

Abstract
Background and purpose
Plasma glutathione peroxidase (GPx-3) deficiency increases extracellular oxidant stress, decreases bioavailable nitric oxide, and promotes platelet activation. The aim of this study is to identify polymorphisms in the GPx-3 gene, examine their relationship to arterial ischemic stroke (AIS) in a large series of children and young adults, and determine their functional molecular consequences.
Methods
We studied the GPx-3 gene promoter from 123 young adults with idiopathic AIS and 123 age- and gender-matched controls by single-stranded conformational polymorphism and sequencing analysis. A second, independent population with childhood stroke was used for a replication study. We identified eight, novel, strongly linked polymorphisms in the GPx-3 gene promoter that formed two main haplotypes (H1 and H2). The transcriptional activity of the two most prevalent haplotypes was studied with luciferase reporter gene constructs.
Results
The H2 haplotype was overrepresented in both patient populations and associated with an independent increase in the risk of AIS in young adults (OR=2.07, 95% CI=1.03–4.47; p=0.034) and children (OR=2.13, 95% CI=1.23–4.90; p=0.027). In adults simultaneously exposed to vascular risk factors, the risk of AIS approximately doubled (OR=5.18, 95% CI=1.82–15.03, p<0.001). Transcriptional activity of the H2 haplotype was lower than that of the H1 haplotype, especially after upregulation by hypoxia (normalized relative luminescence: 3.54±0.32 vs. 2.47±0.26; p=0.0083).
Conclusion
These findings indicate that a novel GPx-3 promoter haplotype is an independent risk factor for AIS in children and young adults. This haplotype reduces the gene’s transcriptional activity, thereby compromising gene expression and plasma antioxidant and antithrombotic activities.
Keywords: thrombosis, platelets, oxidative stress
Introduction
Stroke is the second most common cause of death worldwide1 and the leading cause of long-term disability in developed countries.2 Almost 90% of the burden of cerebrovascular disease stems from arterial ischemic stroke (AIS). On average, 3% to 5% of AIS affect patients under the age of 45 years,3 yet prevalences of over 10% have been reported.4 The prevalence among children is lower then among adults, yet more than half of childhood stroke survivors develop some neurologic or cognitive deficit, 30% have a recurrence, and 5% to 10% of affected children die.5 The most unique feature of stroke in the young is the heterogeneity of underlying etiologies that include non-atherosclerotic vasculopathies, cardioembolism, and hematological and genetic causes.6 Despite extensive investigation, the etiology of this potentially devastating condition remains unknown in approximately one-third of young adults and children.5,6
Plasma glutathione peroxidase (GPx-3) is a major antioxidant enzyme in plasma and, as a member of the selenocysteine-containing GPx family, scavenges hydrogen peroxide and organic (lipid) hydroperoxides produced during normal metabolism or after oxidative insult.7,8 Of the five known GPx isoforms, GPx-3 is the only one found in the extracellular space. It contributes to maintaining the vascular bioavailability of nitric oxide (NO), a major vasorelaxant and inhibitor of platelet function, as NO can be inactivated rapidly by reactive oxygen species (ROS). The reduction of oxidant stress by GPx-3 activity also protects against posttranslational modifications of fibrinogen by ROS and NO-derived oxidants that increase its thrombogenicity.9,10 Consistent with this role, we recently described a novel, functional transcription start site of the GPx-3 gene and showed that GPx-3 gene transcription is regulated by oxygen tension and redox state.11
Two studies of families with idiopathic childhood stroke have provided clinical evidence for the importance of GPx-3 in the modulation of NO bioavailability and thrombosis.12,13 The patients in these studies had hyperreactive platelets, and their plasma impaired the normal inhibition of platelet activation by NO. These findings were determined to be a consequence of a familial reduction in GPx-3 activity that correlated with decreased protein expression by immunoblot analysis.11 We, therefore, hypothesized that mutation(s) or polymorphism(s) in the plasma GPx-3 gene promoter may be responsible for the reduction in enzyme activity and predispose to a thrombotic disorder, thus constituting a genetic risk factor for thrombotic cerebrovascular disease. To address this hypothesis, we investigated the GPx-3 gene in a large series of young adults with AIS of unknown etiology by single-stranded conformational polymorphism (SSCP) and sequencing analysis, followed by risk analysis and functional characterization of the identified genetic variants. We then performed a replication study in a second, independent population with childhood stroke.
SUBJECTS AND METHODS
STUDY SUBJECTS
Two patient populations with AIS, young adults and children, were studied independently; the group with childhood stroke served as the replication study group. Young adults were recruited form the Department of Neurology and the Hematology and Hemotherapy Center of the State University of Campinas, in Brazil. Between January, 1996, and June, 2001, we studied 286 consecutive young (18–45 years) survivors of a first clinically and radiologically diagnosed AIS. The detailed diagnostic and selection criteria have been previously described.14 Briefly, AIS was defined as an acute, focal, neurological deficit lasting >24 hours or symptoms lasting <24 hours if a brain imaging study (computed tomography and/or magnetic resonance imaging) showed an ischemic lesion appropriate to the symptoms. Patients with transient ischemic attacks, cerebral venous thrombosis, and migrainous events were excluded. After undergoing diagnostic work-up, 163 AIS patients for whom an etiology could be determined were excluded, such as cardiac sources of embolism, carotid dissections, illicit drug use, cancer, systemic disease, and hematological disorders.
Unrelated blood donors and volunteers from the same geographical area without a clinical history of coronary, peripheral, or cerebrovascular disease were invited to participate in the study as control subjects. The 123 AIS patients enrolled were matched 1:1 to controls with respect to age (±2 years) and gender. Data on conventional vascular risk factors (hypertension, diabetes mellitus, current or former smoking, and hyperlipidemia) were collected for all study subjects, as previously described.14 Among women, oral contraceptive use, pregnancy, and the postpartum period (the three months after childbirth or abortion) were assessed, and defined as hormonal risk factors. Women were considered to be using oral contraceptives if they had taken them for a minimum of four weeks. A blood sample for laboratory testing and genotyping was obtained. Participation was voluntary, and all study subjects gave informed consent.
Brazil has an ethnically heterogeneous population. In the Campinas area, most of the population descends from Caucasian immigrants from southern and central Europe. In addition, part of the population is of African origin, and a minor portion is of Asian background. We defined ethnic background according to the subjects’ self-determination.15
Children for the replication study were recruited from three institutions: the Institute of Thrombosis and Haemostasis, Sheba Medical Center, Tel Aviv, Israel; the Department of Medical and Surgical Sciences, University of Padua, Padua, Italy; and the Hematology and Hemotherapy Center, State University of Campinas, Brazil. Some of the children from Israel have previously been included in a study addressing the association of thrombophilia risk factors (exclusive of GPx-3) and childhood stroke.16 All children were Caucasian. Among children, AIS was defined as an acute thrombotic cerebrovascular event that manifested as a motor or sensory deficit, aphasia, visual disturbance, ataxia, or seizures. The clinical diagnosis was confirmed with neuroimaging studies in all cases. Children with thromboembolic events, coagulation abnormalities, sepsis, acute febrile disorders, or cancer were excluded. Of the initial 116 pediatric patients who met these criteria, 28 with perinatal stroke and six with congenital hemiplegia were excluded as these conditions are pathophysiologically distinct. Pediatric controls were recruited from the Department of Pediatrics or Hematology Centers from the respective institutions and included children admitted for surgery, trauma, or nonthrombotic conditions. The same matching criteria used for adults were applied to children. Written parental informed consent was obtained for all children. This study was approved by the local medical ethics committees of each institution, as well as the Institutional Review Boards at Boston University School of Medicine and Brigham and Women’s Hospital.
POLYMERASE CHAIN REACTIONS
PCR reactions were performed in a mixture containing 100–200 ng of genomic DNA, 1 μM of each primer, 200 μM dNTP (Gibco, Carlsbad, CA), 10 mM Tris-HCl, 50 mM KCl, 1.5 to 3.5 mM MgCl2 (see Table 1), 1 μl dimethyl sulfoxide, and 0.5 U of high fidelity Taq polymerase (Roche Diagnostics Corporation, Indianapolis, IN) in a final volume of 20 μl. The conditions for PCR amplifications were: initial denaturation at 95°C for 2 min, 35 cycles of 30 sec at 95°C, 30 sec at 52 to 58°C, and 30 sec at 72°C, with a final extension at 72°C for 7 min. PCR reactions were performed in a Bio-Rad iCycler (Bio-Rad Laboratories, Hercules, CA). The annealing temperatures and MgCl2 concentrations were optimized for each primer pair, all of which are listed in Table S1 (online supplement).
Table 1.
Characteristics of study subjects
|
Adults |
Children |
|||||||
|---|---|---|---|---|---|---|---|---|
| Characteristic | Patients1(n=123) | Controls(n=123) | p-value2 | Patients (n=82) | Controls (n=82) | p-value3 | ||
| Age (yr)4 | 36.5 ± 6.6 | 36.8 ± 6.8 | 0.66 | 6.9 ± 5.5 | 7.5 ± 4.7 | 0.36 | ||
| Male | 57 (46.3%) | 57 (46.3%) | 1.0 | 47 (57.3%) | 47 (57.3%) | 1.0 | ||
| Ethnic background: | ||||||||
| Caucasian | 92 (74.8%) | 100 (81.3%) | ) | 82 (100%) | 82 (100%) | ) | ||
| African | 29 (23.5%) | 22 (17.9%) | 0.44 | 0 | 0 | 1.0 | ||
| Asian | 2 (1.6%) | 1 (0.8%) | 0 | 0 | ||||
| Conventional vascular risk factors: | ||||||||
| Hypertension | 50 (40.7%) | 10 (8.1%) | <0.001 | - | - | - | ||
| Smoking | 68 (55.3%) | 45 (36.6%) | 0.003 | - | - | - | ||
| Hyperlipidemia | 43 (35.0%) | 35 (28.5%) | 0.27 | - | - | - | ||
| Diabetes mellitus | 8 (6.5%) | 3 (2.4%) | 0.12 | - | - | - | ||
| No vascular risk factors | 28 (22.7%) | 52 (42.3%) | 0.001 | - | - | - | ||
| Hormonal risk factors:5 | ||||||||
| Oral contraceptive use | 16 (24.2%) | 10 (16.1%) | ) | - | - | - | ||
| Pregnancy | 1 (1.5%) | 0 | 0.087 | - | - | - | ||
| Postpartum period | 2 (3.0%) | 0 | - | - | - | |||
| Inherited prothrombotic risk factors: | ||||||||
| Factor V Leiden6 | 5 (4.1%) | 4 (3.3%) | 0.99 | 5 (6.1%) | 5 (6.1%) | 1.0 | ||
| Prothrombin G20210A6 | 5 (4.1%) | 3 (2.4%) | 0.72 | 3 (3.7%) | 4 (4.9%) | 0.99 | ||
| MTHFR C677T7 | 19 (15.5%) | 14 (11.4%) | 0.35 | 18 (22.0%) | 17 (20.7%) | 0.85 | ||
Some clinical characteristics of adults16 and children have been previously reported.
Adult AIS patients vs. controls;
Childhood stroke patients vs. controls
Plus-minus values are means ± SD
Among 62 female controls (4 postmenopausal controls were excluded from this analysis), 66 female AIS patients, and 16 female CVT patients.
In heterozygosity;
in homozygosity
Table S1.
Oligonucleotide primer sequences, PCR and SSCP running conditions for GPx-3 gene
| PCR Conditions | SSCP Running Conditions | |||||||
|---|---|---|---|---|---|---|---|---|
| Fragment | Size | Primers | [MgCl2] | Annealing temp. (°C) | Gel | Temp. (°C) | Time (Vh) | |
| P.1 | F R |
181 | GTCAGAGAGATTTGAGACTGATTCC CGGGCCTAGATCTCACAAGA |
3.5 | 55 | 8–25% | 15 | 300 |
| P.2 | F R |
120 | CAAAGGACTCCTGGTCTCCGTCT GGCCTCAGTTTCCTCAGCTA |
3.5 | 52 | 20% | 4 | 300 |
| P.3 | F R |
149 | ATCTGACCGAGAGCCAGAAG CCAAACCACCTGGCTTAGAA |
3.5 | 52 | 8–25% | 4 | 300 |
| P.4 | F R |
119 | AGGTGGTTTGGCCACAGATA AATGCGCAGAAATGTGAGTG |
3.5 | 52 | 8–25% | 15 | 300 |
| P.5 | F R |
193 | CACTCACATTTCTGCGCATT CCTCCCCATCATAAGGATCA |
3.5 | 52 | 20% | 15 | 300 |
| P.6 | F R |
200 | TCAGCTGTGATCCTTATGATGG TGGTCCTTCTGGAAATTGAGG |
3.5 | 52 | 20% | 15 | 300 |
| P.7 | F R |
141 | ACCATCGCCAGGTGTTTTTA AAAGTGCGAAAGGAGGCAGA |
3.5 | 55 | 20% | 15 | 300 |
| P.8 | F R |
190 | GCCTCCTTTCGCACTTTG GAGACCCTTGCAGCCAATC |
3.5 | 55 | 8–25% | 15 | 300 |
| P.9 | F R |
260 | GAAATCCCAGCCGCCTA CACTCACCTTCGACTTCTCTTGTC |
2.5 | 58 | 8–25% | 15 | 300 |
SSCP ANALYSIS
Nonradioactive SSCP analysis was performed on an Amersham Pharmacia PhastSystem. The PCR products were diluted 1:2 with denaturing loading buffer (98% formamide, 2% glycerol, 0.05% bromophenol blue, 0.05% xylene cyanol), denatured at 95°C for 5 min, quenched on ice, and electrophoresed on precast non-denaturing acrylamide PhastGels buffered by native agarose strips. The gels used were either 20% homogeneous or 8–25% gradient gels. All gels were pre-run for 100 Vh. Running conditions (time and temperature) were optimized for each primer pair and are shown in Table S1. DNA bands were visualized by automated silver staining with a PhastGel DNA Silver Staining Kit, involving consecutive washes in fixing solution (07% benzene sulfonic acid, 24% ethanol) (10 min at 50°C), staining solution (0.2% silver nitrate, 0.07% benzene sulfonic acid) (30 min at 50°C), distilled water (1 min at 25°C), developing solution (9 min at 30°C), and preserving solution (8 min at 50°C). The PhastSystem, gels, silver staining kits, and buffer strips were purchased from Amersham Pharmacia, Uppsala, Sweden.
DETERMINATION OF THE -568 T→C, -518 T→C, AND -65 T→C POLYMORPHISMS
To genotype the -568 T→C, -518 T→C, and -65 T→C promoter polymorphisms, two fragments were amplified, one containing both the -568 T→C and -518 T→C polymorphisms. As the -518 T→C substitution does not affect any naturally occurring restriction sites, we designed a mutagenic antisense oligonucleotide primer to create selectively a restriction site three bases downstream from nucleotide -518: 5′-GAA AAC CCC ATT CTG GGT AGG GCC T-3′ (with C being the mutated base). The combination of the mutagenic primer with the -518T nucleotide created a restriction site for the endonuclease Dde I. Using the mutagenic primer and primer P.5F (see Table S.1), a fragment of 253 bp was obtained with the PCR conditions described above at 58°C annealing temperature. An additional restriction site for Dde I was present in all fragments at position 40. Thus, when the -518T nucleotide was present, digestion of the PCR product with 5 U Dde I at 37ºC for 2 hr yielded fragments of 186 bp, 40 bp, and 27 bp in length; in the presence of the -518C nucleotide, only two fragments of 213 bp and 40 bp were generated. The -569 T→C substitution contained in the same fragment deletes a normally existing restriction site for the endonuclease Alw I. Thus, when the -569T nucleotide was present, digestion of the product with 5 U Alw I yielded two fragments: 169 bp and 84 bp in length.
The -65 T→C substitution is contained in the 260 bp fragment generated with the P.9 primer pair and deletes a normally existing restriction site for the endonuclease Bsa I. Thus, in the presence of the -65T nucleotide, digestion with Bsa I at 50ºC for 2 hr yielded two fragments: 36 bp and 224 bp in length. All samples were electrophoresed on 2.5% ethidium bromide-stained agarose gels.
SCREENING OF THE GPX-3 GENE
Amplification of genomic DNA was performed using the PCR with the oligonucleotide primers shown in Table S1. Primers were designed to yield partially overlapping fragments covering approximately 1.5 kb of the promoter region and to generate PCR products ≤200bp in size (with one exception: fragment P.9), as the sensitivity of SSCP is dependent upon the size of the DNA fragment.17 The GPx-3 promoter was screened by nonradioactive SSCP analysis, followed by automated sequencing of fragments with abnormal electrophoretic patterns (ABI Prism 3700 Automated DNA Analyzer). In addition, all samples were sequenced analysis for the –568 T→C, –518 T→C, and –65 T→C polymorphisms.
TRANSIENT TRANSFECTIONS OF GPX-3 REPORTER CONSTRUCTS
Two firefly luciferase reporter gene constructs were generated with a pGL3-basic vector (Promega, Madison, WI) containing 1272 bp of the GPx-3 5′ flanking region amplified from genomic DNA obtained from a control subject homozygous for the more common alleles (haplotype H1) and a patient homozygous for the less common risk alleles (haplotype H2). These constructs were transiently transfected into Caki-2 cells, a renal tubular carcinoma cell line that has previously been shown to express GPx-3 in greater quantities than cell lines from other organs,18 as proximal renal tubular cells are the main source of GPx-3 in vivo. Cells were co-transfected with a pRL-CMV vector (Promega), a plasmid expressing Renilla luciferase, to allow for normalization for transfection efficiency. The detailed methodology for generation of the reporter gene constructs, transient cell transfections, and luciferase assays is described elsewhere.11
DETERMINATION OF PROTHROMBOTIC POLYMORPHISMS/MUTATIONS
Factor V Leiden, the prothrombin G20210A mutation, and the C677T substitution in the methylenetetrahydrofolate reductase (MTHFR) gene were detected by PCR and restriction digestion, as previously described.16
CELL CULTURE
Caki-2 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) and grown in McCoy’s 5A medium supplemented with L-glutamine, 25 mM HEPES, and 10% fetal bovine serum (Invitrogen, Carlsbad, CA). Normoxia corresponded to pO2 = 150 mmHg. Hypoxic conditions (pO2 = 35 mmHg) were induced in a modular incubation chamber (Billups-Rothenberg, Del Mar, CA) purged with 95% N2 and 5% CO2 for 30 min; the chamber was then sealed and returned to the incubator.
STASTICAL ANALYSIS
Differences in demographic characteristics and vascular risk factors between patients and controls were initially compared by univariate analysis using Student’s t-test for age and the chi-square test for all categorical variables. The Fisher exact test was used when data cell counts were sparse. Haplotypes were estimated from unphased genotypes by an expectation-maximization (EM) algorithm that assumes random mating and Hardy-Weinberg equilibrium.19 The EM algorithm carries out a series of iterations using observed data and estimated haplotype frequencies from the previous iteration to obtain expected diplotype frequencies, which are then used to update estimated haplotype frequencies. Iterations continue until convergence is reached. To perform this procedure, we used the HelixTree Genetics Analysis Software, version 2.4.0 (Golden Helix Inc, Bozeman, MT). This program was also used to determine allele and genotype frequencies, haplotype frequencies, Hardy-Weinberg equilibrium, and linkage disequilibrium (LD) between polymorphism pairs. We used R as our measure of LD; this value translates the strength of association of two marker loci on the same chromosome without being strongly affected by sample size.
To test whether GPx-3 haplotypes were significantly associated with the risk of AIS, we assigned the most likely haplotype phase to individuals using the results of the EM procedure. For the GPx-3 haplotypes found to have a significant association with the risk of AIS by univariate analysis, we performed multivariate analysis at the diplotype level with logistic regression. Adjustments were made for age, gender, ethnicity, and vascular, hormonal, and inherited prothrombotic risk factors. The extent to which associations with the polymorphisms were modified by other risk factors was assessed through analyses stratified by these risk factors. All reported p-values were two-sided. Statistical analyses were performed with Sigmastat version 3.0.1 (SPSS Inc., Chicago, IL).
RESULTS
ARTERIAL ISCHEMIC STROKE IN THE YOUNG
Table 1 shows the demographic characteristics and risk factor profile of the study subjects. Adult AIS patients and controls were well matched for age, gender, and ethnic background. While all conventional vascular risk factors were more frequent among AIS patients, only hypertension and smoking reached statistically significant differences. Almost 80% of AIS patients had at least one vascular risk factor compared to less than half of the controls (p=0.0011). Hormonal risk factors trended toward a greater prevalence among women with AIS than female controls (28.7% vs. 16.1%; p=0.087). The distribution of known inherited prothrombotic risk factors did not differ significantly between AIS patients and controls.
Analysis by SSCP of GPx-3 promoter fragments obtained from AIS patients and controls revealed abnormal electrophoretic patterns in two of the nine fragments studied: fragments P.3 and P.7. Sequencing of these fragments identified five single base pair changes: -942 A→C, -927 T→C, and -861 A→T in fragment P.3; and -302 A→T and -284 T→A in fragment P.7. Numbering of the nucleotides was performed relative to the recently described, novel transcription start point, defined as +1.11 Sequencing further indicated that the nucleotide substitutions in these fragments were often found together on the same chromosome. As these preliminary results suggested a highly polymorphic promoter with a high degree of LD, and as the sensitivity of SSCP is approximately 85% under ideal running conditions,17 we sequenced the entire GPx-3 5′flanking region (nt −1236 to +158) obtained from a patient homozygous for all of the above single nucleotide polymorphisms (SNPs) to search for base pair changes that may have been undetected by SSCP. Indeed, three additional single base pair transitions were identified: -568 T→C, -518 T→C, and -65 T→C, and these were subsequently assessed in all subjects.
All GPx-3 promoter polymorphisms were in Hardy-Weinberg equilibrium. The distribution of each of the SNPs in patients and controls is shown in Table S2 (online supplement). The rare polymorphic alleles were significantly overrepresented in AIS patients compared to controls. The LD map showed a high degree of linkage between polymorphism pairs, with R values ranging between 0.85 and 1, as shown in Figure 1. In fact, the three most 5′ SNPs (at positions -942, -927, -861), as well as the –568, –518, and -302 polymorphisms, were in complete LD, forming two blocks which, in turn, were linked (R=0.88). An exception to these overall findings was the polymorphism at position –284, which did not have a significantly different distribution between patients and controls and was not linked to the other polymorphisms. Haplotype analysis revealed that of the 28 (=256) possible combinations of these SNPs, only 8 haplotypes were identified, as shown in Table 2.
Figure 1. Linkage disequilibrium (LD) map of the GPx-3 promoter polymorphisms.
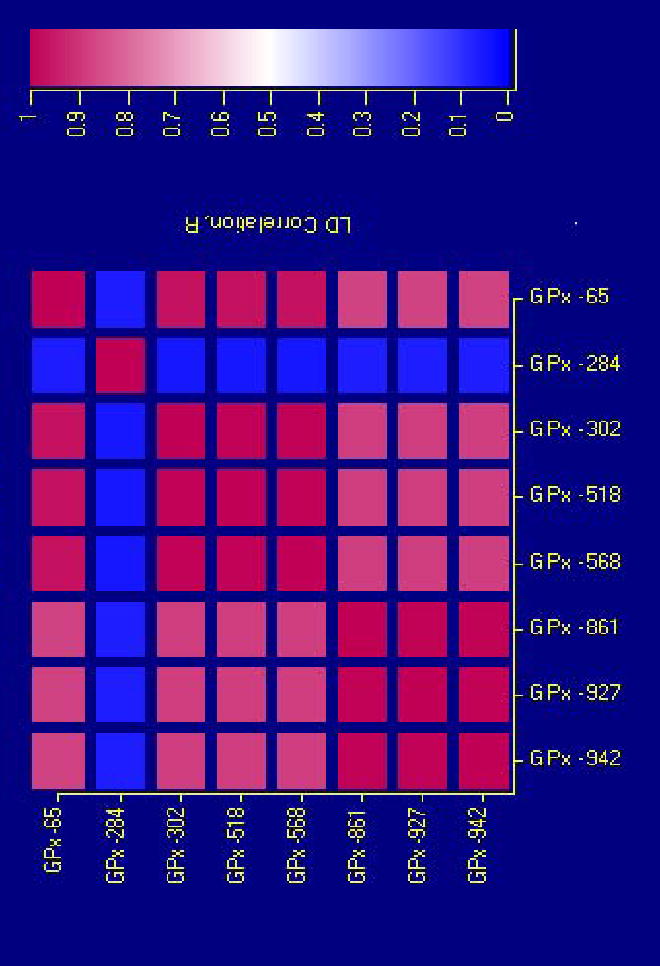
All polymorphisms, with the exception of the –284 T→A, are strongly linked, with R values ranging between 0.85 and 1.0. The polymorphisms at positions -942, -927, and -861, as well as –568, –518, and –302, are in complete LD, forming two blocks.
Table 2.
GPx-3 promoter haplotype frequencies and pairs among study subjects
| Haplotype Frequency | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Adults |
Children |
|||||||||||
| Haplotype | -942 A→C | -927 T→C | -861 A→T | -568 T→C | -518 T→C | -302 A→T | -284 T→A | -65 T→C | Controls (n=123) | Patients (n=123) | Controls (n=82) | Patients (n=82) |
| H1 | A | T | A | T | T | A | T | T | 84.9% | 79.6% | 86.0% | 79.3% |
| H2 | C | C | T | C | C | T | T | C | 7.0% | 13.1% | 7.9% | 13.4% |
| H3 | A | T | A | T | T | A | A | T | 6.2% | 2.9% | 4.3% | 3.7% |
| H4 | C | C | T | T | T | A | T | T | 0.8% | 2.1% | 1.2% | 1.2% |
| H5 | A | T | A | C | C | T | T | C | 0.4% | 1.2% | 0.6% | 1.2% |
| H6 | A | T | A | T | T | A | T | C | 0.4% | 0.4% | 0 | 0.6% |
| H7 | C | C | T | C | C | T | A | C | 0.3% | 0.3% | 0 | 0 |
| H8 | C | C | T | T | T | A | T | C | 0 | 0.4% | 0 | 0.6% |
| The position of each polymorphism is based on the transcription start site being +1. | ||||||||||||
| Haplotype pairs | ||||
|---|---|---|---|---|
| Adults | Children | |||
| Haplotype pair | Controls (n=123) | Patients (n=123) | Controls (n=82) | Patients (n=82) |
| H1/H1 | 89 (72.4%) | 76 (61.8%) | 60 (73.2%) | 48 (58.5%) |
| H1/H2 | 14 (11.4%) | 28 (22.8%) | 12 (14.6%) | 22 (26.8%) |
| H1/H3 | 12 (9.8%) | 6 (4.9%) | 6 (7.3%) | 6 (7.3%) |
| H1/H4 | 2 (1.6%) | 3 (2.4%) | 2 (2.4%) | 2 (2.4%) |
| H1/H5 | 1 (0.8%) | 3 (2.4%) | 1 (1.2%) | 2 (2.4%) |
| H1/H6 | 1 (0.8%) | 1 (0.8%) | 0 | 1 (1.2%) |
| H1/H7 | 0 | 1 (0.8%) | 0 | 0 |
| H1/H8 | 0 | 1 (0.8%) | 0 | 1 (1.2%) |
| H2/H2 | 1 (0.8%) | 1 (0.8%) | 0 | 0 |
| H2/H3 | 2 (1.6%) | 1 (0.8%) | 1 (1.2%) | 0 |
| H2/H4 | 0 | 2 (1.6%) | 0 | 0 |
| H3/H3 | 1 (0.8%) | 0 | 0 | 0 |
Carriers of the GPx-3 promoter H1H1 haplotype pair were used as the reference group.
To assess a potential association of the GPx-3 promoter haplotypes with the risk of AIS among young adults, haplotype frequencies were imputed from the EM algorithm in the patient and control groups (Table 2). Overall, haplotypes H1 and H2 accounted for ~95% of the observed haplotypes in this population; owing to their low frequencies, haplotypes H3 to H8 were not included in further analyses. The frequency of the H2 haplotype was almost twice as high in AIS patients (13.1%) compared to controls (7.0%), yielding an odds ratio (OR) of 2.01 (95% CI=1.04–3.91; p=0.025). Heterozygous and homozygous carriers of the H2 haplotype (H1H2 and H2H2) were overrepresented among AIS patients (23.6% vs. 12.2%), increasing the risk of AIS over two-fold when the H1H1 haplotype pair was used as reference (OR=2.26, 95% CI=1.07–4.81; p=0.019).
The findings from the univariate analyses were further investigated in a multiple logistic regression analysis that included age, gender, ethnicity, the vascular and inherited prothrombotic risk factors, and the GPx-3 H2 haplotype as covariates (Table 3). The association between the H2 haplotype and the risk of AIS remained unchanged, confirming this haplotype as an independent risk factor (OR=2.07, 95% CI=1.03–4.47; p =0.034). Hypertension showed the strongest association with AIS, increasing the susceptibility almost eight-fold (OR=7.67, 95% CI=3.22–18.22; p<0.001), while the risk estimate associated with smoking was 2.30 (95% CI=1.20–4.41; p =0.012).
Table 3.
Multiple logistic regression model incorporating the GPx-3 promoter H2 haplotype, demographic characteristics, and vascular and inherited prothrombotic risk factors of AIS patients and controls
| Adults | Children | |||||
|---|---|---|---|---|---|---|
| OR | 95% CI | p value | OR | 95% CI | p value | |
| GPx-3 H2 haplotype | 2.07 | 1.03 – 4.47 | 0.034 | 2.13 | 1.23 – 4.90 | 0.027 |
| Age | 0.97 | 0.90 – 1.21 | 0.11 | 0.93 | 0.86 – 1.99 | 0.24 |
| Gender | 0.98 | 0.52 – 1.87 | 0.96 | 1.18 | 0.58 – 2.43 | 0.65 |
| Ethnicity | 1.41 | 0.63 – 3.16 | 0.40 | - | - | - |
| Hypertension | 7.67 | 3.22 – 18.22 | <0.001 | - | - | - |
| Smoking | 2.30 | 1.20 – 4.41 | 0.012 | - | - | - |
| Diabetes mellitus | 1.57 | 0.32 – 7.83 | 0.58 | - | - | - |
| Hyperlipidemia | 1.06 | 0.52 – 2.16 | 0.87 | - | - | - |
| Factor V Leiden | 1.28 | 0.25 – 6.54 | 0.77 | 1.10 | 0.25 – 4.84 | 0.90 |
| Prothrombin G20210A | 3.21 | 0.50 – 20.71 | 0.22 | 0.94 | 0.19 – 4.69 | 0.94 |
| MTHFR C677T | 1.15 | 0.46 – 2.87 | 0.76 | 1.12 | 0.93 – 4.90 | 0.79 |
Carriers of the GPx-3 promoter H1H1 haplotype pair were used as the reference group.
Vascular risk factors, including smoking and hypertension, increase oxidant stress.20 To assess if these conditions modified the risk of AIS associated with the GPx-3 promoter polymorphisms, we analyzed the distribution of the H2 haplotype after stratification for smoking and hypertension (Table 4). Smoking alone was associated with an almost two-fold higher risk of AIS (p=0.044), while smokers who were also carriers of the GPx-3 H2 haplotype had an OR for AIS of 4.22 (95% CI=1.48–12.42; p=0.0022). All subjects who carried the H2 haplotype and were also hypertensive had strokes (p<0.001); therefore, no OR could be calculated for this group. Carriership of the H2 haplotype in the presence of at least one vascular risk factor yielded an over five-fold increase in the risk of AIS (OR=5.18, 95% CI=1.82–15.03; p<0.001), suggesting an interaction of this genetic variant with conventional vascular risk factors. Of note, the sample size in some of these subgroup analyses was very small, and, thus, the results must be interpreted with caution.
Table 4.
Effect of the GPx-3 H2 haplotype on the risk of AIS, stratified by vascular risk factors
| Risk factor | H2haplotype | Patients | Controls | OR* | 95% CI | p-value |
|---|---|---|---|---|---|---|
| Smoking: | ||||||
| No | No | 36 | 56 | 1 | – | – |
| No | Yes | 10 | 8 | 1.94 | 0.63 – 6.06 | 0.20 |
| Yes | No | 40 | 33 | 1.89 | 1.01 – 3.69 | 0.044 |
| Yes | Yes | 19 | 7 | 4.22 | 1.48 – 12.42 | 0.0022 |
| Hypertension: | ||||||
| No | No | 46 | 79 | 1 | – | – |
| No | Yes | 18 | 15 | 2.06 | 0.89 – 4.81 | 0.065 |
| Yes | No | 30 | 10 | 5.15 | 2.17 – 12.48 | <0.001 |
| Yes | Yes | 11 | 0 | – | – | <0.001 |
| Any vascular risk factor | ||||||
| No | No | 16 | 36 | 1 | – | – |
| No | Yes | 6 | 5 | 2.70 | 0.61 – 12.32 | 0.17 |
| Yes | No | 60 | 53 | 2.55 | 1.21 – 5.43 | 0.0075 |
| Yes | Yes | 23 | 10 | 5.18 | 1.82 – 15.03 | <0.001 |
All ORs are relative to the reference category: no risk factor and H1H1 haplotype pair.
CHILDHOOD STROKE
To validate the results found among young adults, we performed an independent replication study in a population with childhood stroke. The patient and control groups were well matched for age and gender, and all children were of Caucasian background (Table 1). The distribution of known inherited prothrombotic risk factors was essentially equivalent between groups.
The GPx-3 haplotypes identified among adults were determined in this pediatric population. As in adults, haplotypes H1 and H2 accounted for the majority of haplotypes observed. The GPx-3 H2 haplotype was more prevalent among affected children than among controls (13.4% vs. 7.9%) (Table 2) yielding a significantly increased carrier rate of the H2 haplotype (H1H2 and H2H2) among pediatric patients compared to controls (OR=2.29, 95% CI=1.04–5.51; p=0.039). After adjustment for age, gender, and inherited prothrombotic risk factors, the H2 haplotype remained independently associated with the risk of childhood stroke with an OR of 2.13 (95% CI=1.23–4.90; p=0.027) (Table 3).
ETHNIC HETEROGENEITY
In association studies conducted among subjects from different populations, the concern exists for spurious associations secondary to ethnic variability in allele or haplotype frequencies. Indeed, among adult Brazilian controls, the frequencies of the H1 and H2 haplotypes differed between subjects of Caucasian and African descent (allele frequencies in Caucasians: H1=84.5%, H2=7.0%; African Americans: H1=75.0%, H2=9.1%), albeit not significantly (p=0.13 for H1; p=0.75 for H2). The risk conferred by the H2 haplotype is maintained in an analysis restricted to Caucasian adults (OR=2.45, 95% CI, 1.03–5.92, p=0.026). For the replication study, only Caucasian children were enrolled. The H1 and H2 haplotype frequencies found in the pediatric control group were very similar to those among young adult Brazilian Caucasian controls (allele frequencies in Caucasian adults: H1=84.5%, H2=7.0%; Caucasian children: H1=86.0%, H2=7.9%; p=0.69 for H1; p=0.74 for H2).
FUNCTIONAL GENOMICS
We analyzed the GPx-3 promoter sequence for consensus motifs (MacVector Version 7, Accelyrs, San Diego, CA) and identified that the –943 A→C polymorphism altered the last bp of a redox-sensitive transcription factor, activator protein-1 (AP-1), binding site. To determine the functional effect of the GPx-3 promoter polymorphisms, Caki-2 cells were transiently transfected with firefly luciferase reporter gene constructs containing either the H1 or H2 haplotype. Basal GPx-3 promoter activity of both constructs was measured in 6 hr intervals for 24 hr. At all time points, basal activity of the H2 construct was lower than that of the H1 construct, although this difference was not statistically significant (Figure 2). We have previously reported that hypoxia is a strong transcriptional regulator of GPx-3 expression, leading to an almost three-fold increase in expression levels after 24 hr compared with normoxic conditions.11 When cells transfected with the H2 construct were incubated under hypoxic conditions for 24 hr, we also observed a significant increase in GPx-3 expression compared to normoxic cells (normalized relative luminescence [RL]: 2.47 ± 0.26 vs. 0.96 ± 0.10; p<0.001), albeit not as high as that of the H1 construct (normalized RL: 3.49 ± 0.30). Thus, under hypoxic conditions, the difference in expression between the two constructs was statistically significant (normalized relative luminescence [RL]: 3.54 ± 0.32 vs. 2.47 ± 0.26; p=0.0083).
Figure 2. Regulation of GPx-3 expression by hypoxia.
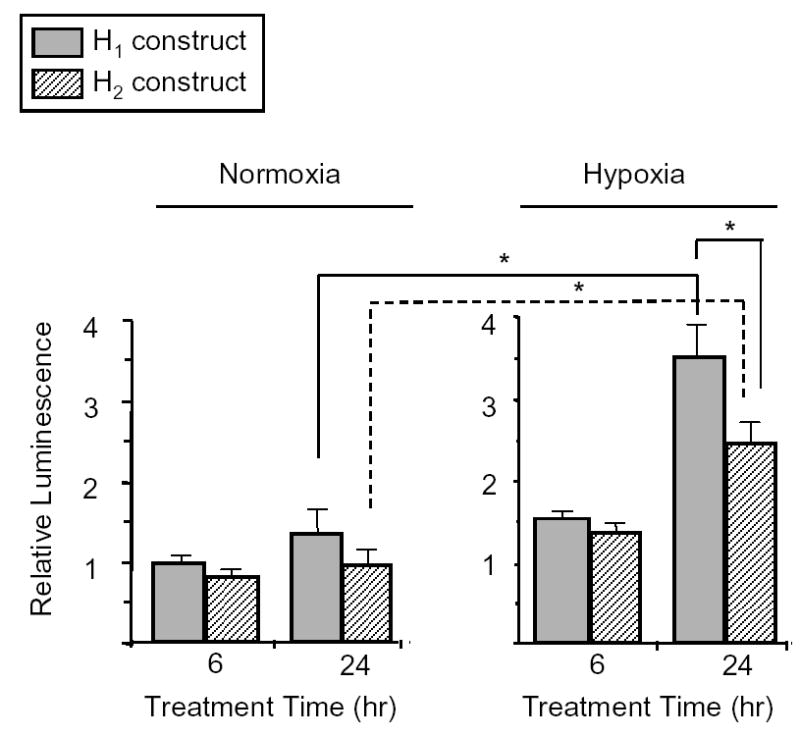
Caki-2 cells transfected with firefly luciferase reporter gene constructs containing either the H1 (gray bars) or H2 (hatched bars) haplotypes were maintained in normoxia or hypoxia for up to 24 hr. Expression levels were measured as the ratio of firefly-to-Renilla luciferase luminescence and normalized to the H1 construct in normoxia at 6 hr. Experiments were run in triplicate and performed a minimum of three times. * p<0.01.
DISCUSSION
The enzymes in the GPx family scavenge ROS in the vasculature and protect the bioavailability of NO, thereby maintaining normal endothelial function and an antithrombotic vascular milieu. Mice lacking the cellular isoform of GPx (GPx-1) have impaired endothelium-dependent vasodilator function,21 and decreased levels of GPx-1 in humans have recently been associated with coronary heart disease in a dose-dependent manner.22 In addition, these antioxidants protect against the free radical-induced brain injury that is a key component in the pathophysiology of ischemic cerebrovascular disease.23 Mice overexpressing the GPx-1 gene and experimental animals treated with the GPx-mimetic ebselen have reduced lesion volumes, less neurological deficits, and improved outcome when compared to control animals after transient or permanent focal cerebral ischemia.24,25 A few studies in humans have also shown a neuroprotective effect of ebselen.26 A familial deficiency of the extracellular isoform GPx-3 has been associated clinically with an increased risk of childhood stroke in small series,12,13 and three reports have demonstrated decreased GPx-3 activity among patients with coronary artery disease, supporting a broader effect of this defect in the vascular system.27–29 Taken together, this evidence points to the GPx-3 gene as a compelling candidate gene for thrombotic cerebrovascular disease risk.
We identified a novel GPx-3 promoter haplotype associated with the risk of AIS among young adults; these findings were replicated in a second, independent patient population with childhood stroke. Eight SNPs, seven of which were in strong LD, formed two main haplotypes that accounted for ~95% of the eight observed haplotypes in our study population. The polymorphisms we identified are in accordance with variations recently described by Rieder and colleagues in GenBank (accession number AY310878).30 Haplotype H2, formed by the combination of the rare alleles of each of the linked polymorphisms, was associated with a 2.1-fold independent increase in the risk of AIS when compared to carriers of the H1H1 haplotype pair. As we performed reporter gene studies only for the two most common haplotypes (H1 and H2), we were conservative in our statistical analysis and restricted it to carriers of these haplotypes.
The polymorphisms at positions – 942, -927, and -861, as well as –568, –518, and –302, were in complete LD, forming two blocks that were, in turn, linked. We can speculate that this high degree of LD may be important for promoter function. Recent investigations have argued that patterns of LD in the human genome occur in block structure; a block is characterized by a low level of recombination among contiguous SNPs within it, marked by surrounding SNPs with a high level of recombination.31,32 Several algorithms have been proposed to determine block size and structure; however, the prevalence of the H1 and H2 haplotypes was sufficiently high in our study to obviate the need to employ these methods. Haplotype studies are likely to represent an advantage over single SNP analyses because they exploit LD information to increase marker informativity, resulting in increased power to detect association.32,33 Furthermore, genetic information may relate to disease status as a functional set of SNPs, rather than any single SNP.34
Thrombotic cerebrovascular disease is multifactorial in etiology and occurs as a complex interaction between environmental and genetic predisposing factors.35 In addition, most prothrombotic risk factors, such as factor V Leiden and the prothrombin G20210A mutation, are not major cardiovascular risk factors, but may assume increased importance in certain patient subgroups, such as children or patients with other conventional vascular risk factors.36 We hypothesized that the GPx-3 H2 haplotype might behave in a similar manner; as conventional vascular risk factors increase oxidant stress,20 an interaction between the GPx-3 H2 haplotype and environmental factors would be plausible. Indeed, the presence of vascular risk factors among carriers of the GPx-3 H2 haplotype more than doubled the risk of AIS when compared to patients who carried only the genetic or environmental risk factor alone. All subjects who carried the H2 haplotype and were also hypertensive had strokes, suggesting that this may be a particularly high-risk combination of factors.
We performed reporter gene studies with the two most common haplotypes in our population and found that the transcriptional activity of the H2 risk haplotype was lower than that of the H1 haplotype, especially under hypoxic conditions. The lower basal expression levels of the GPx-3 H2 haplotype and its compromised upregulation in hypoxia yield less ROS scavenging potential, thereby compromising antithrombotic and neuroprotective function. We have not determined in our study which specific polymorphism(s) is (are) functionally relevant and responsible for the altered transcriptional response. Additional investigation including site-directed mutagenesis would be necessary to answer this question; however, the SNP at position -943 is located within an AP-1 site, suggesting that it may, at least in part, affect the GPx-3 promoter’s ability to respond to changes in redox state and oxidant stress. The AP-1 transcription factor is exquisitely redox-sensitive and regulates antioxidant and proinflammatory genes.37
Some limitations of our study should be noted. First, our ability to correlate the GPx-3 haplotypes with GPx-3 activity levels in vivo was limited by the lack of availability of plasma samples from our study population. Second, we analyzed the effect of the GPx-3 haplotypes on AIS without taking into consideration specific pathogenic subtypes of AIS. Third, this study includes only survivors of thrombotic cerebrovascular disease, and the possibility of survival bias must be considered. Fourth, the sample sizes for some of the subanalyses, particularly the interaction studies, were small, and the associated risk estimates have to be interpreted with caution; the biological plausibility of the results, however, strengthens the validity of these results. Finally, given the ethnic heterogeneity of the study populations, the possibility that these findings are influenced by population stratification cannot be excluded. We did, indeed, find non-significant differences in haplotype frequencies between Brazilians of Caucasian and African descent and, therefore, chose patients and controls of similar ethnic distribution. In addition, we included ethnicity as an independent variable in the logistic regression analysis. The haplotype frequencies among pediatric controls was similar to that found in the adult Caucasians, indicating that the distribution among Caucasians in different countries may be more uniform.
SUMMARY
In conclusion, we have identified novel GPx-3 promoter polymorphisms that form a risk haplotype associated with a significant increase in the risk of AIS among young individuals. These findings, in combination with prior work from our group, support a novel mechanism for thrombotic cerebrovascular disease that involves the antioxidant enzyme GPx-3 and its role in scavenging reactive oxygen species that limit the bioavailability and antiplatelet effects of nitric oxide. This genetic variant interacts with conventional vascular risk factors to potentiate further the risk of AIS. Studies involving a larger number of subjects and patient populations with thrombotic disease in other vascular beds, as well as attempting to correlate the GPx-3 H2 haplotype with enzyme activity in plasma, are necessary to confirm these results and determine the generalizability of this defect.
Supplementary Material
Figure S.1.
Acknowledgments
This work was supported in part by National Institutes of Health Grants HL55993, HL58976, HL61795, HL81587, and HV28718 (to Joseph Loscalzo). The authors thank Professor Annamaria Laverda and Dr. Chiara Gentilomo from the Paediatrics Department at Padua University for access to patients, and Ms. Stephanie Tribuna for excellent editorial assistance.
Footnotes
This is an un-copyedited author manuscript that was accepted for publication in Stroke, copyright The American Heart Association. This may not be duplicated or reproduced, other than for personal use or within the “Fair Use of Copyrighted Materials” (section 107, title 17, U.S. Code) without prior permission of the copyright owner, The American Heart Association. The final copyedited article, which is the version of record, can be found at Stroke. The American Heart Association disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
Role of contributors
B. Voetsch was responsible for the study design, screening of the GPx-3 gene, genotyping and haplotyping of study subjects, construction of GPx-3 vectors, hypoxia and luciferase studies, data analysis and interpretation, and writing of the manuscript. R. Jin performed genotying, and hypoxia and luciferase studies. C. Bierl designed the primers for screening of the GPx-3 gene and participated in the screening of the GPx-3 gene. K. Benke assisted with the statistical analysis and data interpretation, and helped in the preparation of the manuscript. G. Kenet, P. Simioni, and B. Damasceno were involved with recruitment of study subjects and clinical data collection in Israel, Italy, and Brazil, respectively. J. Annichino-Bizacchi assisted with recruitment of study subjects in Brazil. F. Ottaviano assisted with genotyping. D. Handy supervised the functional genomics studies. J. Loscalzo was responsible for the overall study and experimental design, study supervision, data analysis and interpretation, and writing and final critical review of the manuscript.
References
- 1.World Health Organization. The World Health Report 1999. Geneva, Switzerland: World Health Organization; 1999. [Google Scholar]
- 2.American Heart Association. Heart Disease and Stroke Statistics - 2004 Update. Dallas, TX: American Heart Association; 2003. [Google Scholar]
- 3.Gandolfo C, Conti M. Stroke in young adults: epidemiology. Neurol Sci. 2003;24 (Suppl 1):S1–3. doi: 10.1007/s100720300024. [DOI] [PubMed] [Google Scholar]
- 4.Bogousslavsky J, Pierre P. Ischemic stroke in patients under age 45. Neurol Clin. 1992;10:113–124. [PubMed] [Google Scholar]
- 5.Lynch JK. Cerebrovascular disorders in children. Curr Neurol Neurosci Rep. 2004;4:129–138. doi: 10.1007/s11910-004-0027-3. [DOI] [PubMed] [Google Scholar]
- 6.Kittner SJ, Stern BJ, Wozniak M, Bucholz DW, Earley CJ, Feeser BR, Johnson CJ, Macko RF, McCarter RJ, Price TR, Sherwin R, Sloan MA, Wityk RJ. Cerebral infarction in young adults: the Baltimore-Washington Cooperative Young Stroke Study. Neurology. 1998;50:890–894. doi: 10.1212/wnl.50.4.890. [DOI] [PubMed] [Google Scholar]
- 7.Maddipati KR, Marnett LJ. Characterization of the major hydroperoxide-reducing activity of human plasma. Purification and properties of a selenium-dependent glutathione peroxidase. J Biol Chem. 1987;262:17398–17403. [PubMed] [Google Scholar]
- 8.Takahashi K, Avissar N, Whitin J, Cohen H. Purification and characterization of human plasma glutathione peroxidase: a selenoglycoprotein distinct from the known cellular enzyme. Arch Biochem Biophys. 1987;256:677–686. doi: 10.1016/0003-9861(87)90624-2. [DOI] [PubMed] [Google Scholar]
- 9.Upchurch GR, Jr, Ramdev N, Walsh MT, Loscalzo J. Prothrombotic Consequences of the Oxidation of Fibrinogen and their Inhibition by Aspirin. J Thromb Thrombolysis. 1998;5:9–14. doi: 10.1023/a:1008859729045. [DOI] [PubMed] [Google Scholar]
- 10.Vadseth C, Souza JM, Thomson L, Seagraves A, Nagaswami C, Scheiner T, Torbet J, Vilaire G, Bennett JS, Murciano JC, Muzykantov V, Penn MS, Hazen SL, Weisel JW, Ischiropoulos H. Prothrombotic state induced by post-translational modification of fibrinogen by reactive nitrogen species. J Biol Chem. 2004;279:8820–8826. doi: 10.1074/jbc.M306101200. [DOI] [PubMed] [Google Scholar]
- 11.Bierl C, Voetsch B, Jin RC, Handy DE, Loscalzo J. Determinants of human plasma glutathione peroxidase (GPx-3) expression. J Biol Chem. 2004;279:26839–26845. doi: 10.1074/jbc.M401907200. [DOI] [PubMed] [Google Scholar]
- 12.Freedman JE, Loscalzo J, Benoit SE, Valeri CR, Barnard MR, Michelson AD. Decreased platelet inhibition by nitric oxide in two brothers with a history of arterial thrombosis. J Clin Invest. 1996;97:979–987. doi: 10.1172/JCI118522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kenet G, Freedman J, Shenkman B, Regina E, Brok-Simoni F, Holzman F, Vavva F, Brand N, Michelson A, Trolliet M, Loscalzo J, Inbal A. Plasma glutathione peroxidase deficiency and platelet insensitivity to nitric oxide in children with familial stroke. Arterioscler Thromb Vasc Biol. 1999;19:2017–2023. doi: 10.1161/01.atv.19.8.2017. [DOI] [PubMed] [Google Scholar]
- 14.Voetsch B, Benke KS, Damasceno BP, Siqueira LH, Loscalzo J. Paraoxonase 192 Gln-->Arg polymorphism: an independent risk factor for nonfatal arterial ischemic stroke among young adults. Stroke. 2002;33:1459–1464. doi: 10.1161/01.str.0000016928.60995.bd. [DOI] [PubMed] [Google Scholar]
- 15.Style Matters: Ethnicity, race, and culture: guidelines for research, audit, and publication. Br Med J. 1996;312:1094. No authors listed. [PMC free article] [PubMed] [Google Scholar]
- 16.Kenet G, Sadetzki S, Murad H, Martinowitz U, Rosenberg N, Gitel S, Rechavi G, Inbal A. Factor V Leiden and antiphospholipid antibodies are significant risk factors for ischemic stroke in children. Stroke. 2000;31:1283–1288. doi: 10.1161/01.str.31.6.1283. [DOI] [PubMed] [Google Scholar]
- 17.Grompe M. The rapid detection of unknown mutations in nucleic acids. Nat Genet. 1993;5:111–117. doi: 10.1038/ng1093-111. [DOI] [PubMed] [Google Scholar]
- 18.Avissar N, Kerl EA, Baker SS, Cohen HJ. Extracellular glutathione peroxidase mRNA and protein in human cell lines. Arch Biochem Biophys. 1994;309:239–246. doi: 10.1006/abbi.1994.1108. [DOI] [PubMed] [Google Scholar]
- 19.Excoffier L, Slatkin M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- 20.Zalba G, Beaumont J, San Jose G, Fortuno A, Fortuno MA, Diez J. Vascular oxidant stress: molecular mechanisms and pathophysiological implications. J Physiol Biochem. 2000;56:57–64. doi: 10.1007/BF03179777. [DOI] [PubMed] [Google Scholar]
- 21.Forgione MA, Weiss N, Heydrick S, Cap A, Klings ES, Bierl C, Eberhardt RT, Farber HW, Loscalzo J. Cellular glutathione peroxidase deficiency and endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2002;282:H1255–H1261. doi: 10.1152/ajpheart.00598.2001. [DOI] [PubMed] [Google Scholar]
- 22.Blankenberg S, Rupprecht HJ, Bickel C, Torzewski M, Hafner G, Tiret L, Smieja M, Cambien F, Meyer J, Kackner KH for the AtheroGene Investigators. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N Engl J Med. 2003;349:1605–1613. doi: 10.1056/NEJMoa030535. [DOI] [PubMed] [Google Scholar]
- 23.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Weisbrot-Lefkowitz M, Reuhl K, Perry B, Chan PH, Inouye M, Mirochnitchenko O. Overexpression of human glutathione peroxidase protects transgenic mice against focal cerebral ischemia/reperfusion damage. Brain Res Mol Brain Res. 1998;53:333–338. doi: 10.1016/s0169-328x(97)00313-6. [DOI] [PubMed] [Google Scholar]
- 25.Imai H, Graham DI, Masayasu H, Macrae IM. Antioxidant ebselen reduces oxidative damage in focal cerebral ischemia. Free Radic Biol Med. 2003;34:56–63. doi: 10.1016/s0891-5849(02)01180-2. [DOI] [PubMed] [Google Scholar]
- 26.Yamaguchi T, Sano K, Takakura K, Saito I, Shinohara Y, Asano T, Yasuhara H. Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial. Ebselen Study Group. Stroke. 1998;29:12–17. doi: 10.1161/01.str.29.1.12. [DOI] [PubMed] [Google Scholar]
- 27.Porter M, Pearson DJ, Suarez-Mendez VJ, Blann AD. Plasma, platelet and erythrocyte glutathione peroxidases as risk factors in ischaemic heart disease in man. Clin Sci (Lond) 1992;83:343–345. doi: 10.1042/cs0830343. [DOI] [PubMed] [Google Scholar]
- 28.Dogru-Abbasoglu S, Kanbagli O, Bulur H, Babalik E, Ozturk S, Aykac-Toker G, Uysal M. Lipid peroxides and antioxidant status in serum of patients with angiographically defined coronary atherosclerosis. Clin Biochem. 1999;32:671–672. doi: 10.1016/s0009-9120(99)00076-4. [DOI] [PubMed] [Google Scholar]
- 29.Muzakova V, Kandar R, Vojtisek P, Skalicky J, Cervinkova Z. Selective antioxidant enzymes during ischemia/reperfusion in myocardial infarction. Physiol Res. 2000;49:315–322. [PubMed] [Google Scholar]
- 30.Rieder MJ, Livingston RJ, Daniels MR, et al. NIEHS-SNPs, Environmental Genome Project, NIEHS ES 15478. Department of Genome Sciences; Seattle, WA: 2003. [Google Scholar]
- 31.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 32.Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- 33.Johnson GC, Esposito L, Barratt BJ, Smith AN, Heward J, Di Genova G, Ueda H, Cordell HJ, Eavaes IA, Dudbridge F, Twells RC, Payne F, Hughes W, Nutland S, Stevens H, Carr P, Tuomilehto-Wolf E, Tuomilehto J, Gough SC, Clayton DG, Todd JA. Haplotype tagging for the identification of common disease genes. Nat Genet. 2001;29:233–237. doi: 10.1038/ng1001-233. [DOI] [PubMed] [Google Scholar]
- 34.Drysdale CM, McGraw DW, Stack CB, Stephens JC, Judson RS, Nandabalan K, Arnold K, Ruaono G, Liggett SB. Complex promoter and coding region beta 2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci USA. 2000;97:10483–10488. doi: 10.1073/pnas.97.19.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hassan A, Markus HS. Genetics and ischaemic stroke. Brain. 2000;123 (Pt 9):1784–812. doi: 10.1093/brain/123.9.1784. [DOI] [PubMed] [Google Scholar]
- 36.Reiner AP, Siscovick DS, Rosendaal FR. Hemostatic risk factors and arterial thrombotic disease. Thromb Haemost. 2001;85:584–595. [PubMed] [Google Scholar]
- 37.Arrigo AP. Gene expression and the thiol redox state. Free Radic Biol Med. 1999;27:936–944. doi: 10.1016/s0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.