in RA, has provided some additional support to the notion that CII may be a target autoantigen of importance for a substantial group of RA patients. Continued efforts to identify mechanisms behind the general hyporesponsiveness to antigens in RA, as well as the mechanisms behind the potential partial anergy to CII, may provide us with better opportunities to study the specificity and pathophysiological relevance of anti-CII reactivity in RA.
Acknowledgments
Acknowledgement
We thank Associate Professor Robert A Harris for linguistic advice.
References
- Stastny P, Fernandez-Vina M, Cerna M, et al. Sequences of HLA alleles associated with arthritis in adults and children. J Rheumatol Suppl. 1993;37:5–8. [PubMed] [Google Scholar]
- Wooley PH, Luthra HS, O'Duffy JD, et al. Anti-type II collagen antibodies in rheumatoid arthritis. The influence of HLA phenotype. Tissue Antigens. 1984;23:263–269. doi: 10.1111/j.1399-0039.1984.tb00043.x. [DOI] [PubMed] [Google Scholar]
- Morgan K, Clague RB, Reynolds I, Davis M. Antibodies to type II collagen in early rheumatoid arthritis. Br J Rheumatol. 1993;32:333–335. doi: 10.1093/rheumatology/32.4.333. [DOI] [PubMed] [Google Scholar]
- Clague RB, Morgan K, Reynolds I, Williams HJ. The prevalence of serum IgG antibodies to type II collagen in American patients with rheumatoid arthritis. Br J Rheumatol. 1994;33:336–338. doi: 10.1093/rheumatology/33.4.336. [DOI] [PubMed] [Google Scholar]
- Trentham DE, Townes AS, Kang AH. Autoimmunity to type II collagen an experimental model of arthritis. J Exp Med. 1977;146:857–868. doi: 10.1084/jem.146.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery P, Panayi GS, Nouri AM. Interleukin-2 reverses deficient cell-mediated immune responses in rheumatoid arthritis. Clin Exp Immunol. 1984;57:123–129. [PMC free article] [PubMed] [Google Scholar]
- Seitz M, Napierski I, Kirchner H. Depressed PPD and tetanus toxoid presentation by monocytes to T lymphocytes in patients with rheumatoid arthritis: restoration by IFN gamma. Rheumatol Int. 1988;8:189–196. doi: 10.1007/BF00269194. [DOI] [PubMed] [Google Scholar]
- Verwilghen J, Vertessen S, Stevens EA, Dequeker J, Ceuppens JL. Depressed T-cell reactivity to recall antigens in rheumatoid arthritis. J Clin Immunol. 1990;10:90–98. doi: 10.1007/BF00918190. [DOI] [PubMed] [Google Scholar]
- Malmstrom V, Michaelsson E, Burkhardt H, et al. Systemic versus cartilage-specific expression of a type II collagen-specific T-cell epitope determines the level of tolerance and susceptibility to arthritis. . Proc Natl Acad Sci USA. 1996;93:4480–4485. doi: 10.1073/pnas.93.9.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Silverman HA, Johnson JS, Vaughan JH, McGlamory JC. Altered lymphocyte reactivity in rheumatoid arthritis. Arthritis Rheum. 1976;19:509–515. doi: 10.1002/art.1780190301. [DOI] [PubMed] [Google Scholar]
- Mirza NM, Relias V, Yunis EJ, Pachas WN, Dasgupta JD. Defective signal transduction via T-cell receptor-CD3 structure in T cells from rheumatoid arthritis patients. Hum Immunol. 1993;36:91–98. doi: 10.1016/0198-8859(93)90111-d. [DOI] [PubMed] [Google Scholar]
- Allen ME, Young SP, Michell RH, Bacon PA. Altered T lymphocyte signaling in rheumatoid arthritis. Eur J Immunol. 1995;25:1547–1554. doi: 10.1002/eji.1830250612. [DOI] [PubMed] [Google Scholar]
- Rosloniec EF, Brand DD, Myers LK, et al. Induction of autoimmune arthritis in HLA-DR4 (DRB1*0401) transgenic mice by immunization with human and bovine type II collagen. J Immunol. 1998;160:2573–2578. [PubMed] [Google Scholar]
- Nabozny GH, Baisch JM, Cheng S, et al. HLA-DQ8 transgenic mice are highly susceptible to collagen-induced arthritis: a novel model for human polyarthritis. J Exp Med. 1996;183:27–37. doi: 10.1084/jem.183.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson EC, Hansen BE, Jacobsen H, et al. Definition of MHC and T cell receptor contacts in the HLA-DR4 restricted immuno-dominant epitope in type II collagen and characterization of collagen-induced arthritis in HLA-DR4 and human CD4 transgenic mice. . Proc Natl Acad Sci USA. 1998;95:7574–7579. doi: 10.1073/pnas.95.13.7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski A, Klareskog L, Carlsten H, Herberts P, Koopman WJ. Secretion of antibodies to types I and II collagen by synovial tissue cells in patients with rheumatoid arthritis. Arthritis Rheum. 1989;32:1087–1092. doi: 10.1002/anr.1780320906. [DOI] [PubMed] [Google Scholar]
- Ronnelid J, Lysholm J, Engstrom-Laurent A, Klareskog L, Heyman B. Local anti-type II collagen antibody production in rheumatoid arthritis synovial fluid. Evidence for an HLA-DR4-restricted IgG response. . Arthritis Rheum. 1994;37:1023–1029. doi: 10.1002/art.1780370707. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Luthra HS, Moore SB, O'Fallon WM. Serum IgG anti-native type II collagen antibodies in rheumatoid arthritis: association with HLA DR4 and lack of clinical correlation. Clin Exp Rheumatol. 1988;6:373–380. [PubMed] [Google Scholar]
- Huang YH, Ronnelid J, Frostegard J. Oxidized LDL induces enhanced antibody formation and MHC class II-dependent IFN-gamma production in lymphocytes from healthy individuals. Arte-rioscler Thromb Vasc Biol. 1995;15:1577–1583. doi: 10.1161/01.atv.15.10.1577. [DOI] [PubMed] [Google Scholar]
- Coulie PG, Van Snick J. Enhancement of IgG anti-carrier responses by IgG2 anti-hapten antibodies in mice. Eur J Immunol. 1985;15:793–798. doi: 10.1002/eji.1830150810. [DOI] [PubMed] [Google Scholar]
- Hohlfeld R, Conti-Tronconi B, Kalies I, Bertrams J, Toyka KV. Genetic restriction of autoreactive acetylcholine receptor-specific T lymphocytes in myasthenia gravis. J Immunol. 1985;135:2393–2399. [PubMed] [Google Scholar]
- Klareskog L, Forsum U, Scheynius A, Kabelitz D, Wigzell H. Evidence in support of a self-perpetuating HLA-DR-dependent delayed-type cell reaction in rheumatoid arthritis. Proc Natl Acad Sci USA. 1982;79:3632–3636. doi: 10.1073/pnas.79.11.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh S, Deighton J, Rifkin I, Ewan P. Kinetics and functional implications of Th1 and Th2 cytokine production following activation of peripheral blood mononuclear cells in primary culture. Eur J Immunol. 1996;26:1260–1265. doi: 10.1002/eji.1830260612. [DOI] [PubMed] [Google Scholar]
- Sanjeevi CB, Hook P, Landin-Olsson M, et al. DR4 subtypes and their molecular properties in a population-based study of Swedish childhood diabetes. Tissue Antigens. 1996;47:275–283. doi: 10.1111/j.1399-0039.1996.tb02554.x. [DOI] [PubMed] [Google Scholar]
- Sanjeevi CB, Hagopian WA, Landin-Olsson M, et al. Association between autoantibody markers and subtypes of DR4 and DR4-DQ in Swedish children with insulin-dependent diabetes reveals closer association of tyrosine pyrophosphatase autoimmunity with DR4 than DQ8. . Tissue Antigens. 1998;51:281–286. doi: 10.1111/j.1399-0039.1998.tb03103.x. [DOI] [PubMed] [Google Scholar]
- Olerup O, Zetterquist H. HLA-DR typing by PCR amplification with sequence-specific primers (PCR-SSP) in 2 hours: an alternative to serological DR typing in clinical practice including donor-recipient matching in cadaveric transplantation. Tissue Antigens. 1992;39:225–235. doi: 10.1111/j.1399-0039.1992.tb01940.x. [DOI] [PubMed] [Google Scholar]
- Wallstrom E, Khademi M, Andersson M, et al. Increased reactivity to myelin oligodendrocyte glycoprotein peptides and epitope mapping in HLA DR2(15)+ multiple sclerosis. Eur J Immunol. 1998;28:3329–3335. doi: 10.1002/(SICI)1521-4141(199810)28:10<3329::AID-IMMU3329>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Vingsbo C, Larsson P, Andersson M, Holmdahl R. Association of pepsin with type II collagen (CII) breaks control of CII autoimmu-nity and triggers development of arthritis in rats. Scand J Immunol. 1993;37:337–342. doi: 10.1111/j.1365-3083.1993.tb02562.x. [DOI] [PubMed] [Google Scholar]
- Stastny P, Fink CW. HLA-Dw4 in adult and juvenile rheumatoid arthritis. Transplant Proc. 1977;9:1863–1866. [PubMed] [Google Scholar]
- Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- Solinger AM, Stobo JD. Immune response gene control of T dependent reactivity to collagen in man. Adv Exp Med Biol. 1982;150:141–155. doi: 10.1007/978-1-4684-4331-8_8. [DOI] [PubMed] [Google Scholar]
- Solinger AM, Bhatnagar R, Stobo JD. Cellular, molecular, and genetic characteristics of T cell reactivity to collagen in man. . Proc Natl Acad Sci USA. 1981;78:3877–3881. doi: 10.1073/pnas.78.6.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinger AM, Stobo JD. Regulation of immune reactivity to collagen in human beings. Arthritis Rheum. 1981;24:1057–1064. doi: 10.1002/art.1780240812. [DOI] [PubMed] [Google Scholar]
- Kammer GM, Trentham DE. HLA-DR4 is not a requisite for autoim-munity to collagen in rheumatoid arthritis. Arthritis Rheum. 1984;27:489–494. doi: 10.1002/art.1780270502. [DOI] [PubMed] [Google Scholar]
- Strom H, Al-Balaghi S, Moller E. No demonstrable association between HLA DR4 and in vitro collagen reactivity as determined by the production of leukocyte inhibition factor. Tissue Antigens. 1984;24:174–183. doi: 10.1111/j.1399-0039.1984.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Snowden N, Reynolds I, Morgan K, Holt L. T cell responses to human type II collagen in patients with rheumatoid arthritis and healthy controls. Arthritis Rheum. 1997;40:1210–1218. doi: 10.1002/1529-0131(199707)40:7<1210::AID-ART4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Mueller DL, Jenkins MK, Schwartz RH. Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol. 1989;7:445–480. doi: 10.1146/annurev.iy.07.040189.002305. [DOI] [PubMed] [Google Scholar]
- Cook AD, Rowley MJ, Mackay IR, Gough A, Emery P. Antibodies to type II collagen in early rheumatoid arthritis. Correlation with disease progression. Arthritis Rheum. 1996;39:1720–1727. doi: 10.1002/art.1780391015. [DOI] [PubMed] [Google Scholar]
- Taylor PC, Plater-Zyberk C, Maini RN. The role of the B cells in the adoptive transfer of collagen-induced arthritis from DBA/1 (H-2q) to SCID (H-2d) mice. Eur J Immunol. 1995;25:763–769. doi: 10.1002/eji.1830250321. [DOI] [PubMed] [Google Scholar]