


Abstract
The genome sequence framework provided by the human genome project allows us to precisely map human genetic variations in order to study their association with disease and their direct effects on gene function. Since the description of tumor suppressor genes and oncogenes several decades ago, both germ-line variations and somatic mutations have been established to be important in cancer—in terms of risk, oncogenesis, prognosis and response to therapy. The Cancer Genome Atlas initiative proposed by the NIH is poised to elucidate the contribution of somatic mutations to cancer development and progression through the re-sequencing of a substantial fraction of the total collection of human genes—in hundreds of individual tumors and spanning several tumor types. We have developed the CancerGenes resource to simplify the process of gene selection and prioritization in large collaborative projects. CancerGenes combines gene lists annotated by experts with information from key public databases. Each gene is annotated with gene name(s), functional description, organism, chromosome number, location, Entrez Gene ID, GO terms, InterPro descriptions, gene structure, protein length, transcript count, and experimentally determined transcript control regions, as well as links to Entrez Gene, COSMIC, and iHOP gene pages and the UCSC and Ensembl genome browsers. The user-friendly interface provides for searching, sorting and intersection of gene lists. Users may view tabulated results through a web browser or may dynamically download them as a spreadsheet table. CancerGenes is available at http://cbio.mskcc.org/cancergenes.
INTRODUCTION
The completion of a high-accuracy sequence of the human genome will enable significant advances in our understanding of disease-related genetic variation, both somatic and germ-line. Researchers are already using the human genome to study sequence variation in cell populations containing normal or abnormal DNA. Some of these studies have been designed to test hypotheses, for example, through linkage analysis that are concerned with the sequence variations at one locus or a small group of loci. Other studies have been designed to catalog variations across a wide selection of loci or genes, without a particular hypothesis to test, such as the HapMap project (1). If the study focuses on genotyping samples using known variations, these genotyping approaches typically use microarray technology designed to detect hundreds of thousands of single nucleotide polymorphisms (SNPs), or PCR amplification using primers designed to test for a particular set of SNPs. However, when disease-causing somatic mutations are unknown, as is the current case for somatic mutations in nearly all types of cancer, re-sequencing is the state-of-the-art to discover new variations.
Traditional sequencing technology applied to re-sequencing a particular genomic region (such as the exons of a gene) may also be used to detect known mutations. However, because of its higher cost, re-sequencing is generally performed as a discovery tool to screen for new genomic sequence variations and mutations, including base substitutions, insertions and deletions. In particular, gene re-sequencing efforts have recently been undertaken largely to catalog synonymous (not leading to amino acid substitutions) or non-synonymous (leading to amino acid substitutions) substitutions in genetic diseases, such as cancer (2–5).
The Cancer Genome Atlas (TCGA) initiative proposed by the NIH is a large gene re-sequencing effort to take place over the next decade (NIH Press Release, December 13, 2005; available at http://cancergenome.nih.gov). TCGA will involve re-sequencing thousands of human genes from thousands of tumor samples. Without major breakthroughs in sequencing technology, this effort will not be able to re-sequence all genes and will be limited to particular tumor types. This large screening effort will naturally be followed by other smaller efforts undertaken by individual labs and small consortia to fill in the gaps, as well as to verify and validate putative variations on larger sample sizes. A number of these smaller projects will be targeted at particular genes and at particular tumors. In all of these projects, large and small, until sequencing costs drop by several orders of magnitude, funding limits will force decisions about which genes to re-sequence and in what order they will be examined. Therefore, selection and prioritization of genes for re-sequencing is a common first step that will be repeated for each project. With appreciable amounts of sequence and functional data available in public databases, and without tools to navigate these data, the gene selection process can become a painstaking task. The CancerGenes resource will keep up with the results produced by TCGA by utilizing the Catalogue of Somatic Mutations in Cancer (COSMIC) curated resource (6).
Our goal in creating the CancerGenes resource is to simplify the gene selection process commonly encountered in re-sequencing projects, and made difficult in large, geographically dispersed, collaborative groups. Our objective is to provide a resource that supports gene list storage, queries and comparisons. For example, a user of this resource could generate a superset of cancer-related genes listed in Hahn and Weinberg (7) and Vogelstein and Kinzler (8) by unioning these gene list sources, filter this list for tyrosine kinases and then intersect it with the user's own gene list. Because of our interests, we have initially focused on cancer genome re-sequencing projects, such as TCGA. A key requirement for rational, high quality gene selection and prioritization is aggregation of up-to-date gene-centric information, addition of information by domain experts and summarization. Therefore, we automated the data aggregation process, provided a mechanism to add information from domain experts, such as researchers with intimate knowledge of the importance of particular gene sets in certain disease processes, and wrote software to provide convenient web access to frequently updated information. In addition, since lists of genes are a common end-point of many high-throughput studies, such as gene expression profiling using microarrays, we believe this resource will be a useful adjunct to a wide range of cancer-relevant studies outside the scope of gene re-sequencing.
MATERIALS AND METHODS
CancerGenes pulls in gene-centric data from four main sources: NCBI Entrez Gene (9), Ensembl BioMart (10), supplementary data on active promoter regions from Kim et al. (11) and the Sanger Institute COSMIC (Table 1) (6). We use the term ‘gene-centric’ to describe data and resources whose organizing principle is the gene—i.e. the seminal data object is based upon the concept of the gene and their functionalities are based on this concept. In contrast, though it includes gene-centric data, the organizing principle of CancerGenes is based on list of genes and the list comparison operations.
Table 1.
CancerGenes data sources
| Data source | Data type | URL | Number of genes |
|---|---|---|---|
| NCBI Entrez Gene | Names, aliases, GO, gene structure | http://www.Ncbi.nih.gov/entrez/query.fcgi?db=gene | 39 250 |
| Ensembl BioMart | Gene structures, protein domains | http://www.ensembl.org/Homo_sapiens/martview | 20 676 |
| Kim et al. (11) supplementary data | Active TFIID-binding site coordinates | http://www.nature.com/nature/journal/v436/n7052/suppinfo/nature03877.html | 8914 |
| Sanger Institute COSMIC | Mutation frequencies and types, tissue distributions | http://www.sanger.ac.uk/genetics/CGP/cosmic/ | 1172 |
Our process uses all active human gene records in Entrez Gene as the seed data source, and then adds data from the three other sources. Literature source and annotation gene lists are then created from various literature and database sources and queries. These lists are functionally equivalent to database indices. Details of the current aggregation process are given below.
Retrieving all human genes from NCBI Entrez Gene
The NCBI Entrez eUtilities (eSearch, eFetch) provide a means to automate the initial retrieval and future updates of the gene records in the CancerGenes database (9). We retrieve gene report records by a single Entrez eSearch request, which returns numeric Entrez Gene identifiers, followed by a batched eFetch request, which given a list of Entrez Gene ids, returns data-containing gene records. We then parse out the following information from ASN.1 formatted records: Entrez Gene ID, gene name, description, aliases, organism, chromosome number, location, GO terms, gene size, mRNA length, mRNA exon count, peptide length, peptide exon count and transcript count. If more than one protein isoform is reported for a single gene, we use the information pertaining to the longest. If there are two or more protein isoforms of the same length, we use the transcript information for the longest mRNA. The data are then loaded into our MySQL database (see below).
BioMart queries for Ensembl gene structure and InterPro domain data
We retrieve the following gene structure data from the BioMart/EnsMart system (Table 2): coding DNA sequence (CDS) length, protein length and transcript count (10). We handle multiple isoforms as we do with Entrez Gene data (see above).
Table 2.
Ensembl BioMart query parameters for gene structure information
| Step | Field | Settings |
|---|---|---|
| 1. Dataset | Ensembl 39 | |
| Homo sapiens genes (NCBI36) | ||
| 2. Filter | Gene: ID List limit | EntrezGene ID(s) |
| 3. Output | Gene | Ensembl Gene ID |
| Ensembl Peptide ID | ||
| Ensembl CDS length | ||
| Ensembl Peptide length | ||
| Ensembl Transcript count | ||
| External references | EntrezGene ID | |
| Output format | Text, tab separated |
InterPro is a database of protein families, domains and functional sites (12). We retrieve InterPro descriptions from BioMart in a separate query (Table 3) for insertion into the MySQL database.
Table 3.
Ensembl BioMart query parameters for InterPro protein domain information
| Step | Field | Settings |
|---|---|---|
| 1. Dataset | Ensembl 39 | |
| Homo sapiens genes (NCBI36) | ||
| 2. Filter | Gene: ID List limit | EntrezGene ID(s) |
| 3. Output | Protein | InterPro description |
| External references | EntrezGene ID | |
| Output format | Text, tab separated |
Active human promoter sites
In 2005, Kim et al. (11) generated high-throughput experimental ChIP-on chip data using antibodies against the transcription complex component TFIID and reported active promoters (hg16, NCBI Build 34) in the human cell line IMR90 (ATCC). Using the RefSeq indexed results in supplementary Table 2 from that paper, we added these active promoter site coordinates to CancerGenes gene data. This hg16 coordinate data are linked to the UCSC Genome browser's LiftOver utility (http://genome.ucsc.edu/cgi-bin/hgLiftOver), which converts these hg16 coordinates to coordinates of more recent human genome builds. Experimental data on active promoter regions from other groups, as they become available, will be added using a similar process.
COSMIC mutation data from the Sanger Institute
Sanger Institute COSMIC is an ongoing curation effort sponsored by the Sanger Institute to collect mutation data from the scientific literature (6). As of this writing, it includes data on ∼3300 distinct mutations in over 1300 genes from more than 250 000 samples. We downloaded the June 7, 2006 release of COSMIC (v19) (latest release available at ftp://ftp.sanger.ac.uk/pub/CGP/cosmic), parsed files for mutation frequencies and tissue distributions on each of the catalogued genes for inclusion into the MySQL database.
Literature source gene lists
Through both manual and automated methods, we have produced four gene lists, which are based upon peer-reviewed publications that review cancer-related processes. Mitelman (13) reviewed common chromosome aberrations in cancer. Hahn and Weinberg (7) and Vogelstein and Kinzler (8) both review protein and gene pathways important in cancer development, maintenance and metastasis. Futreal et al. (4) performed a census of somatic mutation bearing genes related to cancer. We manually extracted gene symbols from the first three reviews and converted these to Entrez Gene ids. However, since the gene lists given in Futreal et al. formed the basis for the Sanger Institute's Cancer Gene Census (CGC) resource, we have downloaded the current gene list and have used that to populate this list in CancerGenes. All of these literature source lists appear in a multiple-select drop down menu in the web interface (see below) and may be unioned or intersected with each other and/or the annotation gene lists (see below). In Table 4 we indicate the number of genes from these Cancer review sources and compare them to the annotation gene lists.
Table 4.
CancerGenes cancer-related literature and annotation lists
| List type | Description | Number of lists | Number of genes |
|---|---|---|---|
| Cancer review | Peer-reviewed literature sources describing cancer pathways, recurrent aberrations and mutations | 4 | 400 |
| Cellmap.org | Cancer-related, human-curated pathways from Institute of Bioinformatics (Bangalore, India) under contract to MSKCC Computational Biology Center | 9 | 578 |
| Entrez query | Function-related queries to NCBI's Entrez Gene resource | 6 | 1691 |
| Sanger CGC | Cancer mutation categories from Sanger Institute's Cancer Gene Census | 7 | 344 |
Annotation gene lists
We have also derived gene lists from reports, studies and databases that have not undergone the stringent peer-review process; we call these ‘annotation’ gene lists and have separated these in the CancerGenes web interface (below). As of this writing, we have loaded three types of annotation gene lists, including curated cancer-related pathways (from cellmap.org), function-related Entrez Gene queries and Sanger CGC mutation categories (Table 4). Cellmap.org pathways were curated from the scientific literature by the Institute of Bioinformatics (Bangalore, India) under contract to Memorial Sloan-Kettering's Computational Biology Center (cBio) and are available through http://cancer.cellmap.org. We extracted protein RefSeq ids from these BioPAX formatted (http://biopax.org) pathways using custom scripts. We performed several function-based queries of Entrez Gene that are related to categories important in cancer, such as to retrieve all genes containing the tyrosine kinase domain, or oncogenes (Table 5). Finally, we downloaded the latest mutation data available from the CGC website (http://www.sanger.ac.uk/genetics/CGP/Census/) and separated gene lists based on different mutation types (i.e. amplification, frameshift mutation, germ-line mutation, large mutation, missense mutation, nonsense mutation, splicing mutation and translocation). For all of these lists, newline-delimited Entrez Gene ids were generated and subsequently loaded into our MySQL database (described below).
Table 5.
NCBI Entrez Gene active human gene queries performed to generate annotations lists
| List name | Entrez query |
|---|---|
| Oncogene | ‘oncogene’[All Fields] |
| Stability | ‘stability gene’[All Fields] |
| Tumor Suppressor | ‘tumor suppressor’[All Fields] |
| Protein Phosphatase | (cd00047 OR pfam04387 OR pfam00102 OR smart00404 OR smart00194 OR pfam01451 OR cd00115 OR smart00195 OR pfam00782 OR cd00127 OR pfam06617 OR cd01530) AND ‘homo sapiens’[ORGN] |
| Protein kinase | ‘protein kinase’[GO] OR cd00192 [Domain Name] OR ‘serine/threonine kinase’[GO] NOT pseudogene[All Fields] NOT hypothetical |
| Tyrosine kinase | cd00192[Domain Name] |
Database
We have stored all CancerGenes data and gene lists in a MySQL database. We process this data further and then store the results in one production table which is then used by the PHP front-end. We indicate relationships between records in different tables by Entrez Gene ID. Some relationships are one-to-one, and others many-to-one. For example, one Entrez Gene ID may have several corresponding RefSeq ids. We provide validated links to outside resources, including the Entrez Gene, UCSC Genome browser and iHOP (Table 6). An overview of the data sources for CancerGenes are diagrammed in Figure 1.
Table 6.
CancerGenes links to other resources
| Resource | Description | Number of links |
|---|---|---|
| Entrez Gene | Database of aggregated gene-centric resource maintained by NCBI, NIH | 39 250 |
| UCSC Genome browser | Genome browser and database of position-based genome features maintained by UCSC | 32 348 |
| iHOP | Database of concurring gene and protein names in scientific literature abstracts found in PubMed | 22 996 |
| Ensembl Genome browser | Genome browser and database of position-based genome features maintained by EMBL and Sanger Institute | 20 676 |
| COSMIC | Database of somatic mutations in cancer curated from the scientific literature maintained by Sanger Institute | 1172 |
Figure 1.
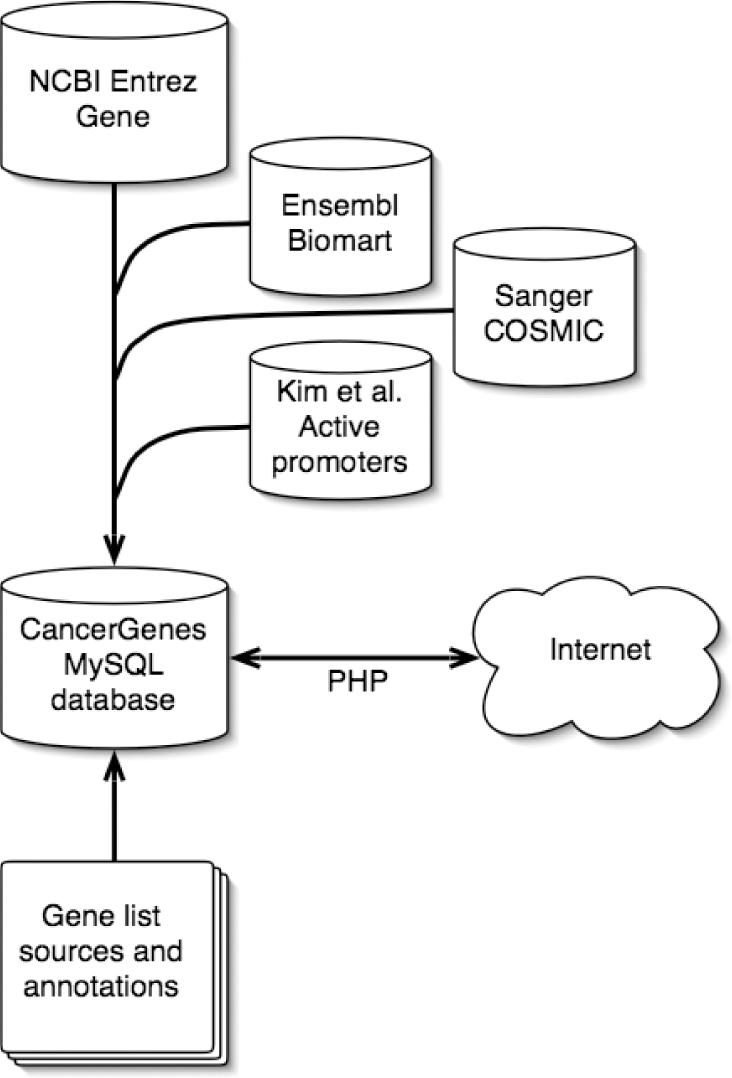
Overview of the data sources for CancerGenes.
Web interface
CancerGenes is available on the World Wide Web at http://cbio.mskcc.org/cancergenes. The CancerGenes web interface is built in PHP and is running under SuSe Linux. The web interface consists of a query section, result summary/export section, and the resultant gene list (Figure 2). The query section allows choices of sort key (i.e. number of sources, number of annotations, COSMIC % mutation, gene symbol, Entrez Gene ID, chromosome, location, number of samples and number of mutations) and sort order. In addition, users may optionally select multiple literature source gene lists and annotation gene lists, as well as upload an arbitrary list of Entrez Gene ids of interest. Union (OR) and intersection (AND) set operations are allowed among and between lists. A gene symbol or Entrez Gene ID can be entered and used to highlight entries in the list based on characters in the gene names, gene symbol or aliases (Figure 3). This feature allows users to quickly find particular genes in the resultant list. Users may choose to display 10 (default), 100 or 1000 tabulated gene data at a time, and may follow links to previous or next pages.
Figure 2.

Screenshot of the CancerGenes web interface at http://cbio.mskcc.org/cancergenes using Apple's Safari web browser.
Figure 3.

Query results for ‘erbb2.’ (A) Query box and result summary with ‘jump to’ buttons and (B) subset of a retrieved gene list showing the highlighted gene ERBB2.
Users perform a query by clicking on the ‘Go’ button, and the following is displayed: total number of genes, page numbers and ‘jump to’ buttons for any highlighted genes, an export button, buttons to page forward and backward, and the tabulated results. Users may click on the export button to download the query results in tab-delimited spreadsheet, readable by any of the common spreadsheet programs.
RESULTS
We have produced a gene list-centric resource called CancerGenes that includes a wide variety of gene-centric data, literature sources and gene lists created by domain experts. The software provides user functionalities focused on user interest driven selection and prioritization of target genes for re-sequencing projects and design of functional experiments.
We have designed CancerGenes for the following primary use cases:
- Retrieve data on genes of interest by
- Uploading a list of Entrez Gene ids
- Selecting preloaded gene list
- Generate intersection or union of lists
Sort genes by occurrence in various published sources
Estimate re-sequencing cost by retrieving the number of exons
Find a gene in the list
Download a list of selected genes and associated data
Table 7 demonstrates some of the pair-wise set operations possible with the preloaded lists, and characterizes the literature source and annotation lists preloaded into CancerGenes.
Table 7.
Numbers of genes in pair-wise set intersections and unions of CancerGenes literature source and annotation lists
 |
The diagonal cells (yellow) contain the number of genes in each list (given in the head row and column). The uppermost, left-hand cell (pink) contains the total number of genes on one or more list. Numbers above and to the right of the diagonal are the number of genes in a pair-wise set intersection (overlap) between two lists (one in the top row, the other in the leftmost column). Numbers below and to the left of the diagonal are the number of genes resulting from a pair-wise set union. Percent overlap is given in parentheses, and is the number of genes in the intersection of two lists divided by the number of genes in the union of two lists. Most overlaps are 10% or less, with the exception of the overlap of Cancer review and Sanger CGC, which is 83%.
DISCUSSION
CancerGenes is a gene list-centric resource, with its intended niche being cancer-associated gene selection and prioritization through aggregation of relevant information, logical operations on lists, selection options and summary presentation. This is in contrast to the large number of bioinformatics resources that support gene-centric queries and retrieve aggregated gene-centric information, or support genome sequence browsing. In fact, we have retrieved data from a number of these gene-centric resources (e.g. NCBI Entrez Gene, Ensembl BioMart, Sanger COSMIC). However, as CancerGenes's intended user population deals in gene lists, rather than individual gene records, we have focused on gene list-based functionalities, such as union and intersection set operations, rather than single gene-based queries. In addition, we have preloaded lists of genes that we anticipate our target audience will find useful, including list provided by domain experts. In addition to the list-centric operations, as all active human genes from Entrez Gene are included in CancerGenes, and as it supports a number of use cases, projects that requires gene-centric data may also find this resource useful.
Because of our interest in cancer, we have loaded cancer-focused data and lists into CancerGenes. It is our desire to support cancer genome sequencing projects, such as the TGCA with this resource. However, there is no reason why CancerGenes could not support other re-sequencing projects, which target other diseases with a genetic component.
Our intent with the four types of preloaded gene lists—Cancer review, Cellmap.org Pathways, Entrez query and Sanger CGC—was to cover all genes likely to be related to cancer through mutation. Table 7 shows the sizes and numbers of genes resulting from set intersection (overlap) and union operations between these four types of preloaded gene lists. Most relative overlaps between lists (i.e. intersection size divided by union size) are ∼10%, with the exception of the overlap between Cancer review and Sanger CGC, which is 83%. We are not surprised by these results, because of our criteria used to generate each list (see Materials and Methods and Tables 4 and 5).
There are a total of 2274 unique genes in our preloaded lists, which means annotation coverage of all 39 250 genes in CancerGenes is 5.8%. If the Sanger CGC list is considered definitive in regard to current state of knowledge about mutated genes in cancer, then 15% (344/2274) of this 5.8% is known to be due to mutation. Confirmation of a mutation-based association to cancer for the remaining 85% of genes in our preloaded lists (and ∼5% of all genes), will have to await the conclusion of the many cancer genome sequencing initiatives.
CancerGenes consolidates information from several gene-centric resources of use to those embarking on gene re-sequencing efforts large or small. Our focus in developing this resource has been on simple gene list-centric query tools and the tabular display of information. The simple and flexible architecture we have developed for storing and annotating gene lists will allow us to keep pace with the currently rapid development of gene lists, sets and signatures, and provide an up-to-date resource for our users.
FUTURE DEVELOPMENTS
We have planned several developments to make CancerGenes more useful to cancer genome projects, as well as other gene re-sequencing initiatives that aim to survey somatic or germ-line genetic variation of genes. These plans include adding gene-centric data, such as PCR primer and amplicon predictions, and a re-sequencing ‘difficulty’ score (perhaps based on regional genomic GC-content variations, amplicon sizes and presence of pseudogenes), We also plan to add preloaded gene lists for genetic diseases other than cancer (e.g. Kegg pathways), and adding functionalities, such as sequence retrievals and selected field downloads. Finally, we plan to collaborate with additional disease domain experts for inclusion of particular gene lists or particular disease annotations into the database underlying CancerGenes.
Acknowledgments
We gratefully acknowledge the efforts of Marc Ladanyi and Doron Betel in the critical review of a preliminary implementation of CancerGenes and their helpful suggestions. In addition, we thank Marc Ladanyi for contributing his domain knowledge. Funding to pay the Open Access publication charges for this article was provided by Bristol-Myers Squibb Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bignell G., Smith R., Hunter C., Stephens P., Davies H., Greenman C., Teague J., Butler A., Edkins S., Stevens C., et al. Sequence analysis of the protein kinase gene family in human testicular germ-cell tumors of adolescents and adults. Genes Chromosomes Cancer. 2006;45:42–46. doi: 10.1002/gcc.20265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies H., Hunter C., Smith R., Stephens P., Greenman C., Bignell G., Teague J., Butler A., Edkins S., Stevens C., et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005;65:7591–7595. doi: 10.1158/0008-5472.CAN-05-1855. [DOI] [PubMed] [Google Scholar]
- 4.Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nature Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stephens P., Edkins S., Davies H., Greenman C., Cox C., Hunter C., Bignell G., Teague J., Smith R., Stevens C., et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nature Genet. 2005;37:590–592. doi: 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- 6.Forbes S., Clements J., Dawson E., Bamford S., Webb T., Dogan A., Flanagan A., Teague J., Wooster R., Futreal P.A., et al. COSMIC 2005. Br. J. Cancer. 2006;94:318–322. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hahn W.C., Weinberg R.A.. Modeling the molecular circuitry of cancer. Nature Rev. Cancer. 2002;2:331–341. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- 8.Vogelstein B., Kinzler K.W. Cancer genes and the pathways they control. Nature Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 9.Wheeler D.L., Barrett T., Benson D.A., Bryant S.H., Canese K., Chetvernin V., Church D.M., DiCuccio M., Edgar R., Federhen S., et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2006;34:D173–D180. doi: 10.1093/nar/gkj158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasprzyk A., Keefe D., Smedley D., London D., Spooner W., Melsopp C., Hammond M., Rocca-Serra P., Cox T., Birney E. EnsMart: a generic system for fast and flexible access to biological data. Genome Res. 2004;14:160–169. doi: 10.1101/gr.1645104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim T.H., Barrera L.O., Zheng M., Qu C., Singer M.A., Richmond T.A., Wu Y., Green R.D., Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulder N.J., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bradley P., Bork P., Bucher P., Cerutti L., et al. InterPro, progress and status in 2005. Nucleic Acids Res. 2005;33:D201–D205. doi: 10.1093/nar/gki106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitelman F. Recurrent chromosome aberrations in cancer. Mutat. Res. 2000;462:247–253. doi: 10.1016/s1383-5742(00)00006-5. [DOI] [PubMed] [Google Scholar]